Autoantikörper bei Hodgkin-Lymphom-Patienten mit und ohne begleitende Paraneoplastische Neurologische Syndrome
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Autoantikörper bei Hodgkin-Lymphom-Patienten
mit und ohne begleitende Paraneoplastische
Neurologische Syndrome
Inauguraldissertation
zur Erlangung des Grades eines Doktors der Medizin
des Fachbereichs Medizin
der Justus-Liebig-Universität Gießen
vorgelegt von
Julia Huwe, geb. Scharfenberger
aus Worms
Gießen 2009Aus dem Zentrum für Neurologie und Neurochirurgie
Klinik für Neurologie
der Universitätsklinikum Gießen und Marburg GmbH, Standort Gießen
Leiter: Prof. Dr. med. M. Kaps
Gutachter: PD Dr. Blaes
Gutachter: PD Dr. Rummel
Tag der Disputation: 28.07.2009Inhaltsverzeichnis
Zusammenfassung .......................................................................................................i
Summary .................................................................................................................... iii
1. Einleitung................................................................................................................ 1
1.1 Morbus Hodgkin ............................................................................................... 1
1.2 Paraneoplastische neurologische Syndrome (PNS)......................................... 5
1.3 Paraneoplastische neurologische Syndrome bei M. Hodgkin..........................12
1.4 Fragestellung...................................................................................................15
2. Patienten und Methoden........................................................................................16
2.1 Patienten .........................................................................................................16
2.1.1 Patienten mit M. Hodgkin (Hodgkin-Gruppe)............................................16
2.1.2 Hodgkinpatienten mit PNS (Hodgkin/Para-Gruppe) .................................16
2.1.3 Gesunde Kontrollen (Kontroll-Gruppe) .....................................................17
2.2 Methoden ........................................................................................................17
2.2.1 Zellkultur...................................................................................................17
2.2.2 Autoantikörperdiagnostik..........................................................................18
2.2.3 Isolierung von Auerbachplexus ................................................................23
2.3 Statistik............................................................................................................24
3. Ergebnisse.............................................................................................................25
3.1 Klinische Daten ...............................................................................................25
3.1.1 Fallbeschreibungen der 5 Hodgkin-Patienten mit PNS (Hodgkin/Para-
Gruppe) .............................................................................................................25
3.1.2 Patienten mit M. Hodgkin (Hodgkin-Gruppe)............................................29
3.2 Autoantikörperergebnisse................................................................................30
3.2.1 Immunfluoreszenz ....................................................................................30
3.2.2 Durchflußzytometrie ................................................................................40
3.2.3 Western Blot.............................................................................................43
4. Diskussion .............................................................................................................48
4.1 Antineuronale Autoantikörper bei PNS ............................................................48
4.2 Antineuronale Autoantikörper bei M. Hodgkin .................................................49
4.3 Oberflächen-bindende Autoantikörper bei M. Hodgkin....................................53
4.4 Antinukleäre Antikörper bei M. Hodgkin ..........................................................54
5. Literaturverzeichnis ...............................................................................................57
6. Anhang ..................................................................................................................68Zusammenfassung Einleitung: Paraneoplastische neurologische Syndrome (PNS) sind Erkrankungen, die mit einem Tumor assoziiert sind, aber nicht durch diesen selbst oder seine Metastasen hervorgerufen werden. Es wird eine autoimmune Genese der PNS vermutet, nachdem mehrere onkoneuronale Autoantikörper identifiziert werden konnten. Beim Hodgkin-Lymphom wurden anti-Tr-Autoantikörper im Zusammenhang mit paraneoplastischer Kleinhirndegeneration (PCD) identifiziert. Antineuronale Autoantikörper konnten niedrigtitrig ebenfalls bei Tumorpatienten ohne PNS festgestellt werden und scheinen mit einer verbesserten Prognose einherzugehen. Wir untersuchten Hodgkin-Lymphom-Patienten ohne und mit PNS auf das Vorkommen antineuronaler Autoantikörper und ob bei vorhandenen Autoantikörpern daraus eine verbesserte Prognose resultiert. Methoden: Die Seren von 67 Hodgkin-Patienten ohne bekanntes PNS und 5 Seren von Hodgkin-Patienten mit bekanntem PNS (3 PCD; 1 limb. Enzephalitis; 1 Polyneuropathie) wurden untersucht. Seren von gesunden Erwachsenen dienten als Kontrollen. Die Autoantikörperdiagnostik wurde mittels Immunfluoreszenz, Western Blot und Durchflusszytometrie durchgeführt. Resultate: 4 der 5 Hodgkin-Patienten mit PNS wiesen antineuronale Autoantikörper auf. 2 PCD-Patienten waren anti-Tr positiv. Die Patienten mit Polyneuropathie und limb. Enzephalitis zeigten beide einen atypischen neuronalen Antikörper. Bei den Hodgkin-Patienten ohne PNS wiesen 6/67 (8.9%; Kontrollen: 2.2%; n.s.) einen antineuronalen Antikörper auf. Davon könnte einer ein anti-Tr Antikörper sein. 10/67 (14.9%) zeigten einen antinukleären Antikörper (Kontrollen: 23.9%; n.s.). Durchflußzytometrisch hatten mehr Hodgkin-Patienten (29.9%; 20/67) einen Antikörper gegen ubiquitäre Oberflächenepitope im Vergleich zu den Kontrollen (8.7%; 4/46) (p
Antikörpern (wie z.B. für Anti-Hu bekannt), konnte in dieser Studie nicht festgestellt
werden. Als weitere Tumor-assoziierte Antikörper wurden antinukleäre Antikörper
(ANA) in vorhergehenden Studien beschrieben. Diese können ebenfalls mit einer
verbesserten Prognose einhergehen. Auch in dieser Studie zeigten sich bei den
Hodgkin-Patienten ohne PNS antinukleäre Antikörper und auch Antikörper gegen
ubiquitäre Oberflächenepitope. Es wurde auch für diese Antikörper keine Korrelation
zu einem prognostischen Effekt festgestellt. Die erhöhte unspezifische AK-
Produktion könnte speziell beim Hodgkin-Lymphom auch durch eine Dysregulation
der entarteten B-Zellen verursacht sein, wofür allerdings keine weiteren Hinweise
gefunden werden konnten.
iiSummary Introduction: Paraneoplastic neurologic syndromes (PNS) are disorders associated with cancer which are not caused by a local effect of the tumour or its metastases. In supposition of autoimmunity, different onconeuronal autoantibodies have been found. Hodgkin’s disease with paraneoplastic cerebellar degeneration (PCD) is associated with the anti-Tr-antibody. Low titers of antineuronal autoantibodies could also be found in cancer-patients without PNS. These patients had a better clinical outcome. We tested sera of patients with hodgkin’s disease with and without PNS and looked for antineuronal autoantibodies and an association to a better clinical outcome. Methods: 67 sera of patients with hodgkin’s disease without PNS and 5 sera of patients with hodgkin’s disease with PNS (3 PCD, 1 limbic enzephalitis, 1 polyneuropathy) were tested. Sera of healthy adults were used as controls. Autoantibodies were detected by indirect immunofluorescence, western blot analysis and flow cytometry. Results: 4/5 patients with hodgkin’s disease and PNS had an antineuronal antibody, 2 with PCD had the anti-Tr-antibody. These with polyneuropathy and limbic encephalitis had both an atypical antineuronal antibody. 6/67 (8.9%) patients with hodgkin’s disease without PNS had an antineuronal antibody (controls: 2.2%; n.s.), 1 could be anti-Tr. 10/67 (14.9%) were ANA-positive (controls: 23.9%; n.s.). 20/67 (29.9%) patients had an antibody against indifferent epitope-antigenes in flow cytometry but only 4/46 (8.7%) of the healthy controls (p
malignant B-cells may cause a dysregulation of antibody production and a higher
incidence but there was no further indication for this.
iv1. Einleitung
1.1 Morbus Hodgkin
Der Morbus Hodgkin gehört zu den malignen Lymphomen. Er wird synonym auch als
Hodgkin-Lymphom, Lymphogranulomatose oder Hodgkin’s disease bezeichnet.
Erstmals beschrieben wurde er 1832 von Thomas Hodgkin (1798-1866) (Hodgkin
1832). Carl Sternberg und Dorothy Reed beschrieben unabhängig voneinander die
charakteristischen Zellen, die danach als Sternberg-Reed-Zellen bezeichnet wurden
(Sternberg 1898, Reed 1902).
Die Inzidenz liegt bei 2-4:100 000 pro Jahr. Männer sind im Verhältnis 5:3 häufiger
betroffen als Frauen. Die Altersverteilung zeigt einen zweigipfligen Verlauf. Der erste
Gipfel liegt in den industrialisierten Ländern um das 30. Lebensjahr, in
Entwicklungsländern bereits im Kindesalter. Der zweite Gipfel findet sich bei einem
Alter von 70 Jahren (Wolf et al. 1999).
Das charakteristische Merkmal des M. Hodgkin ist eine kleine klonale Population von
großen Zellen. Besitzt diese Zelle nur einen Kern, wird sie als Hodgkin-Zelle
bezeichnet, ist sie multinukleär, wird sie Reed-Sternberg-Zelle genannt (Küppers et
al. 1998, Thomas et al. 2004).
Umgeben werden diese Zellen von nichtmalignen T-Lymphozyten (hauptsächlich), B-
Lymphozyten, Histiozyten, Eosinophilen, Neutrophilen und Plasmazellen (Burke
1992). Das Verhältnis von HRS-Zellen zu den umgebenden, nichtmalignen Zellen
liegt bei 1:1000 (Wolf et al. 1999, Cossmann 2001).
Früher wurde zur histologischen Einteilung die Rye-Klassifikation angewendet, seit
2001 ist die neue WHO-Klassifikation für Lymphome gültig (Chan 2001). Danach
werden verschiedene Formen des M. Hodgkin unterschieden (Tab. 1).
1Tabelle 1: WHO-Klassifikation des Hodgkin-Lymphoms
1. Klassisches Hodgkin-Lymphom - Lymphozytenreich
- Nodulär Sklerosierend
- Mischzellig
- Lymphozytenarm
2. Nodulär Lymphozytenreiches Hodgkin-Lymphom
Die eigenständige Betrachtung des Nodulär Lymphozytenreichen Hodgkin-
Lymphoms ergab sich durch den Nachweis von morphologischen Unterschieden
zwischen den HRS-Zellen des klassischen M. Hodgkin und denen des Nodulär
Lymphozytenreichen M. Hodgkin. Erstere tragen als Oberflächenantigene die
Merkmale CD30, CD15 und selten CD20 und entstehen aus B-Lymphozyten des
Keimzentrums/Follikels, haben aber die Fähigkeit zur Immunglobulinbildung verloren
(Küppers et al. 1998, Küppers et al. 2002). Bei den letzteren dominiert CD20. Diese
Zellen entstehen ebenfalls aus B-Lymphozyten des Keimzentrums, die Fähigkeit zur
mRNA-Bildung für Antikörper ist aber noch vorhanden (Küppers et al. 1998, Chan
2001). In seltenen Fällen gibt es eine Variante, wobei die HRS-Zellen beim
klassischen M. Hodgkin aus T-Lymphozyten generiert sind (Küppers et al. 1998).
Der Mutationsvorgang ist bisher unbekannt. Ebenfalls unbekannt ist der Vorgang,
wie diese mutierten Zellen der Apoptose entgehen, da normalerweise Zellen im
Keimzentrum mit verlorener Fähigkeit zur Antikörper-Bildung eliminiert werden
(Cossmann 2001, Jarrett 2002).
Als eine Theorie des Mutationsvorgangs wird der Einfluss von Viren, besonders des
Epstein-Barr-Virus (EBV), diskutiert. EBV als onkogenes Agens ist z.B. auch in
Zusammenhang mit dem isolierten cerebralen Lymphom bei HIV-Patienten bekannt.
Dort konnte in fast allen Zellen aktives EBV-Genom nachgewiesen werden
(Camilleri-Broet et al. 1997).
Im Genom der HRS-Zellen konnte in den Industriestaaten bei ca. 50% und in den
Entwicklungsländern bei über 90% der Erkrankungen DNS des Epstein-Barr-Virus
nachgewiesen werden (Wolf et al. 1999, Gandhi et al. 2004). Diese Zellen
exprimieren als Oberflächenmerkmal EBV-codierte Proteine: Latent membrane
protein-1 (LMP1), Epstein-Barr nuclear antigen-1 (EBNA1) und Latent membrane
protein-2 (LMP2) (Herbst 1996, Jarrett 2002). Diese könnten durch Veränderung der
2Interaktionen im Keimzentrum eine Rolle im Überleben dieser Zellen spielen (Jarrett
2002). EBV-positive Fälle kommen besonders häufig bei Kindern, älteren
Erwachsenen (>45Jahre) und beim Mischzelligen M. Hodgkin vor (Gandhi et al.
2004).
Bei einer Untersuchung zum Zusammenhang zwischen M. Hodgkin und dem
Masernvirus konnten bei der Hälfte der Fälle eine Bindung von anti-Masern-
Antikörpern an HRS-Zellen nachgewiesen werden (Benharroch et al. 2004). Die
klinisch-pathologischen Zusammenhänge werden noch diskutiert, wobei in einer
anderen Studie keine Zusammenhänge zwischen einer Masern-Infektion und dem
gehäuften Auftreten von M. Hodgkin festzustellen war (Wilson et al. 2007).
Als erstes Leitsymptom eines M. Hodgkin gilt die schmerzlose
Lymphknotenschwellung, meist zervikal, seltener auch mediastinal, axillär oder
inguinal. Zusätzliche Symptome sind häufig Leistungsverlust/Müdigkeit, Fieber
>38°C, Nachtschweiß und Gewichtsverlust von >10% des Körpergewichts innerhalb
von 6 Monaten.
In der Regel beginnt das Hodgkin-Lymphom in einer Lymphknotengruppe und breitet
sich zuerst lymphogen, später auch hämatogen aus. Letztlich kommt es zu einer
Systemerkrankung, die sich auch in extralymphatischen Organen manifestiert.
Zur Diagnose und histologischen Einteilung wird eine Extirpation/Biopsie der
vergrößerten oder verdächtigen Lymphknoten durchgeführt.
Darüber hinaus wird ein Staging durchgeführt. Dazu müssen die befallenen
Lymphknoten und/oder die Hodgkinherde außerhalb von Lymphknoten gefunden
werden. Je nach Anzahl und Lokalisation wird dann das Krankheitsstadium
festgelegt (Tab. 2) (Wolf et al. 1999).
3Tabelle 2: M. Hodgkin – Stadieneinteilung nach der (modifizierten) Ann-Arbor-
Klassifikation
Stadium Merkmale
I Befall einer einzigen Lymphknotenregion (I/N) oder Vorliegen eines
einzigen extranodalen Herdes (I/E)
II Befall von 2 oder mehr Lymphknotenregionen auf einer Seite des
Zwerchfells (II/N) oder Vorliegen lokalisierter extranodaler Herde mit
Befall einer oder mehrerer Lymphknotenregionen auf einer Seite des
Zwerchfells (II/E)
III Befall von 2 oder mehr Lymphknotenregionen beiderseits des
Zwerchfells (III/N) oder Befall lokalisierter extranodaler Herde und
Lymphknoten beiderseits des Zwerchfells (III/E)
III1: Subphrenisch beschränkt auf Milz und/oder zöliakale/portale LK
III2: Subphrenisch unterhalb des Truncus coeliacus
IV Disseminierter Befall eines oder mehrerer extralymphatischer Organe
mit oder ohne Lymphknotenbefall.
Zusatz: A: Ohne Allgemeinsymptome
B: Mit Allgemeinsymptomen (z.B. Fieber, Gewichtsverlust)
Als Therapieziel wird die komplette Remission angestrebt. Die Behandlung erfolgt
meist im Rahmen kontrollierter klinischer Studien je nach Stadium und Klassifikation
mit intensivierter Polychemotherapie in Kombination mit Radiotherapie (Diehl et al.
2004). Ein anderer Ansatz stellt die Immuntherapie dar. Dabei will man z. B.
cytotoxische T-Zellen gegen das latente EBV-Antigen in den Sternberg-Reed-Zellen
einsetzen. Diese Methode ist noch in der Erprobung (Übersicht in (Wiedemann et al.
2002)). Die Prognose ist unterschiedlich, je nach Stadium und Vorliegen von
weiteren Symptomen. Durch die Risiko-adaptierte Behandlung konnten sehr gute
Überlebensraten erreicht werden (Diehl et al. 2004). Die 5-Jahres-Überlebensrate
liegt zur Zeit zwischen 80 und 95%. Diese insgesamt günstige Prognose wird aber
auch durch die Langzeittoxizität der Radio- und Chemotherapie, die zu
Zweitneoplasien führen kann, getrübt. Die häufigsten soliden Zweittumoren (relatives
Risiko: 2.4%) sind Lungenkarzinome, kolorektale Karzinome und Mammakarzinome
(Behringer et al. 2004).
41.2 Paraneoplastische neurologische Syndrome (PNS)
Unter Paraneoplasien versteht man allgemein Syndrome, die mit einem Tumor
assoziiert sind, jedoch nicht durch seine lokale Wirkung, seine Metastasen oder die
angewendete Therapie und deren Nebenwirkungen entstehen („the remote effect of
cancer“) (Henson 1982).
Paraneoplastische neurologische Syndrome sind erst in den letzten Jahren
zunehmend in den Mittelpunkt des wissenschaftlichen Interesses gerückt. Erstmals
beschrieb Denny-Brown 2 Patienten mit sensorischen Symptomen, bei denen post-
mortem ein bronchogenes Karzinom entdeckt wurde (Denny-Brown 1948). Später
veröffentlichte Wilkinson 5 Fälle von Tumor-assoziierter sensorischer
Neuromyopathie (Wilkinson 1964) und fand 1965 mit Zeromski bei diesen Patienten
einen antineuronalen Antikörper in der indirekten Immunfluoreszenz
(Wilkinson/Zeromski 1965).
Insgesamt leiden wenige Tumorpatienten an einer Paraneoplasie (weniger als 15%).
Es wurden schon annähernd alle Organe als Ziel von Paraneoplasien beschrieben,
die meisten finden sich im endokrinen System, im Nervensystem, Hämatologisch
oder Kutan (Übersicht in (Agarwala 1996)). Paraneoplastische neurologische
Syndrome kommen insgesamt mit einer Häufigkeit zwischen 0.01-2% aller
Tumorpatienten vor und sind meistens mit einem kleinzelligen Bronchialkarzinom,
gynäkologischen Tumoren oder Lymphomen assoziiert (Grisold et al. 1995, Blaes
2002). Der Zeitpunkt des Auftretens ist variabel und kann dem Tumor vorangehen,
gleichzeitig mit ihm auftreten oder nach der Tumorentdeckung in Erscheinung treten
(Agarwala 1996). Dabei gehen 50-60% der PNS der Entdeckung des Tumors voraus
(Dropcho 1995, Blaes 2002).
Paraneoplastische neurologische Syndrome können im gesamten zentralen und
peripheren Nervensystem vorkommen (Tab. 3). Außerdem können mehrere
Syndrome gleichzeitig vorliegen, z.B. bei paraneoplastischer Enzephalomyelitis mit
sensorischer Neuronopathie (Übersicht in (Posner/Dalmau 2000)).
5Tabelle 3: Paraneoplastische neurologische Syndrome
Fett: klassische paraneoplastische neurologische Syndrome nach Graus et al. 2004; ZNS: Zentrales
Nervensystem; PNS: Peripheres Nervensystem
ZNS PNS Neuromuskulär/ Muskel
• Enzephalomyelitis • Motorische • Myasthenia gravis
• Limbische Neuropathie • Lambert-Eaton
Enzephalitis • Sensorische myasthenes
• Hirnstammenzephalitis Neuropathie Syndrom
• Paraneoplastische • Autonome • Neuromyotonie
Kleinhirndegeneration Neuropathie/ • Dermatomyositis
• Opsoklonus- Gastrointestinale
Myoklonus-Syndrom Pseudoobstruktion
• Stiff-Person-Syndrom • Sensomotorische
Neuropathie
• Guillain-Barré-
Syndrom
• Paraneoplastische
Retinopathie
2004 wurden von der Arbeitsgruppe um Graus diagnostische Kriterien festgelegt,
welche die Diagnose eines sicheren oder möglichen PNS ermöglichen. Diese
berücksichtigen das neurologische Syndrom, den Tumor und onkoneuronale
Antikörper. (Tab. 4)
Dabei wurden von der Autorengruppe die PNS in klassische und nicht-klassische
paraneoplastische neurologische Syndrome aufgeteilt (Tab. 3) (Graus et al. 2004).
6Tabelle 4: Diagnostische Kriterien für paraneoplastische neurologische Syndrome
Sichere PNS:
1. Ein klassisches Syndrom mit einem Tumor, welcher innerhalb von 5 Jahren
nach Diagnose der neurologischen Erkrankung in Erscheinung tritt.
2. Ein nicht-klassisches Syndrom, das nach der Tumorbehandlung (ohne
begleitende Immuntherapie) verschwindet bzw. sich signifikant verbessert.
Vorrausgesetzt, dieses Syndrom neigt nicht zu spontaner Remission.
3. Ein nicht-klassisches Syndrom mit onkoneuronalem Antikörper und einem
Tumor, welcher innerhalb von 5 Jahren nach Diagnose der neurologischen
Erkrankung in Erscheinung tritt.
4. Ein neurologisches Syndrom (klassisch oder nicht) mit gut charakterisierten
onkoneuronalen Antikörpern (anti-Hu, -Yo, -CV2, -Ri, -Ma2 oder -
Amphiphysin) und keinem Tumor.
Mögliche PNS:
1. Ein klassisches Syndrom, kein onkoneuronaler Antikörper, kein Tumor, aber
ein hohes Risiko an einem zugrundeliegenden Tumor zu leiden.
2. Ein neurologisches Syndrom (klassisch oder nicht) mit teilweise
charakterisiertem onkoneuronalem Antikörper und keinem Tumor.
3. Ein nicht-klassisches Syndrom, keine onkoneuronalen Antikörper und ein
Tumor, welcher innerhalb von 2 Jahren nach Diagnose in Erscheinung tritt.
Die Entstehung einer Paraneoplasie generell kann verschiedene Ursachen haben.
Eine Möglichkeit besteht in der Hormonproduktion von neuroendokrin differenzierten
Tumoren. So führt zum Beispiel eine Produktion von Adrenocorticotropem Hormon
(ACTH) beim kleinzelligen Bronchialkarzinom zu einem paraneoplastischen Cushing-
Syndrom (Übersicht in (Terzolo et al. 2001)).
Die Pathogenese der paraneoplastischen neurologischen Syndrome ist noch nicht
sicher geklärt, es finden sich jedoch deutliche Hinweise auf eine
Autoimmunpathogenese. Dabei wird davon ausgegangen, dass Tumor und Neurone
identische Antigene exprimieren. Gegen diese “onkoneuronalen“ Antigene wird dann
eine Immunreaktion generiert. Dadurch kommt es zur klinischen Manifestation des
paraneoplastischen Syndroms (Dropcho 1998, Posner/Dalmau 2000).
Die Art der autoimmunen Reaktion wird kontrovers diskutiert. Es werden zelluläre
(z.B. zytotoxische T-Zellen) und humorale (Autoantikörper) Auslösungsmechanismen
7vorgeschlagen. Tatsächlich wurden verschiedene Autoantikörper gegen neuronale
Strukturen im Serum und/oder Liquor von Patienten mit Paraneoplasien gefunden
(Tab. 5). Mittlerweile konnten für einen großen Teil von ihnen auch die Antigene in
Nervensystem und Tumorgewebe identifiziert werden (Übersicht in (Blaes 2002)).
Tabelle 5: PNS, assoziierte Autoantikörper und assoziierte Tumore
Benennung der Antikörper nach Indexpatienten: Hu, CV2, Yo, Tr, Ri, Ma; SCLC: Small cell lung
cancer (Kleinzelliges Bronchialkarzinom); ANA: Antinukleärer Antikörper; ANNA: Antineuronal
nukleärer Antikörper; AchR: Acetylcholinrezeptor; VGCC: Voltage gated calcium channel
(spannungsabhängiger Kalziumkanal); GAD: Glutamatdecarboxylase; PCA: Purkinjezell Antikörper;
PNS Autoantikörper Assozierte Tumore
Paraneoplastische Neuropathie Anti-Hu Verschiedene (meist SCLC)
ANA Verschiedene (meist gynäk.)
ANNA-3 SCLC
Anti-CV2 Lunge
Paraneoplastische Anti-Hu SCLC
Kleinhirndegeneration Anti-ANNA3 SCLC
Anti-Yo Ovar, gynäkologische
Anti-Tr Morbus Hodgkin
Opsoclonus-Myoclonus Syndrom Anti-Ri Brustdrüse
Anti-Hu/atypisch Neuroblastom, SCLC
Limbische Enzephalitis Anti-Hu SCLC
Anti-Ma Testis
Anti-PCA-2 SCLC
Paraneoplastische Retinopathie Anti-Recoverin SCLC, andere
Gastrointestinale Anti-Hu SCLC
Pseudoobstruktion
Stiff-man Syndrom Anti-GAD
Anti-Amphiphysin Brustdrüse
Myasthenia gravis Anti-AchR
Anti-Titin Thymom
LEMS Anti-VGCC SCLC
8Als erster paraneoplastischer Autoantikörper wurde z.B. 1985 anti-Hu beschrieben
(Graus et al. 1985). Anti-Hu ist der am häufigsten gefundene Antikörper bei PNS und
ist meist mit einem kleinzelligen Bronchialkarzinom und einer paraneoplastischen
Enzephalomyelitis oder der subakuten sensiblen Neuropathie (PEM/SSN-
Paraneoplasie) assoziiert (Graus et al. 2001, Sillevis Smitt et al. 2002).
Bei etwa 2/3 aller Patienten mit paraneoplastischen Erkrankungen können im Serum
spezifische antineuronale Antikörper nachgewiesen werden. Diese treten praktisch
nie bei gesunden Kontrollen oder Patienten mit nicht-paraneoplastischen
Erkrankungen auf, so dass diese Antikörper bei mäßiger Sensitivität eine hohe
Spezifität für das Vorliegen eines PNS haben (Blaes et al. 1998a). Wird ein solcher
Antikörper (AK) nachgewiesen, ohne dass ein Tumor bisher bekannt ist, sollte eine
intensive Tumorsuche durchgeführt werden (Voltz 2002). Eine Ausnahme bilden die
Antikörper bei neuromuskulären Erkrankungen (Blaes 2002). Beim Lambert-Eaton-
myasthenen-Syndrom (LEMS) wurden Antikörper gegen den spannungsabhängigen
Kalziumkanal (Voltage-gated calcium channel/ VGCC) vom P/Q-Typ gefunden, was
zu einer Störung der Übertragung an der motorischen Endplatte führt (Lang et al.
1981, Motomura et al. 1997, O’Suilleabhain et al. 1998). Hierbei ist der
präsynaptische Anteil der neuromuskulären Synapse betroffen. Diese Antikörper
wurden bei der paraneoplastischen und nicht-paraneoplastischen Erkrankung
gefunden. Ähnlich ist das Bild bei der Myasthenia gravis (MG). Hier rufen Antikörper
gegen den Acetylcholinrezeptor (anti-AchR) der Muskelzelle die Krankheit hervor
(Vincent 1980). Antikörper gegen den spannungsabhängigen Kaliumkanal (voltage-
gated K+ channel/VGKC) sind die Ursache der Neuromyotonie (NMT) (Vincent et al.
1998). Diese Antikörper sind sehr spezifische Marker für das Vorliegen einer MG
oder NMT, allerdings unterscheiden diese nicht zwischen der paraneoplastischen
und nicht-paraneoplastischen Form der Erkrankung (Voltz 2002).
Bei den autoimmunen Erkrankungen der neuromuskulären Synapse ist die
pathogenetische Wirkung der Autoantikörper nachgewiesen und kann im Rahmen
eines PNS ausgelöst werden. Die Pathogenität anderer antineuronaler Antikörper
(z.B. anti-Hu, anti-Yo, anti-Ri) konnte bisher nicht vollständig geklärt werden
(Brashear et al. 1991, Blaes 2002, Voltz 2002). Einen Hinweis auf die Wirkung von
Autoantikörpern auf neuronale Zellen zeigte die Untersuchung von Greenlee et al.
Sie konnten in vitro eine Zelllyse der zerebellären Granularzellen von Ratten
nachweisen, nachdem diese mit anti-Hu-Serum inkubiert wurden (Greenlee et al.
91993). Ebenso fanden Schäfer et al. einen zytotoxischen Effekt von PNS-Seren und
deren IgG auf kultivierten Plexus myentericus (Schäfer et al. 2000). Verschuuren et
al. untersuchten die Zytotoxizität von anti-Hu-Serum gegen Tumorzelllinien. Sie
konnten feststellen, dass zwar das Serum, nicht aber das reine IgG einen
zytotoxischen Effekt zeigte (Verschuuren et al. 1997). Weitere Versuche, über die
Immunisierung mit onkoneuronalen Antigenen (HuD-Antigen, (Sillevis Smitt et al.
1995), Yo-Antigen (Tanaka et al. 1995)) ein PNS auszulösen, waren nicht
erfolgreich. Bei einer DNA-Immunisierung von Mäusen mit cdr2(PCD17)-cDNA
bildeten diese Antikörper und zytotoxische T-Lymphozyten gegen Purkinjezellen.
Allerdings kam es auch hier zu keiner klinischen Symptomatik oder histologischen
Schädigung (Sakai et al. 2001). Einzig für Antikörper gegen das retinale Protein
Recoverin konnte in vivo ein Beweis der Pathogenität an Ratten erbracht werden.
Diese dringen in Photorezeptor- und Bipolarzellen ein und führen zur Apoptose der
Zellen (Polans et al. 1991, Adamus et al. 1998).
Es wird nun postuliert, dass neuromuskuläre PNS (LEMS, MG, NMT) meist
Antikörper-vermittelt zu sein scheinen, während PNS des ZNS vermutlich T-Zell
vermittelt sind, mit fraglichem zusätzlichem Effekt der Antikörper (Blaes 2002).
Es wurden post mortem Zellinfiltrationen im Nervengewebe von Patienten gefunden,
die einen Anhaltspunkt für eine zelluläre Beteiligung darstellten (Sutton et al. 2001).
Dabei handelte es sich hauptsächlich um CD8+ T-Lymphozyten, die in Gehirn und
hinteren Grenzstrangganglien beschrieben wurden (perivaskulär und interstitiell)
(Sutton et al. 2002). Ein weiterer Hinweis brachte die Analyse der T-Zell-Rezeptoren
im Gehirn anti-Hu-positiver Patienten, die eine oligoklonale Expansion von
zytotoxischen T-Zellen zeigte (Voltz et al. 1998). Die Induktion von zytotoxischen T-
Lymphzyten gegen Purkinjezellen konnte nach einer Immunisierung mit
cdr2(PCD17)-Antigen festgestellt werden, allerdings ohne eine
Purkinjezellschädigung zu beobachten (Sakai et al. 2001).
Pellkofer et al. lieferten einen Beweis der Beteiligung von T-Zellen bei der
Entstehung von PNS. Nach einer Immunisierung von Ratten mit PNMA1 (Ma1)-
Protein konnten von diesen CD4+ T-Helferzellen entnommen werden. Nach Transfer
der Zellen in andere Ratten lösten diese dann eine perivaskuläre inflammatorische
Reaktion im ZNS in für PNS typischen Regionen aus, allerdings ohne dass die Tiere
erkrankten (Pellkofer et al. 2004). Sowohl Autoantikörper als auch T-Lymphzyten
scheinen also in der Pathogenese eine Rolle zu spielen.
10Warum bei manchen Tumorpatienten eine Autoimmunreaktion ausgelöst wird, ist
noch fraglich. Bei 85% der Patienten mit einem Tumor werden potentiell
immunogene Antigene exprimiert ohne eine Antitumor-Immunantwort auszulösen.
Bei 15-20% entwickelt sich eine Antitumor-Immunantwort, allerdings ohne ein PNS
als Folge (Übersicht in (Albert et al. 2004)). Alle kleinzelligen Bronchialkarzinome
exprimieren Hu-Proteine. Dabei finden sich bei 16% der neurologisch unauffälligen
Patienten niedrigtitrig Hu-Antikörper, während bei Patienten mit SCLC und PNS die
Antikörper in höherer Konzentration vorkommen (Dalmau et al. 1990). Es scheint
ebenfalls die Expression von Major Histocompatibility complex 1 (MHC1) neben der
Expression der Hu-Proteine bei SCLC bedeutsam zu sein. Es zeigte sich ein
erhöhtes Auftreten von Hu-Antikörpern bei diesen SCLC-Patienten, die ebenfalls
MHC1 exprimierten (Dalmau et al. 1995).
Dass die Immunantwort gegen den Tumor für den Patienten von prognostischer
Bedeutung ist, zeigte eine Studie an 196 Patienten mit SCLC. Bei ebenfalls 16%
konnten niedrigtitrige Anti-Hu-Antikörper gefunden werden, wobei kein Patient
klinisch neurologische Symptome aufwies. Die AK-positiven Patienten im Vergleich
mit den AK-negativen Patienten wiesen im Durchschnitt ein weniger fortgeschrittenes
Tumorstadium und eine höhere Sensitivität für die Chemotherapie auf (Graus et al.
1997a). Blaes et al. untersuchten die Seren von Patienten mit Non-SCLC auf
antineurale und antinukleäre Autoantikörper. Auch bei diesen AK-positiven Patienten
zeigte sich, unabhängig vom Tumorstadium, eine bessere Überlebensrate im
Vergleich zu Antikörper-negativen Patienten (Blaes et al. 2000).
Es scheint die folgende Regel zu gelten: Patienten mit hohen Antikörper-Titern zu
spezifischen Antigenen haben meist eine neurologische Dysfunktion, sind aber nur
niedrige Antikörper-Titer vorhanden, besteht ein Einfluss auf das Tumorwachstum
ohne unbedingt mit neurologischen Symptomen einherzugehen (Posner/Dalmau
2000).
111.3 Paraneoplastische neurologische Syndrome bei M. Hodgkin
Paraneoplastische neurologische Syndrome (PNS) kommen auch bei M. Hodgkin
vor. Am häufigsten findet sich eine paraneoplastische Kleinhirndegeneration (PCD)
(Horwich et al. 1966, Hammack et al. 1992, Cehreli et al. 1995, Graus et al. 1997b,
Benzing et al. 1998, Hahn et al. 2000, Spyridonidis et al. 2002). Umgekehrt findet
sich ein M. Hodgkin als Auslöser bei der PCD nach Lungen- und gynäkologischen
Tumoren an dritter Stelle (Hammack et al. 1992, Shams’ili et al. 2003). PCD-
Patienten leiden unter einem sich subakut (innerhalb von wenigen Wochen)
entwickelnden panzerebellären Syndrom mit Nystagmus, okulomotorischer Ataxie,
Dysarthrie, Dysmetrie und Rumpf- oder Extremitätenataxie. Selten können zusätzlich
andere zentralnervöse oder periphernervöse Störungen auftreten (Al-Lozi et al.
2003). Der häufigste neuropathologische Befund ist ein diffuser Purkinjezellverlust
(McCrystal et al. 1995, Bartos et al. 2002).
Trotter beschrieb 1976 erstmals einen immunologischen Zusammenhang zwischen
PCD und M. Hodgkin (Trotter et al. 1976). Später fand man bei einer Studie über 21
Hodgkin-Patienten mit PCD bei 6 Patienten immunhistochemisch anti-Yo ähnliche
Autoantikörper (Hammack et al. 1992). Anti-Yo färbt das Purkinjezellzytoplasma in
einem charakteristischen granulären Muster (Altermatt et al. 1991). Der bei M.
Hodgkin und PCD gefundene Autoantikörper zeigte ebenfalls eine Bindung an
Purkinjezellzytoplasma, allerdings weniger inhomogen als durch anti-Yo verursacht
(Hammack et al. 1992).
1997 beschrieben Graus et al. einen Antikörper, assoziiert mit einer
paraneoplastischen Kleinhirndegeneration und einem Hodgkin-Lymphom, und
nannten ihn anti-Tr nach Trotter (Graus et al. 1997b). Eine Klonierung des Antigens
gelang nicht; das Protein konnte weder im Western Blot dargestellt, noch über ein
cDNA-Bank-Screening kloniert werden (Graus et al. 1997b; Graus, Verschuuren,
persönliche Mitteilung). Die Lokalisation des Antigens zeigte sich im Zytosol und
außen am endoplasmatischen Retikulum der Perikaryen in der Molekularschicht und
im Zellkörper und den Dendriten der Purkinjezellen. Immunhistochemisch bindet anti-
Tr an Purkinjezellzytoplasma mit einem charakteristischen gepunktetem Muster der
Purkinjezelldentriten. Zusätzlich findet sich ein gepunktetes Muster in der
Molekularschicht (Graus et al. 1998). Eine weitere Analyse von 28 Patienten mit anti-
Tr Antikörpern zeigte, dass anti-Tr ein sehr spezifischer Marker für die Diagnose
12einer M. Hodgkin-assoziierten PCD ist. 27 Patienten davon litten an einem
cerebellären Syndrom und 25 Patienten davon an einem M. Hodgkin (Bernal et al.
2003).
Außer anti-Tr gibt es noch 2 weitere Autoantikörper, die mit PCD und M. Hodgkin
assoziiert werden. Bataller et al. fanden bei 11 von 19 anti-Tr-positiven PCD
Patienten zusätzlich Antikörper gegen das MAZ-Protein (Myc-associated zinc-finger
protein) (Bataller et al. 2003). Bei 2 Patienten mit cerebellärer Ataxie wurden
Antikörper gegen den extrazellulären Teil des metabotropen Glutamatrezeptors (anti-
mGluR1) gefunden. Die Patienten litten zwar an einem M. Hodgkin, der
Zusammenhang als PNS konnte allerdings aufgrund der langen Zeitdauer zwischen
Beginn der Ataxie und des M. Hodgkin (2 und 9 Jahre) nicht gesichert werden
(Sillevis Smitt et al. 2000). In einer anderen Studie fanden sich aber ebenfalls bei 2
PCD-Patienten anti-mGluR1-Antikörper in Assoziation mit dem Hodgkin-Lymphom
(Shams’ili et al. 2003).
Neben der PCD finden sich weitere paraneoplastische neurologische Syndrome bei
M. Hodgkin. Hier wurden sowohl verschiedene Polyneuropathieformen, wie auch
limbische Enzephalitiden beschrieben. Unter einer Polyneuropathie wird die
Erkrankung mehrerer peripherer Nerven verstanden, wobei je nach Art der
betroffenen Nerven motorische, sensible oder vegetative (autonome) Ausfälle
vorliegen können. Die Symptome sind je nachdem Parästhesien,
Tiefensensibilitätsstörungen mit sensibler Ataxie, strumpf- bzw. handschuhförmige
Oberflächensensibilitätsstörungen, distale Paresen und Reflexabschwächung. So
wurden schon Hodgkin-Patienten mit sensorischer (Horwich et al. 1977, Plante-
Bordeneuve et al. 1994, Maslovsky et al. 2001, Oh et al. 2004), sensomotorischer
(Blaes et al. 1998b, Lahrmann et al. 2001), motorischer (Younger 1991) und
autonomer (Van Lieshout et al. 1986, Turner et al. 1993) Neuropathie beschrieben.
Ebenfalls gibt es mehrere Fallbeschreibungen über Hodgkin-Patienten mit
Polyradikulitis in Form des Guillain-Barré-Syndroms (GBS) (Julien et al. 1980,
Maslovsky et al. 2001) und der chronisch inflammatorischen demyelinisierenden
Polyneuropathie (CIDP) (Navellou et al. 2001).
Das Guillain-Barré-Syndrom wird selten paraneoplastisch verursacht, als assoziiertes
Neoplasma steht das Hodgkin-Lymphom an erster Stelle (Julien et al. 1980). Die
autonome Polyneuropathie scheint eine häufige PNS bei Hodgkin-Patienten zu sein.
13Während Van Lieshout et al. von einem Fall von akuter Dysautonomie (Van Lieshout
et al. 1986) berichteten, fanden Turner et al. in einer prospektiven Studie bei 80%
ihrer Hodgkin-Patienten eine autonome Dysfunktion, die als paraneoplastisch
angesehen wurde (Turner et al. 1993). Bei der subakuten sensorischen Neuropathie
(SSN) ist der Nachweis von anti-Hu Autoantikörpern charakteristisch (Dalmau et al.
1992), wobei der assoziierte Tumor dann meist ein kleinzelliges Bronchialkarzinom
ist (Anderson/Posner 1988). In Zusammenhang mit M. Hodgkin wurden bisher
diesbezüglich keine Autoantikörper beschrieben. Es fanden sich auch bei keiner der
anderen paraneoplastischen Polyneuropathien neuronale Autoantikörper.
Als ein weiteres PNS bei M. Hodgkin wurden Fälle von Limbischer Enzephalitis
beschrieben (Deodhare et al. 1996, Kung et al. 2002). Autoantikörper wurden nicht
gefunden. Die limbische Enzephalitits ist Bestandteil verschiedener
paraneoplastischer Syndrome und kann isoliert oder in Kombination mit anderen
Syndromen als multifokale paraneoplastische Enzephalomyelitis (PEM) auftreten
(Dropcho 1998). Assoziierte Tumoren sind das kleinzellige Bronchialkarzinom,
Hodentumoren und Mammatumoren (Gultekin et al. 2000), selten auch Thymome
oder Hodgkin-Lymphome (Al-Lozi et al. 2003). Weiterhin ist auch die Neuromyotonie
als paraneoplastisches Syndrom beschrieben. In der Literatur finden sich 2 Fälle
assoziiert mit einem Hodgkin-Lymphom (Caress et al. 1997, Lahrmann et al. 2001).
Als Auslöser findet man Antikörper gegen Kaliumkanäle. (anti-VGKC Antikörper/
voltage-gated K+ channels) (Vincent et al. 1998).
Verschiedenste andere neurologische Paraneoplasien in Zusammenhang mit dem
Hodgkin-Lymphom sind als Fallbeschreibungen publiziert worden. Mehrere Fälle von
paraneoplastischer Myelopathie wurden in Zusammenhang mit M. Hodgkin gefunden
(Hughes et al. 1992, Dansey et al. 1988). Kay et al. beschrieben ein Opsoklonus-
Myoklonus Syndrom (Kay et al. 1993) und Batchelor et al. eine Chorea (Batchelor et
al. 1998) als Paraneoplasie bei M. Hodgkin. Es wurden auch Fälle von nicht-
neurologischen Paraneoplasien bei M. Hodgkin beschrieben, wie z.B. ein
nephrotisches Syndrom (Utsch et al. 1999, Spyridonidis et al. 2002).
Ein Patient mit M. Hodgkin zeigte Zeichen einer paraneoplastischen entzündlich-
rheumatischen Erkrankung. Dieser Patient litt an Arthralgien, Myalgien, Arthritis und
Polyneuritis. Im Serum dieses Patienten fand sich neben zirkulierenden
Immunkomplexen ein antinukleärer Antikörper (ANA) mit einem Titer von 1:400 (Miro
et al. 1982). Berichte über ANA beim Hodgkin-Lymphom finden sich sonst nicht in
14der Literatur. Bei einer Studie über 347 Patienten mit Non-Hodgkin-Lymphom wurden
bei 19% Antinukleäre Antikörper gefunden (Guyomard et al. 2003).
Bisherige Autoantikörperuntersuchungen bei M. Hodgkin wurden fast ausschließlich
in Zusammenhang mit vorhandener Paraneoplasie durchgeführt. In einer Studie
wurde bei Hodgkin-Patienten ohne PNS Autoantikörper gegen ssDNA, anti-RNP und
anti-Sm (ribonukleare Protein-Antigene) gefunden (Swissa et al. 1992). Es ist aber
bekannt, dass auch bei neurologisch unauffälligen Patienten mit Malignom
onkoneuronale Autoantikörper gebildet werden können (Dalmau et al. 1990,
Übersicht in (Albert et al. 2004)).
Es konnte bisher bei Hodgkin-Patienten kein Zusammenhang zwischen
epidemiologischen Daten und der Neigung zur Autoantikörperbildung festgestellt
werden. Bei 21 untersuchten Patienten mit PCD unterschieden sich die anti-Tr-
positiven Patienten nicht hinsichtlich Alter, Geschlecht, Hodgkin-Subtyp oder
Tumorstadium von den anti-Tr-negativen Patienten (Hammack et al. 1992). Auch bei
einer Studie mit 25 Hodkgin-Patienten zeigten sich keine signifikanten Unterschiede
zwischen Alter, Geschlecht, Tumorklassifikation und Stadium der anti-Tr-positiven
und -negativen Patienten in Bezug auf die Antikörperbildung (Bernal et al. 2003).
Auch konnten keine Schlüsse aus dem Auftreten der PCD im Verhältnis zur
Diagnose des Hodgkin-Lymphoms gezogen werden. Die zeitlichen Abstände
variierten dabei von 0-24 Monaten vor und 1-120 Monaten nach der Lymphom-
Diagnose (Hammack et al. 1992, Bernal et al. 2003).
1.4 Fragestellung
Wie häufig finden sich Autoantikörper bei M. Hodgkin-Patienten mit und ohne
bekanntes paraneoplastisches neurologisches Syndrom?
Korreliert das Vorhandensein von Autoantikörpern mit klinischen oder
epidemiologischen Daten und sind daraus Aussagen bezüglich der Prognose
abzuleiten?
152. Patienten und Methoden
2.1 Patienten
Insgesamt wurde Serum von 72 Patienten untersucht.
Nach Aufklärung und Zustimmung zur Studienteilnahme wurde den Patienten bzw.
Probanden Blut abgenommen und das Serum abzentrifugiert. Bis zur Verwendung
wurde das Serum bei –20 °C tiefgefroren.
57 Serumproben der M. Hodgkinpatienten wurden uns freundlicherweise von der
German Hodgkin Study Group aus Köln zur Verfügung gestellt. Die Zustimmung zur
Verwendung dieser Seren wurde von der Ethikkomission der Universität zu Köln
erteilt.
Die Patienten mit M. Hodgkin wurden in zwei Gruppen unterteilt. Patienten mit M.
Hodgkin und bekanntem PNS wurden gesondert von Patienten mit M. Hodgkin ohne
bekannte Paraneoplasie betrachtet.
2.1.1 Patienten mit M. Hodgkin (Hodgkin-Gruppe)
In diese Gruppe wurden Patienten aufgenommen, bei denen ein Hodgkin-Lymphom
diagnostiziert und histologisch gesichert und bei denen keine neurologische
Krankheit bekannt war. Insgesamt wurden 67 Patientenproben untersucht, von
denen 23 weiblich und 44 männlich waren. Das mittlere Alter in dieser Gruppe betrug
37.3 ± 15.8 Jahre (Mittelwert ± Standardabweichung).
2.1.2 Hodgkinpatienten mit PNS (Hodgkin/Para-Gruppe)
Es wurden 5 Hodgkin-Patienten (4m / 1w, mittleres Alter 33.8 ± 11.9 Jahre) mit
bekanntem PNS mituntersucht (Tab. 6). Die Diagnose des M. Hodgkin wurde
histologisch gesichert. Die Diagnose des PNS wurde anhand der Richtlinien für
Paraneoplastische Neurologische Syndrome von 2004 (Graus et al. 2004) gestellt.
Diese Patienten werden gesondert von der Hodgkin-Gruppe als Fallbeschreibungen
aufgeführt.
16Tabelle 6: Daten der Hodgkinpatienten mit paraneoplastischem neurologischem
Syndrom
PCD: Paraneoplastische Kleinhirndegeneration
Patient Alter/Geschlecht PNS
1 30m Polyneuropathie
2 46m PCD
3 34w PCD
4 43m PCD
5 16m Limbische Enzephalitis
2.1.3 Gesunde Kontrollen (Kontroll-Gruppe)
In diese Gruppe wurden 46 gesunde Probanden aufgenommen. Es waren 28
weibliche und 18 männliche Probanden mit einer Altersverteilung von 38.9 ± 15.3
Jahre.
2.2 Methoden
2.2.1 Zellkultur
Für unsere Versuche verwendeten wir die Zelllinie SKN-SH (Neuroblastomzelllinie)
und die Zelllinie HEK 293 (Nierenfibroblasten, Human embryonic kidney). Das
Kulturmedium bestand aus RPMI 1640 (Sigma, Steinheim) mit 10% fetalem
Kälberserum (FKS, HyClone, PerbioScience, USA), L-Glutamin (4mmol/l, Sigma),
Streptomycin (20mg/l, Sigma) und Penicillin (20.000IE/l, Sigma). Darin wurden die
Zellen bei 37°C und 5% CO2 kultiviert. Jeden 2. Tag wurde das Medium erneuert.
Jeden 4. Tag wurden die Zellen im Verhältnis 1/4 gesplittet. Dazu wurden die Zellen
mit Trypsin (2.5 g/l, Gibco-Invitrogen, USA) für 5 Minuten bei 37°C inkubiert.
172.2.2 Autoantikörperdiagnostik
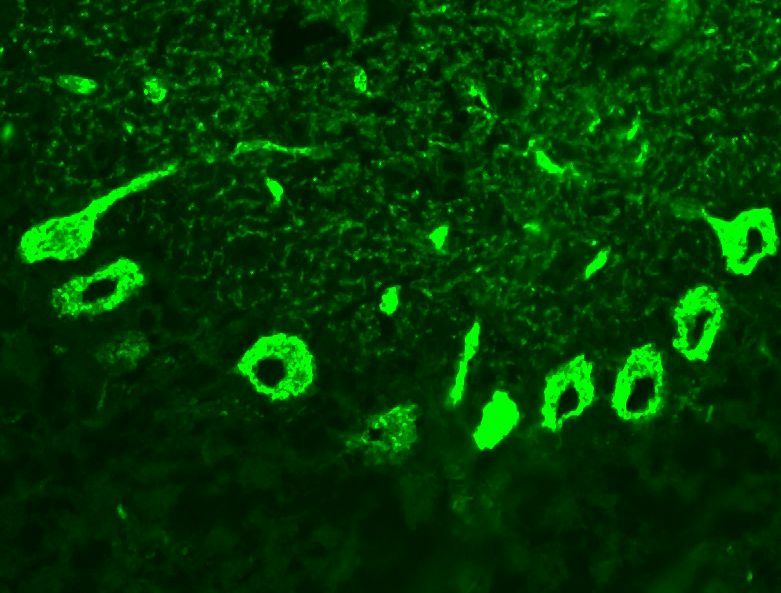
2.2.2.1 Indirekte Immunfluoreszenz
Bei der Immunfluoreszenz werden Antikörperbindungen von Patientenserum an
Zellstrukturen untersucht. Dazu werden histologische Gewebeschnitte zuerst mit
Patientenserum (sind Antikörper vorhanden, findet eine Bindung statt) und
anschließend mit einem monoklonalen anti-human IgG Zweitantikörper inkubiert.
Dieser bindet an die Antikörper der Patienten. Durch einen fluoreszierenden
Farbstoff an diesem Zweitantikörper können diese Bindungen dann mikroskopisch
untersucht und ausgewertet werden. Mit diesem Verfahren wurden alle Proben
ausgetestet, um vorhandene Antikörper im Serum zu finden. Es wurde ein
kommerziell erhältlicher Assay zur Diagnostik antineuronaler Autoantikörper
(Euroimmun, Lübeck) verwendet. Auf den Objektträgern waren unfixierte
Gefrierschnitte von Kleinhirn, humanen Epithelzellen (Hep2-Zellen) und Darmwand
mit Auerbachplexus aufgebracht.
Die Objektträger wurden 30 Minuten mit 65 µl Patientenserum (Verdünnung 1/32 in
phosphatgepufferter NaCl-Lösung (Phosphate buffered saline (=PBS) Tween 0.2%)
bei Raumtemperatur inkubiert. Danach wurden die Objektträger dreimal in PBS
Tween 0.2% gewaschen. Im nächsten Schritt wurden die Objektträger mit 67µl
Fluoreszeinthiocyanat- (FITC-) markiertem anti-human IgG (Verdünnung 1/75 in PBS
Tween 0.2%, Dako, Hamburg) bei Raumtemperatur 30 Minuten inkubiert. Nach
einem weiteren Waschvorgang mit PBS Tween 0.2% wurden die Objektträger mit
Eindeckmedium und Deckgläschen versehen.
Unter einem Fluoreszenzmikroskop (Zeiss) wurden die Schnitte ausgewertet. Als
Positivkontrolle wurde ein anti-Hu positives Serum mitgetestet, wobei das typische
antineuronal nukleäre Bindungsmuster eindeutig zu erkennen sein musste. Als
Negativkontrolle dienten Puffer und Serum von gesunden Probanden.
182.2.2.2 Western Blot
Im Western Blot werden Proteine bestimmter Gewebe nach Größe aufgetrennt.
Anschließend können Antikörperreaktivitäten der Patientenseren gegen diese
Proteinfraktionen dargestellt werden und die Größe der Proteinfraktion in kD
(kiloDalton) gemessen werden. Zur Messung werden die dargestellten Bindungen mit
einem Standardstreifen verglichen und dadurch deren Größe bestimmt.
Als Antigengewebe in unseren Versuchen dienten die löslichen Proteinfraktionen von
Kleinhirn (Ratte), SKN-SH Zellen, HEK 293-Zellen und Auerbachplexus (Ratte).
1. Herstellung der Proteinfraktionen
Zur Herstellung der Proteinfraktionen wurde Kleinhirn von Ratten, sofort post-
mortem, und Auerbachplexus von Ratten, der nach unten beschriebener Methode
gewonnen wurde, verwendet. SKN-SH und HEK 293-Zellen sind im eigenen Labor
etabliert. Das entsprechende Gewebe oder die Zellen wurden zunächst in PBS im
Glaszylinder durch einen Mörser zerkleinert. Die Probe wurde danach 1min. bei 4000
Umdrehungen zentrifugiert und der Überstand abpipettiert. Danach wurde die Probe
mit dem Ultraschallhomogenisator (Bandelin HD2070) in 4 Zyklen von je 15 sek.
Dauer homogenisiert und dann erneut 5min. zentrifugiert (4000 rpm, RT). Der
Überstand, der jetzt die lösliche Proteinfraktion enthält, wurde mit LDS sample buffer
(Invitrogen) 4/1 verdünnt und bei 90°C für 5 Minuten erhitzt. Abschließend wurde
Reducing Agent (Invitrogen) im Verhältnis 9/1 dazugegeben und die Probe bis zur
Weiterverwendung bei –24°C tiefgefroren.
2. Elektrophorese
Es wurde eine Elektrophoresekammer (XCell SureLock, Invitrogen) und ein 4-12%
Bis-Tris Polyacrylamid 2D-Well Gel (NuPage, Invitrogen) verwendet.
Die Proteinprobe wurde für 5 Minuten bei 70°C erwärmt, anschließend wurden 100µl
in die größere Geltasche pipettiert. Als Standard wurde 20µl SeaBlue oder Mark12
(Invitrogen) in die kleinere Geltasche pipettiert. Als Puffer wurde Running Buffer
(Invitrogen) verwendet, dem 500µl Antioxidans (Invitrogen) zugesetzt wurde. Es
wurde über 10 Minuten ein Strom von 20 mA angelegt, danach 100mA mit einer
Dauer von ca. 1 Stunde. Dadurch wurde die Proteinprobe in die einzelnen
Proteinfraktionen aufgetrennt.
193. Blotting
Es wurde eine Semidry-Blotkammer (XCell II Blotmodule, Invitrogen) und als Puffer
Transfer Buffer (Invitrogen) verwendet. Für 2 Stunden wurde ein Strom von 150 mA
angelegt. Dabei wurden die Proteinfraktionen vom Elektrophoresegel auf eine
Nitrocellulose-Blotfolie (Protan Nitrocellulose Transfer Membrane,
Schleicher&Schuell) übertragen. Die Blotfolie wurde anschließend mit Ponceaurot
(Sigma) angefärbt, um die aufgetrennten Proteine sichtbar zu machen. Danach
wurde die getrocknete Folie in 3mm breite Streifen geschnitten. Jeder Streifen
enthält also die komplette aufgetrennte Proteinfraktion.
4. Austestung
Zum Austesten der Streifen wurden diese 10 Minuten mit je 1ml Mischpuffer (PBS
Tween 0.5% mit 11g/l Trockenmilchpulver) getränkt. Nach Abschütten des Puffers
wurde zu jedem Streifen jeweils eine Serumprobe (10µl in 1ml Mischpuffer) gegeben
und eine Stunde bei Raumtemperatur inkubiert. Nach diesem Schritt wurden die
Streifen 3mal jeweils 5 Minuten mit 1ml Mischpuffer gewaschen.
Danach wurde je 1ml mit Alkalischer Phosphatase- (AP-) markierter anti-human IgG
(1/1000 in Mischpuffer, Dako) zu den Streifen pipettiert. Nach 30 Minuten erneuter
Inkubationszeit wurde wieder 3mal gewaschen. Zur Visualisierung möglicher
Reaktivitäten wurde zu den Streifen je 1ml 5Bromo - 4chloro - 3indolylphoshat -
nitroblue - tetrazoliumchloride (BCIP-NBT, Sigma) gegeben und 10 min inkubiert.
Zur Auswertung der Banden wurden die Streifen eingescannt. Mit der GelScan-
Software (BioSciTec, Frankfurt) erfolgte die Auswertung. Durch Vergleich mit dem
Standardstreifen konnte die Größe des Proteins, an das eine Bindung erfolgte,
abgelesen werden. Ebenso wurde semiquantitativ die Bindungsstärke abgeschätzt.
Als Positivkontrolle verwendeten wir dabei anti-Hu positives Serum, wobei die
typische Bindung an ein 35-40 kD Protein im Kleinhirn-Western Blot vorhanden sein
musste. Als Negativkontrolle verwendeten wir Puffer und Serum von gesunden
Probanden.
202.2.2.3 Durchflusszytometrie/ Fluorescent activated cell sorting (FACS)
Mit dem Verfahren der Durchflußzytometrie können Antikörperbindungen an
extrazelluläre Epitope gemessen werden. Dazu werden einzelne Zellen mit Serum
inkubiert (vorhandene Antikörper binden an die Zellen). Anschließend wird ein
fluoreszierender Zweitantikörper eingesetzt, dieser bindet an die gebundenen
Serumantikörper. Anschließend wird die Fluoreszenz gemessen und somit
festgestellt, ob Antikörper gegen diese Zellen im Serum vorhanden sind.
In der Durchflusszytometrie wurden alle Proben mit SKN-SH Zellen und HEK 293
Zellen inkubiert und gemessen. Zeigten sich in einem Serum Antikörperbindungen an
beide Zelllinien oder eine Bindung nur an die HEK 293 Zellen, wurde dies als
Bindung gegen ubiquitäre Autoantigene gewertet. Bei einer ausschließlichen
Antikörperbindung an SKN-SH Zellen wurde dies als neuronal-spezifische Bindung
interpretiert.
1. Inkubation
Als Puffer wurde PBS mit 1% FKS (HyClone) und 0.1% Natriumazid (NaN3, Sigma)
vermischt, im folgenden als FACS Puffer bezeichnet. In eine 96-Well Platte wurden
pro Well 105 Zellen pipettiert. Die Zellen wurden mit Serum (1/50 in FACS Puffer) 30
Minuten bei 4°C inkubiert. Nach zweifacher Waschung mit FACS Puffer wurde FITC-
markiertes anti-human IgG (1/75 in FACS Puffer, Dako) dazugegeben und ebenfalls
30 Minuten bei 4°C inkubiert. Nach einem erneuten Waschschritt wurden die Zellen
in Messröhrchen gegeben.
2. Messung
Mit dem FACSCalibur (Becton-Dickinson) wurde die Fluoreszenz der Zellen jeder
Serumprobe mit der CellQuest®-Software gemessen. Als Positivkontrollen dienten
anti-CD 56 (bindet spezifisch an ein neuronales Adhäsionsmolekül (N-CAM 123C3),
1/100 in FACS Puffer, Dako) und anti-MHC1 (bindet an den Major Histocompatibility
Complex, 1/100 in FACS Puffer, Dako). Zur Negativkontrolle wurden die Zellen nur
mit FACS-Puffer inkubiert. Zusätzlich wurden bei jedem Versuch 10 Seren gesunder
Probanden inkubiert. Bei einer starken Antikörperbindung an die Zellen wurde eine
hohe Rate an stark fluoreszierenden Zellen gemessen. (Abb. 1) Eine geringe Anzahl
21von fluoreszierenden Zellen wurde hingegen gemessen, wenn keine
Antikörperbindung an die Zellen stattgefunden hatte.
Abbildung 1: FACS-Messung; Unterschied schwach – stark fluoreszierende Zellen.
X-Achse: Fluoreszenzstärke der einzelnen Zellen; Y-Achse: Anzahl der gemessen Zellen. Linkes Bild:
unspezifische Hintergrundbindung eines Kontrollserums; Rechtes Bild: Bindung eines Antikörper-
postiven Serums
3. Auswertung
Es wurde nun ausgewertet, ob aus einem Serum eine Antikörperbindung an die
Zellen stattgefunden hatte.
Es zeigte sich bei allen Proben eine leichte Fluoreszenz der Zellen. Zur
Unterscheidung zwischen unspezifischen Bindungen und spezifischer positiver
Antikörperbindung wurden zunächst die Ergebnisse der 10 Probandenseren
ausgewertet. Von diesen wurde jeweils die Fluoreszenzstärke (mean fluorescence
intensity (mfi)) gemessen und aus den Ergebnissen ein Mittelwert mit
Standardabweichung errechnet. Dieser Mittelwert wurde als Grundlage zur
Auswertung verwendet und jeweils mit den Ergebnissen der übrigen Proben
verglichen.
Dabei wurde einem Serum eine positive Antikörperbindung zugeschrieben, wenn
eine Fluoreszenzstärke gemessen wurde, die über dem Cut-off (Mittelwert + 2,5-
facher Standardabweichung) lag.
222.2.3 Isolierung von Auerbachplexus
Der Auerbachplexus wurde aus Wistar-Ratten gewonnen. Die Tiere wurden zur
Präparation durch Decapitation getötet und das Peritoneum eröffnet. Es wurde der
gesamte Darm herausgetrennt und auf Eis gelegt. Weiterhin wurde der Darm in
Kolon und Dünndarm unterteilt und unter mikroskopischer Sicht das Bindegewebe
und die Gefäße abgetrennt. Nach Verkleinerung in 3-5 Teilstücke wurde das
Peritoneum viscerale strumpfförmig abgezogen (Stripping) und das verbleibende
Material in ein Eppendorfgefäß gegeben. Im Verhältnis 1:1 wurde nun Stammlösung
(Dulbecco’s modified Eagle’s medium (DMEM, ICN Biomedicals, Inc., USA) +
Gentamycin (PAA Laboratories GmbH, Cölbe) + Metronidazol (Bayer AG,
Leverkusen) + Glutamin (Sigma-Aldrich Chemie GmbH, Taufkirchen)) mit
Kollagenase Typ II (2mg/ml, Gibco Invitrogen GmbH, Karlsruhe) zugegeben und bei
37°C inkubiert. Dabei wurde für ein 3 Tage altes Tier die Inkubationszeit von 1
Stunde gewählt und für jeden weiteren Lebenstag 10 Minuten hinzuaddiert.
Nach der Inkubation wurden die angefallenen Netze abpippetiert und in eisgekühltes
DMEM gegeben. Mit dem restlichen Gewebe wurde dann ein weiterer
Inkubationsschritt mit Kollagenase (1mg/ml DMEM) für 30 Minuten durchgeführt und
danach je nach Lösungseigenschaft weitere Inkubationsschritte mit einer
Inkubationszeit von jeweils 15-20 Minuten. Die Netze wurden in ein Eppendorfgefäß
gesammelt und 10 Minuten bei 800 Umdrehungen zentrifugiert. Der Überstand
wurde abpippetiert und der Rest mit einer Lösung aus 400µl Start V (serumfreies
Fertigmedium, speziell für neuronale Zellen, Biochrom AG, Berlin) + FKS (10%,
Biochrom AG) resuspendiert. Danach wurde erneut zentrifugiert und der Überstand
abpippetiert. Die so verbleibenden Zellen wurden bis zur Weiterverarbeitung bei
-82°C tiefgefroren.
23Sie können auch lesen

















































