Der Fibroblast als Unterhändler der rheumatoiden Arthritis
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Der Fibroblast als Unterhändler der rheumatoiden Arthritis Medizinische Klinik 3 – Rheumatologie und Immunologie Direktor: Prof. Dr. univ. Georg Schett Der Medizinischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades Dr. med. vorgelegt von Benjamin Wirth

Als Dissertation genehmigt von der Medizinischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg Tag der mündlichen Prüfung: 11. Februar 2020 Vorsitzender des Promotionsorgans: Prof. Dr. med. Markus F. Neurath Gutachter: PD Dr. rer. nat. Markus Hoffmann PD Dr. med. Christian Beyer
Abstract 1 Abstract Background: Rheumatoid Arthritis (RA) affects up to 1% of the world population. Patients suffer from a high disability due to severe joint damage. Systemic effects of the disease, such as vasculitis and heart disease as a result cause a shorter life span. The local connective tissue, especially synovial fibroblasts (SFB) are more and more put into the spotlight of research regarding the pathogenesis of RA. Due to their ability to actively modulate inflammation and destructive processes in cartilage and bone they are a key player in the development of the disease. Objective: Objective of this thesis is a deeper understanding of the phenotypical changes that SFB undergo during different stages of RA and their ability to maintain inflammation and thus mediate chronification. Methods: Five SFB cell lines from human patients with longstanding RA as well as five SFB cell lines from very-early RA were analysed against five cell lines from a healthy control group. A 3D organoid system (micromass) was established and follow up immunofluorescent staining was used to investigate change in morphology of the synovial lining. Cytokine measurement was done for all SFB cell lines. Furthermore, the invasive potential of all SFB cell lines was quantified via Matrix-associated transepitelial resistance and invasion (MATRIN)-Assay and proliferation and migration via a wound healing assay. Results: RA-SFB proved to be significantly more invasive than the VERA cell lines and the control group. RA-SFB also showed tendencies towards a pathological phenotype in the micromass system. Migration/Proliferation did not differ between the cell lines.
Abstract 2 Conclusion: The results indicate a phenotype switch of RA-SFB compared to the control group, most likely because of imprinted intrinsic alterations. Follow up studies with higher case numbers are required to substantiate results. Keywords: synovial fibroblasts, rheumatoid arthritis, phenotype switch, organoid, synovial membrane
Kurzfassung 3 Kurzfassung Hintergrund: Die rheumatoide Arthritis (RA) ist eine chronische Erkrankung die bis zu 1% der Weltbevölkerung betrifft. Durch Gelenkbefall kann es zu starken Beeinträchtigungen der Patienten kommen. Systemische Begleiterkrankungen wie Vaskulitis und kardiovaskuläre Ereignisse bedingen zudem eine kürzere Lebenserwartung. Das lokale Bindegewebe rückt immer stärker in den Fokus mit seiner Rolle zur Krankheitsaufrechterhaltung. Speziell synoviale Fibroblasten (SFB) und ihre entzündlichen Eigenschaften sowie ihre Fähigkeit zum Knorpel- und indirekt Knochenabbau sind bedeutende Zellen in der Pathogenese. Zielsetzung: Ziel der vorliegenden Arbeit ist das tiefere Verständnis von Veränderungen des Phänotyps der synovialen Fibroblasten als Teil des lokalen Bindegewebes in verschiedenen Krankheitsstadien der rheumatoiden Arthritis sowie mögliche Mechanismen zur Aufrechterhaltung und Chronifizierung der Entzündung. Methoden: Es standen jeweils 5 Zelllinien von humanen synovialen Fibroblasten aus Patienten mit sehr kurz (very-early-RA, VERA) sowie lang andauernder, etablierter RA im Vergleich zu gesunden Patienten zur Verfügung. Es erfolgte die Analyse von 3D-Organoiden (Micromass) durch Immunfluoreszenz und Betrachtung der Morphologie. Messung der Zytokin/Chemokin-Ausschüttung der verschiedenen Zelllinien wurde durchgeführt. Des Weiteren diente ein sog. Matrix-associated transepithelial resistance and invasion (MATRIN)-Assay zur Analyse des invasiven Potentials sowie ein Scratch wound healing Assay zur Beurteilung der Migration und Proliferation von RA-SFB im Vergleich zu Zellen aus gesunden Individuen. Ergebnisse: Es ließ sich eine signifikant stärkere Invasivität von RA-SFB gegenüber VERA und normalen SFB feststellen. Bezüglich der Micromass-Färbungen konnten den RA-SFB lediglich Tendenzen eines aggressiveren Phänotyps zugeschrieben werden. Bezüglich Migration/Proliferation zeigten sich keine Unterschiede.
Kurzfassung 4 Schlussfolgerung: Die Ergebnisse lassen, verglichen mit normalen SFB, auf eine Phänotypänderung von RA-SFB rückschließen. Der Wandel ist primär durch intrinsische Veränderungen bedingt. Zur Bestätigung ist eine größere Fallzahl in zukünftigen Studien erforderlich. Schlagwörter: synoviale Fibroblasten, rheumatoide Arthritis, Phänotypänderung, Organoid, Synovialmembran
Inhaltsverzeichnis 5 Inhaltsverzeichnis Abstract ............................................................................................................................ 1 Kurzfassung ..................................................................................................................... 3 Inhaltsverzeichnis............................................................................................................ 5 1 Einleitung.............................................................................................................. 7 1.1 Arthritis und Arthrose ............................................................................................ 7 1.2 Rheumatoide Arthritis............................................................................................ 7 1.2.1 Diagnosekriterien und Evaluationsscores .............................................................. 9 1.2.2 Einteilung der rheumatoiden Arthritis und serologische Auffälligkeiten ............ 12 1.2.3 Pathogenese der rheumatoiden Arthritis .............................................................. 13 1.2.4 Therapie der rheumatoiden Arthritis.................................................................... 20 1.2.5 Rolle der synovialen Fibroblasten in der rheumatoiden Arthritis........................ 23 2 Zielsetzung .......................................................................................................... 26 3 Material und Methoden..................................................................................... 27 3.1 Zellgewinnung ..................................................................................................... 27 3.2 Zellkultivierung ................................................................................................... 28 3.2.1 Auftauprozess ...................................................................................................... 28 3.2.2 Mediumwechsel ................................................................................................... 28 3.2.3 Passagieren der Zellen ......................................................................................... 28 3.2.4 Cryokonservierung............................................................................................... 29 3.2.5 Zellzählung .......................................................................................................... 30 3.3 Experimente ......................................................................................................... 31 3.3.1 Micromass ............................................................................................................ 31 3.3.2 Rasterelektronenmikroskopie .............................................................................. 33 3.3.3 Färbungen ............................................................................................................ 34 3.3.4 IBIDI-Wound-Healing and Migration Assay ...................................................... 37 3.3.5 Matrix-Associated Transepithelial Resistance Invasion (MATRIN) Assay........ 38 3.4 Statistische Auswertung....................................................................................... 39 4 Ergebnisse ........................................................................................................... 40 4.1 Patientenkollektiv ................................................................................................ 40 4.2 Micromasses und Färbungen ............................................................................... 43 4.3 Zytokinmessung in 3D-Organoiden..................................................................... 48 4.4 IBIDI-Wound-Healing and Migration Assay ...................................................... 50
Inhaltsverzeichnis 6 4.5 Messung der Invasivität durch MATRIN-Assay ................................................. 51 5 Diskussion ........................................................................................................... 53 5.1 Der Fibroblast als Unterhändler ........................................................................... 53 5.2 Von der Maus in den Menschen .......................................................................... 54 5.3 Persistenz des aggressiven Phänotyps ................................................................. 55 5.4 Schlussfolgerung.................................................................................................. 59 5.5 Ausblick ............................................................................................................... 60 6 Appendix ............................................................................................................. 63 6.1 Literaturverzeichnis ............................................................................................. 63 6.2 Materialienliste .................................................................................................... 70 6.2.1 Lösungen und Puffer ............................................................................................ 70 6.2.2 Verbrauchsmaterialien ......................................................................................... 72 6.2.3 Geräte ................................................................................................................... 73 6.2.4 Programme ........................................................................................................... 74 6.3 Abkürzungsverzeichnis ........................................................................................ 74 6.4 Tabellenverzeichnis ............................................................................................. 76 6.5 Abbildungsverzeichnis......................................................................................... 76 7 Danksagung ........................................................................................................ 78
1 Einleitung 7 1 Einleitung 1.1 Arthritis und Arthrose Pathologische Gelenkveränderungen können entzündlicher (Arthritis) und degenerativer (Arthrose) Natur sein [1]. Beide Gelenkspathologien gehen einher mit Dolor (Schmerz) und Functio laesa (Funktionseinschränkung). Die entzündlichen Arthritiden treten zusätzlich durch Rubor (Rötung) Calor (Überwärmung) und Tumor (Schwellung) in Erscheinung [2]. Arthrose entsteht durch Über-/Fehlbelastung und fällt durch Knorpelveränderungen auf. Im Verlauf entwickeln sich deformierte Gelenke und Gelenkflächen. Primär nicht entzündlich ist ein fließender Übergang in eine Arthritis und somit einer sekundären Entzündung möglich. [1] Die Arthritis per se ist, wie oben beschrieben, definiert durch die Entzündung eines Gelenkes. Dabei ist vor allem das dünnschichtige Synovium der Gelenkkapsel betroffen. Ätiologisch wird unterschieden, ob dabei eine durch Arthrose bedingte sekundäre Arthritis vorliegt oder eine Arthritis mit primärer Entzündung. [1] Bei primär entzündlichen Gelenkerkrankungen unterscheidet man zwischen vielen Formen wie der rheumatoiden Arthritis, Spondyloarthritis oder Psoriasis-Arthritis. Besonders die rheumatoide Arthritis hat viele Unter- und Sonderformen (bei außergewöhnlich schwerem Verlauf oder atypischem Befall) und kann sich auf unterschiedlichste Weise präsentieren. [1] 1.2 Rheumatoide Arthritis Die rheumatoide Arthritis (RA) kann zusätzlich zur Gelenksentzündung auch diverse extraartikuläre Manifestationen aufweisen und gleicht somit eher einem Syndrom [3]. Eine Rolle bei der Entstehung der Krankheit spielen nach heutigen Erkenntnissen Genetik im Zusammenspiel mit diversen Umweltfaktoren, wie z.B. Rauchen und einem niedrigen sozioökonomische Status [4, 5]. Die Krankheit manifestiert sich typischerweise um das 55. Lebensjahr, und es sind mehr Frauen als Männer betroffen
1 Einleitung 8 [6]. Im Zentrum der Pathologie der RA steht die Entwicklung einer Autoimmunreaktion, mit der Folge, dass Gelenke angegriffen werden. Im Zusammenspiel mit Veränderungen des Bindegewebes wird diese Entzündung vor Ort unterhalten. [7] Das Befallsmuster der entzündeten Gelenke ist typischerweise symmetrisch, sich zentripetal ausbreitend. Ausgangspunkt sind oftmals die Gelenke der Finger sowie die Handgelenke. Pathognomonisch bleiben jedoch die distalen Interphalangealgelenke ausgespart. Ein Befall der großen Gelenke (z.B. Knie, Ellbogen, Schulter) ist nicht ausgeschlossen. [1] Patienten können sich außerdem mit systemischen Symptomen wie Nachtschweiß, Gewichtsverlust und Abgeschlagenheit präsentieren [8]. Hinzu kommt eine Tendenz zu kognitiven Auffälligkeiten sowie Depression [9, 10]. Es ist zudem eine kürzere Lebenserwartung für Patienten mit einer RA festzustellen, vor allem durch ein erhöhtes kardiovaskuläres Risiko. Dies wird einer Vaskulitis als extraartikulärer Manifestation der RA zugeschrieben [11]. Speziell bei schlecht kontrollierten Krankheitsverläufen entstehen mit der Zeit, am Übergang von der Gelenkkapsel zum Knochen, Knorpel- und Knochendefekte. Diese führen zu den RA-typischen Deformitäten, wie der Ulnar-Deviation der Metacarpophalangealgelenke oder Schwanenhals- sowie Boutonnièredeformitäten (Knopflochdeformitäten). [12] [7] Abbildung 1: Klinisches Bild der Hand eines RA-Patienten mit: a | Linien: Achsendarstellung und Ulnardeviation Pfeil: Subluxation im MCP-Gelenk II. b | Pfeil: Schwellung über dem MCP II rechts. c | Pfeil: Knopflochdeformität des Pollex rechts. (Patientenbilder aus der Medizin 3 des Universitätsklinikums Erlangen mit freundlicher Genehmigung von Dr. med. Jürgen Rech)
1 Einleitung 9 Die Erkrankung stellt eine gravierende Einschränkung der Lebensqualität dar. Chronische Schmerzen und eine eingeschränkte Funktionalität können chirurgisch/orthopädische Interventionen notwendig machen. Hinzu kommt eine enorme sozioökonomische Belastung. Weltweit sind 0,5% - 1% der adulten Bevölkerung betroffen. [13] Der chronische Verlauf der Krankheit erfordert eine dauerhafte Therapie, welche sich sehr kostenintensiv gestaltet. Den größten Anteil haben Medikamente, die ambulante Versorgung sowie stationäre Krankenhausaufenthalte. Dabei sind direkte Kosten zu berücksichtigen, die sich 2010 im Schnitt auf über 6000 Euro pro Patient beliefen (die altersentsprechende Kontrollgruppe lag dabei knapp über 3000 Euro) [14]. Die in der Therapie immer mehr eingesetzten Biologika zählen zu den neueren, aber auch teureren Medikamenten. Patienten, welche mit diesen Medikamenten behandelt werden, beanspruchen durchschnittlich über 15000 Euro Behandlungskosten pro Jahr. [14]Indirekte Kosten werden über den Arbeitsausfall und verminderte Erwerbstätigkeit berechnet und belaufen sich je nach Studie auf bis zu 38000 Euro pro Patient pro Jahr. Differenzen innerhalb der Studien sind erklärbar über Unterschiede der Länder und abweichende Gesundheitssysteme. [15] 1.2.1 Diagnosekriterien und Evaluationsscores Die Diagnosekriterien für die RA werden vom American College of Rheumatology (ACR) in Zusammenarbeit mit der European League against Rheumatism (EULAR) festgelegt. In einem Konsens wurden die Kriterien nach 1987 zuletzt 2010 neu evaluiert, um auch ein früheres Stadium einer RA sensibler zu diagnostizieren. Die regelmäßige Neuevaluation ist notwendig, da immerfort neue Erkenntnisse zur RA gewonnen werden, welche grundlegende Veränderung der Diagnostik und Therapie mit sich bringen. Während 1987 noch der Fokus auf den eher spezifischen Kriterien einer RA, wie Rheumaknoten, mehr als 3 geschwollene Gelenke oder bereits radiologisch sichtbare Gelenkserosionen lag, wird heutzutage mit einem sensibleren Scoring System gearbeitet [16]. Dabei wird mehr Abstand von der manifesten Klinik genommen, und es wird zunehmend auf die Serologie geachtet. Dies spiegelt auch die Erkenntnis der Wichtigkeit einer konsequenten, frühen Behandlung wieder [17].
1 Einleitung 10 Nach den Klassifikationskriterien von 2010 gilt bereits ein geschwollenes Gelenk sowie eine fehlende plausible Differenzialdiagnose als verdächtig, und es wird ein Score erhoben. Dieser gilt als positiv zur Klassifikation einer RA, wenn man 6 von möglichen 10 Punkten erreicht. Diese Klassifikation, ergänzt durch das bestätigte klinische Bild durch einen Arzt, führt letztendlich zur Diagnose einer RA. [18] Tabelle 1 Klassifikationskriterien des American College of Rheumatology/European League Against Rheumatism für rheumatoide Arthritis aus 2010 Punkte Zielgruppe (Wer sollte getestet werden?): Patienten welche 1) mindestens ein Gelenk mit einer eindeutig klinischen Synovitis aufweisen (Schwellung) 2) Die Synovitis, durch eine andere Krankheit, nicht plausibler erklärbar ist Klassifikationskriterien für die RA (Punkte-basierter Algorithmus; es werden die Punkte der Kategorien A-D zusammengezählt; ein Punktewert von ≥6/10 wird benötigt für die Klassifikation eines Patienten mit definitiver RA) ‡ A. Gelenkbeteiligung 1 großes Gelenk¶ 0 2−10 große Gelenke 1 1−3 kleine Gelenke (mit oder ohne Gelenkbeteiligung großer Gelenke)# 2 4−10 kleine Gelenke (mit oder ohne Gelenkbeteiligung großer Gelenke) 3 ˃10 Gelenke (mit Beteiligung mindestens eines kleinen Gelenks)** 5 B. Serologie† Negativer RF und negative ACPA 0 Schwach-positiver RF oder schwach-positive ACPA 2 Stark-positiver RF oder stark-positive ACPA 3 C. Akut-Phase Reaktionen† Normales CRP und normale ESR 0 Abnormales CRP oder abnormale ESR 1 D. Symptomdauer§§ ˂ 6 Wochen 0 ≥ 6 Wochen 1 ‡ Obwohl Patienten mit einen Punktewert ˂ 6/10 nicht mit RA klassifiziert werden, kann ihr Status neuevaluiert werden. Es ist möglich die Kriterien kumulativ über einen Zeitraum zu erfüllen. ¶ “Große Gelenke” bezieht sich auf Schultern, Ellenbogen, Hüften, Knie, und Sprunggelenke. # “Kleine Gelenke” bezieht sich auf Metacarpophalangealgelenke, proximale Interphalangealgelenke, zweite bis fünfte Metatarsophalangealgelenke, Daumen, Interphalangealgelenk und Handgelenke. ** In dieser Kategorie muss eins der betroffenen Gelenke ein kleines Gelenk sein; die weiteren Gelenke können jegliche Kombination von großen und kleinen Gelenken sein, sowie weitere nicht explizit aufgeführte (z.B. Temporomandibulargelenk, Acromioclaviculargelenk, Sternoclaviculargelenk etc.) † Es wird mindestens ein Ergebnis für die Klassifikation benötigt §§ Die Symptomdauer bezieht sich auf die Auskunft des Patienten über die Dauer von Symptomen einer Synovitis eines Gelenks (z.B. Schmerzen, Schwellung, Schmerzempfindlichkeit), welches zum Zeitpunkt der Bestandsaufnahme klinisch betroffen ist – unabhängig des Behandlungsstatus. [18]
1 Einleitung 11 Für die Verminderung von Langzeitfolgen ist eine Krankheitsremission anzustreben. Das Ziel ist die Remissionserhaltung durch konsequente Evaluation der Krankheitsaktivität und gegebenenfalls eine Therapieeskalation. Dafür dient der Simplified Disease Activity Index Score (SDAI). Dieser definiert eine Remission der RA ab einem Punktewert von ≤ 3,3. Dabei werden objektive Faktoren wie die Anzahl der geschwollenen Gelenke (Swollen Joint Count = SJC), Anzahl der schmerzempfindlichen Gelenke (Tender Joint Count = TJC) aus 28 vordefinierten Gelenken, das Serum-Level des C-reaktiven Proteins (in mg/dl) mit subjektiven Faktoren gewichtet und verrechnet. Die subjektiven Faktoren beinhalten eine Patienten- sowie ärztliche Einschätzung der Krankheitsaktivität (auf einer Skala von 1-10). [19] = 28 + SJC28 + ℎä + ℎä + In 2011 wurde diesbezüglich in Zusammenarbeit von ACR/EULAR eine Remission der RA definiert, falls alle Punkte der Definition auf Boolean-Basis erfüllt sind, sowie ein SDAI ≤ 3,3 Punkten besteht. Tabelle 2 American College of Rheumatology/European League Against Rheumatism Definition der klinischen Remission in rheumatoider Arthritis Definition auf Boolean-Basis Zu jeder Zeit müssen alle der folgenden Punkte erfüllt sein: Anzahl der schmerzempfindlichen Gelenke ≤ 1† Anzahl der geschwollenen Gelenke ≤ 1† C-reaktives Protein ≤ 1mg/dl Globale Patienten Einschätzung ≤ 1(von 0−10) Definition auf Index-Basis Zu jeder Zeit muss der Patient einen Simplified Disease Activity Index Score ≤ 3,3 aufweisen †Für die Anzahl der schmerzempfindlichen und geschwollenen Gelenke wird eine Summe aus 28 vordefinierten Gelenken genutzt. Da dies limitierend ist wird für die Evaluation einer Remission empfohlen weitere auffällige Gelenke mit einzubeziehen, speziell Füße und Sprunggelenke [19] Bis 2011 wurde mit dem modified Disease Activity Score (DAS28) gearbeitet. Ähnlich aufgebaut wie der SDAI, bezieht der DAS28 die Blutkörperchensenkungsgeschwindigkeit in die Berechnung mit ein (statt CRP-Level) und beinhaltet nicht die ärztliche Einschätzung der Krankheitsaktivität. [20] Die Patienten, deren Fibroblasten in dieser Dissertation untersucht worden sind, wurden ebenfalls mittels DAS28 bezüglich der Krankheitsaktivität eingestuft.
1 Einleitung 12 1.2.2 Einteilung der rheumatoiden Arthritis und serologische Auffälligkeiten Die RA wird primär in 2 Formen eingeteilt, einerseits die seropositive RA und andererseits die seronegative RA. Bei der seropositiven RA, im Gegensatz zur seronegativen RA, finden sich die sogenannten Rheumafaktoren (RF) und/oder Anti-citrullierte-Peptid-Antikörper (ACPAs). [1] Früh gefunden (1940) und mit einer Präsenz von bis zu 75% bei etablierter Krankheit, kann der Rheumafaktor bereits hinweisend auf eine RA sein [21]. Es ist ein Auto- Antikörper, welcher aus verschiedenen Subklassen wie IgG, IgM oder IgA bestehen kann und sich gegen den Fc-Teil humaner IgG-Antikörper richtet [1]. RA-Patienten mit positivem Rheumafaktor haben eine schlechtere Prognose bezüglich Funktionalität und Mortalität. Die Spezifität bezüglich RA leidet jedoch, da dieser Antikörper bei vielen rheumatischen Erkrankungen positiv ist - sogar mit bis zu 5% in der Normalbevölkerung vorliegend - und somit nicht beweisend für die Krankheit ist. [22, 23] Eine weitere große Rolle in der Diagnose der seropositiven RA spielen Antikörper gegen citrullierte Proteine (ACPAs). Die prominentesten Tests sind auf Antikörper gegen cyclic citrullated Protein (anti-CCP) und mutated citrullinated Vimentin (anti-MCV) [18]. Die Citrullinierung von Proteinen ist ein physiologischer Vorgang im Körper, bei dem die Aminosäure Arginin eines Proteins enzymatisch in Citrullin umgewandelt wird [24]. Durch die hohe Spezifität der ACPA-Tests von über 95% sind diese nicht nur ein wichtiges Standbein der Diagnostik, sondern haben auch eine starke Vorhersagekraft für das Auftreten einer RA in Patienten mit undifferenzierter Arthritis [25, 26]. Abhängig vom Test konnte z.B. für anti-MCV auch eine Korrelation mit der Krankheitsaktivität festgestellt werden [26, 27].Durch die im Vergleich zum RF hohe Spezifität - bei ähnlicher Sensitivität - wurde der RF als der wichtigste diagnostischen Antikörper durch ACPAs abgelöst [26]. Jedoch sind die Kosten der ACPA-Tests höher als die der Messung des RF. Zudem besteht eine Korrelation zwischen dem RF und extraartikulären Manifestationen der RA. Somit behält der RF einen Platz in der Diagnostik der RA. [28] Außerdem zu nennen ist das C-reaktive Protein (CRP), welches in der Leber gebildet wird und ein Akut-Phase Protein darstellt. Dieses wird unter Stress (Infektion oder im Falle der RA durch Entzündung) gebildet und ist laborchemisch nachzuweisen. Die
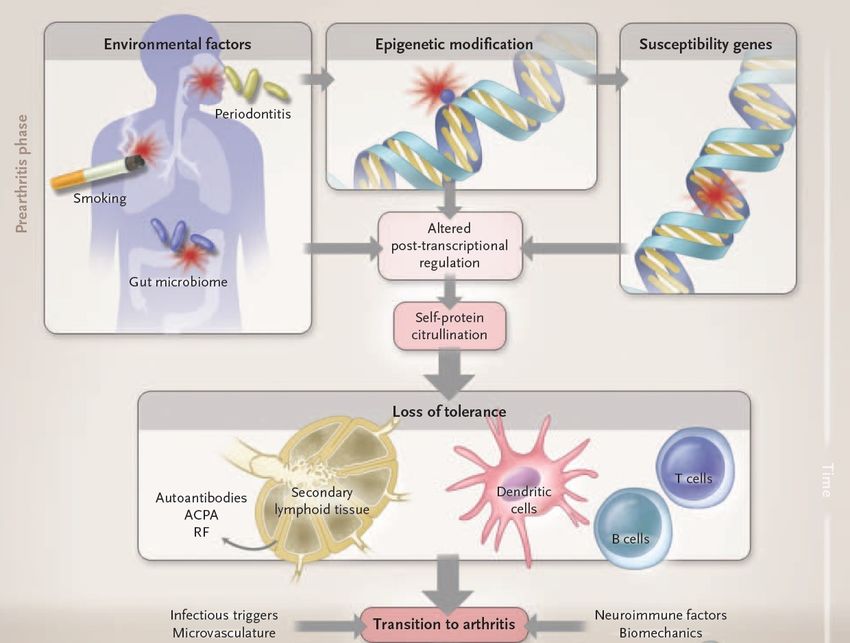
1 Einleitung 13 Blutkörperchensenkungsgeschwindigkeit (BSG, ESR) ist ein indirekter Nachweis für Akut-Phase-Proteine (dazu zählen unter anderem CRP, Ferritin, Haptoglobin) [29]. Man benutzt, mit Citrat versetztes, nicht gerinnbares Blut und misst, wie schnell es sich in Erythrozyten und Plasma differenziert. Diverse, in der Entzündung erhöhte Proteine (Akut-Phase-Proteine) verursachen einen Ladungswechsel der Erythrozyten und somit eine Pseudoagglutination. Durch eine Oberflächenverringerung im Vergleich zu einzelnen Erythrozyten führt dies zu einer schnelleren Sedimentation [30]. Bestimmung des CRPs sowie die BSG weisen eine Infektion/Entzündung nach und können hinweisend für eine RA sein. Mit einer geringen Spezifizität sind diese Verfahren nicht beweisend, allerdings dienen sie maßgeblich der Krankheitsaktivitätskontrolle [18, 20]. 1.2.3 Pathogenese der rheumatoiden Arthritis Die Pathogenese der RA ist noch nicht komplett erforscht und verstanden. Die gut erforschten Zusammenhänge und grundlegende Mechanismen werden in den folgenden Punkten dargelegt. Für die RA wichtige Einflüsse sind die genetische Prädisposition sowie Umweltfaktoren [17]. Diese Trigger bringen den „Stein ins Rollen“, und es entwickelt sich über Monate bis Jahre eine Immunantwort, welche letztendlich Gewebe angreift [31]. Die daraus resultierende Entzündung wird durch sich gegenseitig verstärkende Mechanismen vor Ort nicht nur unterhalten, sondern auch verstärkt [17]. Die Krankheit wird in ihrem Verlauf in Stadien eingeteilt, welche sich nach Antikörperstatus und Klinik richten [7]. Die Gliederungspunkte dieses Kapitels entsprechen der Phase der Isolierung, der in dieser Arbeit untersuchten Zellen. 1.2.3.1 Prä-RA Im Stadium der Prä-RA weist der Patient noch keine klinischen Symptome wie z.B. geschwollene Gelenke auf. Im Blut sind aber bereits Auto-AK nachzuweisen. Das zukünftige Auftreten einer RA ist wahrscheinlicher. [7] Die zwei Arme des menschlichen Immunsystems, das erworbene oder spezifische sowie das angeborene oder unspezifische, arbeiten eng zusammen. Die Leukozyten des erworbenen Immunsystems, hier speziell B- und T-Lymphozyten, erkennen Antigene
1 Einleitung 14 und reagieren spezifisch auf sie. Dabei findet vor der vollen Einsatzfähigkeit dieser Zellen eine Selektion dieser statt, damit körpereigene Zellen von ihren Abwehrmechanismen verschont bleiben. Das angeborene Immunsystem besteht aus Zellbarrieren und Membranen sowie einer zellvermittelten Gegenwehr, die hauptsächlich über antigen-unspezifische Mechanismen wie Phagozytose oder Peptide mit antimikrobiellen Eigenschaften vermittelt wird. Bei Aktivierung der Mechanismen entsteht eine allgemeine Entzündung in Verbindung mit dem Komplementsystem des Körpers. Bestimmte weiße Blutkörperchen, wie die dendritischen Zellen, schlagen die Brücke zwischen den beiden Immunsystemen. Sie gehören zum angeborenen Immunsystem und kommunizieren mit dem erworbenen Immunsystem. Dies erfolgt durch Präsentation von Antigenen phagozytierter Zellen gegenüber T-Zellen. [32] Durch Zwillingsstudien ist ein deutlicher Zusammenhang einer RA im Sinne einer genetischen Prädisposition belegt [4]. Dabei sehr gut erforscht sind diverse Variabilitäten im Gen HLA-DRB1. Das von diesem Gen kodierte Protein ist Teil des MHC II Moleküls. MHC II dient als Oberflächenprotein auf körpereigenen weißen Blutkörperchen zur Präsentation von Antigenen gegenüber T-Zellen. Es werden in der RA eine erhöhte Bindungsaffinität bei bestimmten Abfolgen (sog. Shared Epitopes, SE), oder eine fehlgesteuerte negative Selektion im Thymus vermutet, was zu einer veränderten Immunantwort von T-Zellen führen kann [33, 34].
1 Einleitung 15 Abbildung 2: Pathogenese der rheumatoiden Arthritis, modifiziert nach [17] Zudem sind diverse Umweltfaktoren zu nennen, die im Verdacht stehen - vor allem zusammen mit einer genetischen Prädisposition - eine Auto-Immunantwort zu provozieren. [35]. Bronchialer Stress und speziell Rauchen fördern die Entstehung einer RA. Obwohl der dafür zuständige Pathomechanismus nicht gänzlich geklärt ist, konnte gezeigt werden, dass Rauchen das Immunsystem moduliert. Es ist verantwortlich für eine Reduktion natürlicher Killerzellen und der Aktivität des Zell-vermittelten Immunsystems sowie für dysfunktionale T-Zellen. Das sind Faktoren, welche in Zusammenhang mit der Entstehung und Aufrechterhaltung der RA stehen [17, 36]. Des Weiteren wurde festgestellt, dass Periodontitis, unspezifische Entzündungen/Infekte (speziell durch E.coli, EBV und Proteus-Spezies) sowie das Mikrobiom des Darms eine Rolle bei der Entstehung spielen. Erklärt wird das durch molekulares Mimikri. E. coli besitzt das Hitzeschockprotein (HSP) 65 und EBV das Peptid p107, welche köpereigenen Proteinen ähneln und somit eine Autoimmunität fördern. P. gingivalis besitzt nicht nur eine Enolase, die der des menschlichen Körpers ähnlich ist, sondern durch die Peptidyl Arginin Deiminase (PAD) auch die Fähigkeit, Citrullierungen von Mucosa-Proteinen zu verändern. Toleranzverlust gegenüber diesen neuen Epitopen
1 Einleitung 16 führt zur Bildung von ACPAs [37]. Einige Studien fanden auch in Rauchern eine verstärktes Auftreten von citrullinierten Proteinen und eine erhöhte Expression von PADs in der Lunge, was aber in anderen Studien nicht bestätigt werden konnte [38, 39]. 1.2.3.2 Early-RA Die Früh-RA weist im Vergleich zur Prä-RA zusätzlich zu serologischen Auffälligkeiten auch erstmalig entzündliche Gelenkmanifestationen auf. Die Translationsphase von Prä-RA hin zur Early-RA ist bis heute nicht gänzlich verstanden. Es werden gelenklokale Mikrotraumata sowie mikrovaskuläre Mechanismen diskutiert. [7] Die Entzündung eines Gelenkes entspricht einer Entzündung der Gelenksynovialis (Synovitis). Als Ausgang der Entzündungseskalation sind proinflammatorische Cytokine zu sehen [3]. Eine erhöhte Expression von Integrinen (Transmembranproteine) und Cytokinen bewirkt eine endotheliale Aktivierung, welche Leukozyten eine vermehrte Einwanderung ins Gewebe ermöglicht [17]. Cytokine sind Proteine, die die Aufgabe von Botenstoffen und Attraktantien erfüllen. Zudem regulieren sie Zellproliferation und Differenzierung [40]. Diese Cytokine und lokal hypoxische Zustände induzieren eine Neoangiogenese. Somit sind die Kennzeichen einer Synovitis erfüllt: Einwanderung von Zellen, Cytokinüberexpression, Hypervaskularität und somit Hypertrophie [41]. Mit diesen Voraussetzungen entsteht durch Reorganisation des lokalen Bindegewebes und Aktivierung der Fibroblasten innerhalb des Synoviums das klassische Bild eines entzündlichen Synovialgewebes der RA (Pannus) [17].
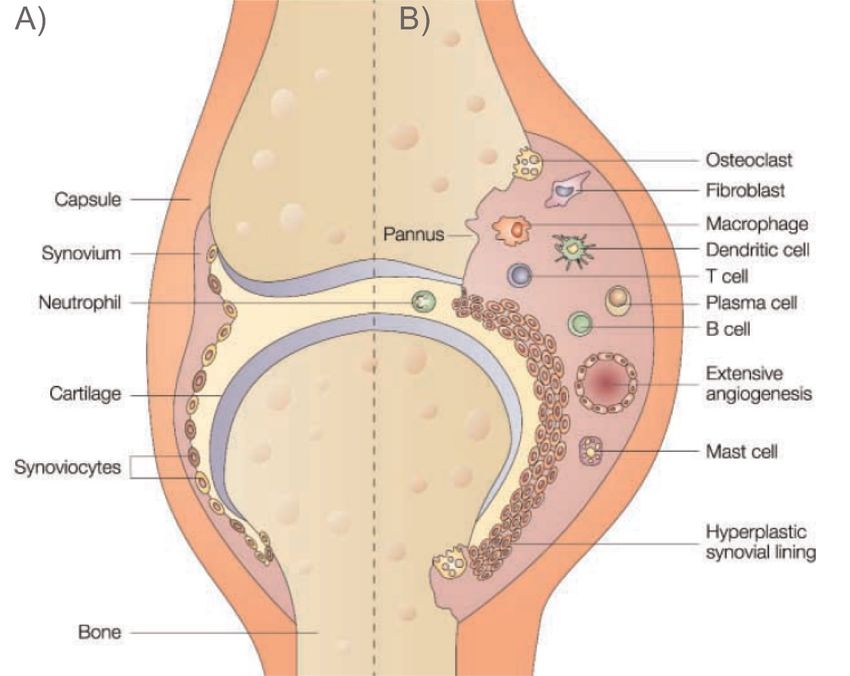
1 Einleitung 17 Abbildung 3: Schema eines Gelenks, modifiziert nach [42] mit A | Querschnitt eines gesunden Gelenkes und B | eines entzündlich veränderten Gelenkes mit hypertrophem Synovium im Sinne einer RA 1.2.3.3 Established-RA Für die manifeste Entzündungsreaktion des Gelenkes spielen T-Zellen eine wichtige Rolle [43]. T-Zellen differenzieren sich in einer frühen Form im Thymus und übernehmen unterschiedliche Aufgaben [32]. Für Autoimmunkrankheiten, speziell die RA, sind zwei Subtypen wichtig: Th1-Zellen, welche eine zellvermittelte Immunantwort bedingen, und Th17-Zellen [44]. Sie wirken proinflammatorisch und beeinflussen B-Zellen und ihre AK-Produktion [45]. Zudem gibt es regulierende T-Helfer Zellen, welche eine Immunsuppression und -toleranz fördern [46].
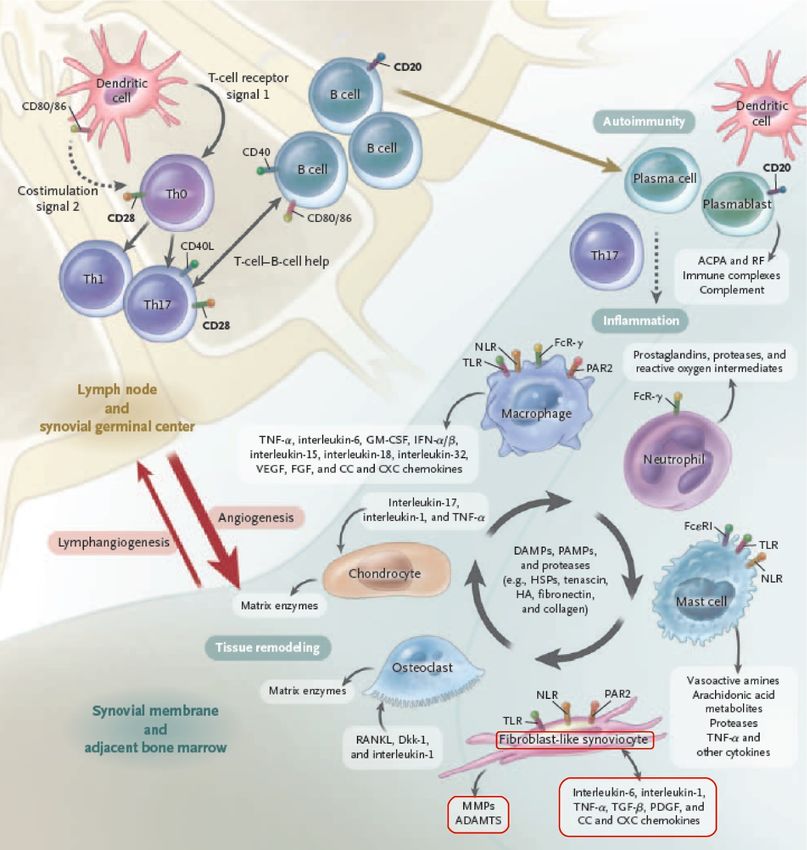
1 Einleitung 18 Abbildung 4: Zelldifferenzierung von T-Zellen, modifiziert nach [47] Die Kommunikation zwischen Leukozyten erfolgt über Interleukine, eine Gruppe der Cytokine [32]. Die nach der Translationsphase von prä-RA auf early-RA aktivierten synovialen Zellen exprimieren vermehrt Interleukin (IL) 1, 6, 21 und 23 sowie Tumornekrosefaktor α (TNF α) [17]. Dieses Milieu fördert vor Ort einerseits eine T-Zell Differenzierung zu proinflammatorischen Th17 und hemmt andererseits die Ausbildung von antiinflammatorischen regulierenden T-Helfer Zellen (Tregs) [46]. Th17 Zellen wiederum sezernieren IL 17A, IL 17F, IL 21, IL 22 und TNF α [17]. Mit Einwanderung von T- und B-Zellen durch o.g. Mechanismen legen sich diese in Aggregaten innerhalb des Synoviums zusammen und bilden teilweise sogar ektope Lymphfollikel [48]. IL 17A, IL 1 und TNF α wirken synergistisch und stimulieren synoviale Fibroblasten (SFB) und Chondrozyten. Eine Produktion von Cytokinen durch die stimulierten Synovialzellen lässt einen sich selbst unterhaltenden Kreislauf entstehen [49].
1 Einleitung 19 Abbildung 5: Pathogenese der Inflammation und die sich unterhaltenden Mechanismen innerhalb der synovialen Membran, modifiziert nach [17] Durch die fortwährende Entzündung und die stimulierten Zellen im Synovium (hauptsächlich bestehend aus SFB und Makrophagen), gehen die protektiven Effekte des Synoviums verloren. Somit steuert die entzündete Gelenkhaut einen großen Beitrag an entstehenden Knorpelschäden bei [50]. Radiologisch bereits früh nachweisbar ist eine Verschmälerung des Gelenkspaltes, die dem Knorpelabbau entspricht. Zudem ist der Abbau von Knochenstruktur zu beobachten, welcher durch gelenksnahe osteolytische Zonen sichtbar wird [51].
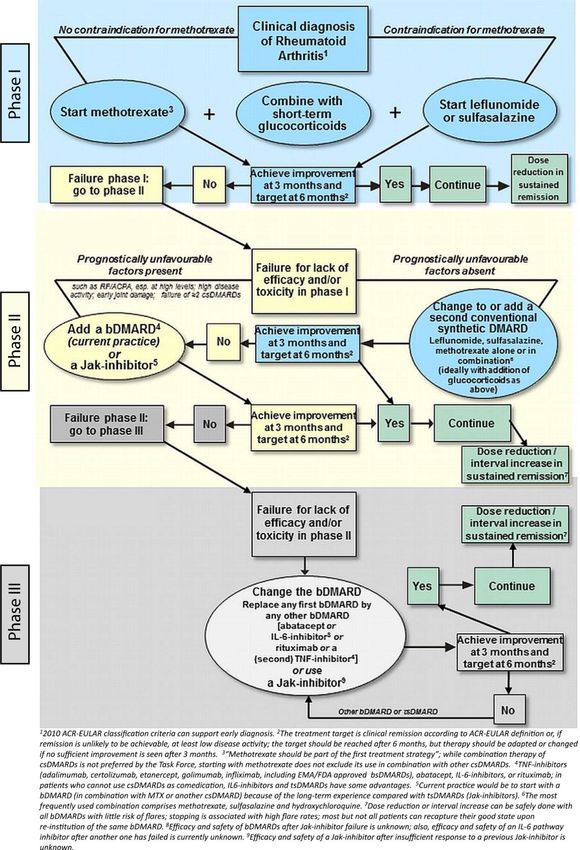
1 Einleitung 20 1.2.4 Therapie der rheumatoiden Arthritis Die Therapie der rheumatoiden Arthritis hat sich in den letzten 30 Jahren drastisch geändert [52]. Durch bessere Erforschung der Pathogenese fanden sich auch zusätzliche therapeutische Angriffspunkte [53]. Wenn früher eine Remission unter Therapie als ehrgeizig erachtet wurde, ist sie heutzutage das primäre Ziel [54]. Die in 1.2.1 genannten Klassifikationskriterien der ACR/EULAR von 2010 erlauben eine frühere Diagnose und damit eine frühere Therapie. Die Empfehlung zum Therapiestart ist ab Diagnosestellung [52]. Eine klinische Remission mittels Boolean Definition oder SDAI ist elementar, um den Progress von Erosionen zu stoppen [55]. Der Patient gewinnt dadurch Lebensqualität, die Arbeitsfähigkeit wird gefördert, und Komorbiditäten werden gesenkt [56, 57]. Dafür werden diverse Medikamente eingesetzt, welche immunsuppressive und/oder antiinflammatorische Eigenschaften haben [52]. Dabei sind die conventional synthetic (cs) disease modifying anti-rheumatic drugs (DMARDs) als Basis der Therapie zu sehen [52]. Methotrexat (MTX) als Vertreter der cs DMARDs zeichnet sich durch eine sehr gute Wirksamkeit aus und wird als „Anchor Drug“ bezeichnet. Die langjährige Erfahrung gewährt zudem eine Sicherheit der Therapie [58].Mit den Zulassungen der biological (b) DMARDs wurden sehr effektive Medikamente in den Markt eingeführt, um vor allem schwere Krankheitsverläufe zu therapieren. Zusätzlich gibt es seitdem eine Möglichkeit der Therapieeskalation. [59] Des Weiteren sind neuerdings zwei target synthetic (ts) DMARDs in Europa zugelassen, die erstmals keine Immunzellen oder deren Botenstoffe generell inhibieren, sondern direkt - auf Zellebene - in die Entzündungskaskade eingreifen. Dadurch ist eine neue Eskalation in der Therapie möglich geworden [60, 61].
1 Einleitung 21 Tabelle 3 Medikamente Medikamentengruppe Wirkstoffbeispiele Wirkmechanismus in Bezug auf die rheumatoide Arthritis (cs) DMARDs Methotrexat (MTX) Folsäure Antagonist conventionall synthetic Leflonumid Pyrimidin Stoffwechsel Inhibitor Sulfasalazin Immunsuppressiv * Hydroxychloroquine Immunmodulation † (GC) Glucocorticoide Immunsuppresiv # (b) DMARDs TNF-α Blocker § TNF- α Inhibitor biological Abatacept T-Zell Inhibitor Rituximab B-Zell Inhibitor Anakinra IL-1 Inhibitor Tocilizumab IL-6 Inhibitor Clazakizumab IL-6 Inhibitor Sarilumab IL-6 Inhibitor Sirukumab IL-6 Inhibitor (ts) DMARDs Tofacitinib Janus Kinase Inhibitor target synthetic Baricitinib Janus Kinase Inhibitor * Wirkmechanismus multifaktoriell über Hemmung von IL-1, TNF- α und Induktion der Apoptose in Entzündungszellen † Wirkmechanismus über Hemmung der Komplement- und Ag-AK-Reaktion sowie Hemmung bestimmter Proteinaktivität (Kollagenasen; Proteasen) # Wirkmechanismus über eine Steigerung der Expression immunsuppressiver Proteine § dazu zählen Adalimumab, Certolizumab pegol, Etanercept, Golimumab, Infliximab Das Schema der Therapie wird wie auch die Klassifikationskriterien von ACR/EULAR regelmäßig evaluiert und angepasst. Mit der letzten Veröffentlichung 2016 ist der folgend dargestellte Algorithmus aktuell.
1 Einleitung 22 Abbildung 6: Therapieschema nach den EULAR Empfehlungen 2016, modifiziert nach [52] Trotz der sich mehrenden Therapieoptionen bleibt die Remission als oberstes Ziel oft unerreicht. Das zeigt das nicht-Ansprechen, trotz einer Therapieeskalation auf (b)DMARDs in vielen RA-Patienten. Der Bedarf von weiteren stark wirksamen oder
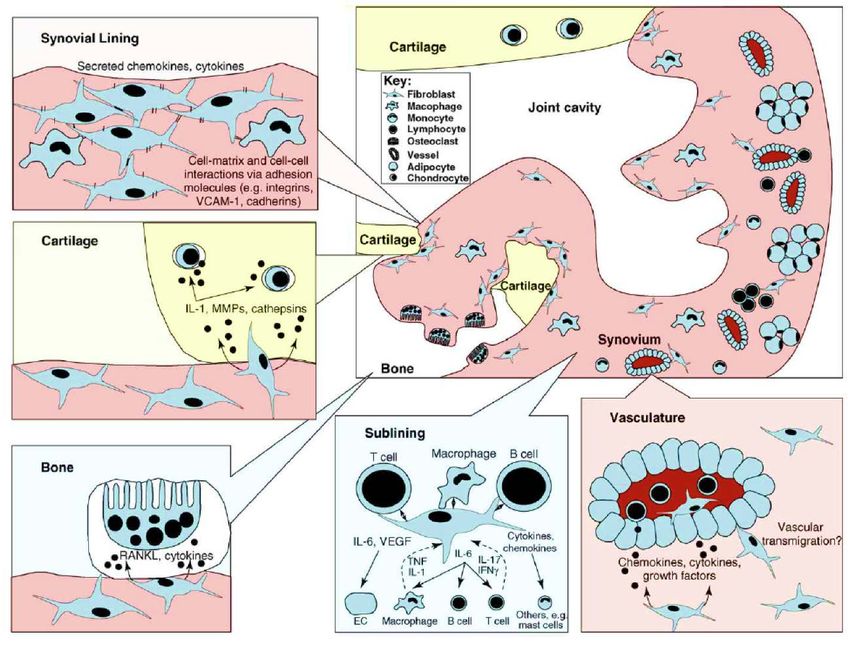
1 Einleitung 23 synergistischen Therapien ist die Folge. [62] Des Weiteren ist es nicht möglich, die Krankheit nach dem heutigen Stand zu heilen. Das verdeutlicht ebenfalls, dass andere krankheitsunterhaltenden Mechanismen als Ansatzpunkt der Therapie dienen müssen. Solch ein neuer Ansatzpunkt für die Therapie ist der synoviale Fibroblast, der die Fähigkeit besitzt, die Entzündung vor Ort zu unterhalten. Zudem ist er der Ausgangspunkt für Knochen- sowie Knorpeldegeneration [63]. 1.2.5 Rolle der synovialen Fibroblasten in der rheumatoiden Arthritis Die synovialen Fibroblasten (SFB) sind erst seit Kurzem als wichtige Vermittler der rheumatoiden Arthritis erkannt worden. Bei stetig steigendem Verständnis der Pathogenese rücken sie weiter in den Fokus [64]. Grundlegend für die Pathogenese ist der anatomische Aufbau des Gelenks und Synoviums sowie die Lage und Typisierung der SFB. Ein Gelenk bildet sich aus den beiden angrenzenden Knochen, die an der in Kontakt stehenden Seite, mit weichem gleitfähigem Knorpel überzogen sind. Umschlossen und verstärkt durch eine bindegewebsderbe Schale (Gelenkkapsel) liegt auf deren Innenseite eine dünne Zellschicht auf (Synovium). Das Synovium ist unterteilt in eine zellarme, hauptsächlich aus Extrazellularmatrix bestehende, äußere Schicht und eine dünnschichtige, innere Membran. Die innere Schicht ist für die Homöostase der Gelenkflüssigkeit verantwortlich und ernährt darüber hinaus gleichzeitig den nicht vaskularisierten Knorpel. Die Zelltypen dieses Synoviums werden unterteilt in Typ A Zellen (Makrophagen-ähnliche Synovialzellen) und Typ B Zellen (Fibroblasten- ähnliche Synovialzellen / synoviale Fibroblasten). [49] Es kommt durch eine vorangehend beschriebene Gelenkentzündung zu einer Zellaktivierung und Zelleinwanderung in das Synovium [3]. Eine Zellaggregation an der knochennahen Übergangszone ist zu beobachten und wird als Pannus beschrieben. Dieser Zellhaufen besteht primär aus Makrophagen und SFB [65], es kommt aber auch zur Einwanderung von anderen Zellen des angeborenen (wie etwa neutrophilen Granulozyten) und des adaptiven Immunsystems (wie etwa versch. Arten von T-Zellen). Durch diese, durch Cytokine stimulierten (speziell IL-1 und TNFα) SFB und Chondrozyten werden Matrix-Metalloproteasen (MMPs) gebildet, die Knorpel zersetzen und abbauen [66]. Vor allem aktivierte synoviale Fibroblasten, Makrophagen und T-Zellen sezernieren das Protein receptor activator of nuclear factor κ B ligand
1 Einleitung 24 (RANKL) [67]. Dies induziert eine Zelldifferenzierung von prä-Osteoklasten zu Osteoklasten und veranlasst einen Knochenabbau der Übergangszone [68]. Diese Läsionen führen zu starken Schmerzen und ultimativ zu gravierenden Funktionalitätseinbußen für die Patienten [3]. Abbildung 7: Pathomechanismen der rheumatoiden Arthritis auf Zellebene innerhalb der synovialen Membran, modizifiert nach [69]
1 Einleitung 25 Aktivierte Fibroblasten bilden einen eigenen aggressiven Phänotyp, der oft über längere Zeiträume aufrechterhalten wird. Bottini et al. konnten bereits zeigen, dass aktivierte humane synoviale Fibroblasten in kompletter Abwesenheit von autoimmunen Triggern selbst nach Monaten noch Knorpel abbauen. Sie handeln nach dem Muster eines „imprinted agressor“ und sind keine „passive responders“, unterhalten somit aktiv den Entzündungsstatus im Gelenk. Die gleiche Forschungsgruppe veranschaulichte ebenso eine sogenannte Metastasierung dieses aggressiven Phänotyps [70]. Lefevre et al. konnten in einem ähnlichen Versuchsaufbau zusätzlich noch die Hypothese stützen, dass zumindest teilweise SFB die Krankheit auf bisher nicht betroffene Gelenke ausbreiten können. Die Erklärung liegt in einer Migration der aktivierten SFB [71]. SFB können nicht nur in ihrem Phänotyp persistieren, sondern, wie Valencia et al. zeigen konnten, es in vitro intuitiv schaffen ihre Formation wie in vivo einzunehmen. Die Fibroblasten legen sich in diesen in vitro-Versuchen in ähnlich dünnen Schichten, gleich dem Synovium, zusammen. Diese Eigenschaft wurde in den Versuchen dieser Doktorarbeit in einer 3D Zellkultur genutzt. Das transmembrane Schlüsselprotein für diese Eigenschaft ist Cadherin-11, welches Zelladhäsion vermittelt. In Abwesenheit dieses Proteins konnte bei Tierversuchen eine Knochendestruktion verhindert werden. [72, 73] Mit den dargestellten Eigenschaften des angeborenen Immunsystems kommt die Vorstellung, dass nur das adaptive Immunsystem ein Gedächtnis ausbilden kann, ins Wanken. Speziell Fibroblasten sind in der Lage, sich durch Entzündung langfristig zu verändern und in einem aggressiven Phänotyp zu persistieren. Grundlage dafür sind epigenetische Veränderungen. Dieser Vorgang wird als „trained immunity“ bezeichnet. Das ist eine schwerwiegende Folge für RA Patienten, bei denen ein Progress der Krankheit unter anderem auf diesem Pathomechanismus beruht. SFB als ein zentraler Punkt der Pathogenese bedürfen weiterer intensiver Forschung, um die Aufrechterhaltung von Entzündung und Gelenksdestruktion zu verstehen und effektiver zu verhindern. [74]
2 Zielsetzung 26 2 Zielsetzung Das Ziel der vorliegenden Arbeit ist das tiefergehende Verständnis der Pathogenese der rheumatoiden Arthritis in Bezug auf die synergistischen Mechanismen einer Entzündungsunterhaltung. Als potentielle Ursache der daraus resultierenden Chronifizierung der Entzündung wurden molekulare Mechanismen und die Veränderung des Phänotyps synovialer Fibroblasten, als Bestandteil des angeborenen Immunsystems, untersucht. Dazu wurden humane synoviale Fibroblasten aus verschiedenen Phasen der rheumatoiden Arthritis isoliert und analysiert. Dies bietet die Möglichkeit, Morphologie, Funktionalität und veränderte Expressionsmuster zu vergleichen. Ein tieferes Verständnis in Folge von Zellexperimenten kann Aufschluss über mögliche neue Therapiekonzepte liefern, welche einen denkbare Chronifizierung verhindern könnten.
3 Material und Methoden 27 3 Material und Methoden Die im Folgenden verwendeten Materialien sind dem Appendix zu entnehmen. 3.1 Zellgewinnung Es standen 15 SFB Zellreihen der Rheumatology Research Group des Institute of Inflammation and Ageing (IIA) der University of Birmingham des Queen Elizabeth Hospital in Birmingham in Passage 4 oder 5 zur Verfügung. Diese wurden von 3 Patientengruppen gewonnen. Patienten mit erst kurzzeitig symptomatischer RA (Very early RA/ VERA) und seit langen bestehender symptomatischer RA sowie aus Patienten ohne diagnostizierter RA (normal). Wir erhielten 2x105- 5x105 Zellen pro Zellreihe, welche auf Trockeneis verschickt wurden. Die Zellen kamen in eingefrorenem Zustand an. Tabelle 4 Kategorisierung der Fibroblasten Normal VERA RA BX070 BX003 BX093 BX089 BX063 BX118 BX095 BX141 BX150 BX096 BX084 BX151 n=5 n=5 n=5 Zudem wurden SFB aus Gelenkpunktaten der medizinischen Klinik 3 des Klinikums Erlangen gewonnen (zur Verfügung gestellt von Dr. Kleyer). Das Punktat wurde im ambulanten oder stationären Rahmen gewonnen. Es wurde Synovialflüssigkeit bei akutem Gelenkerguss steril abpunktiert. Einschlusskriterium war eine Weiterverarbeitung innerhalb von 4 h. Die angezüchteten SFB wurden in Vorversuchen verwendet. Zellgewinnungs- und Kultivierungsmethoden fanden unter sterilen Bedingungen auf einer Reinraumwerkbank statt. Das Gelenkpunktat wurde mit im Wasserbad auf 37 °C erwärmten Fibroblastenmedium (FB-Medium, Zusammensetzung siehe Punkt 3.2.2) im Verhältnis 1:1 verdünnt und mit 1800 rpm für 5 min zentrifugiert. Der Überstand wurde abgenommen, das entstandene Zellpellet in FB-Medium aufgenommen und, abhängig von der Zellzahl, in eine T25 oder T75 Zellkulturflasche überführt. Danach wurde die Zellkulturflasche bei 37 °C und 5% CO2 mit angefeuchteter Luft in einem Brutschrank kultiviert.
3 Material und Methoden 28 3.2 Zellkultivierung Folgende Abschnitte beziehen sich auf untenstehende Flaschengrößen und dementsprechende Füllmengen sowie Grenzen zur Flaschenteilung Tabelle 5 Zellkulturflaschen Flasche Fläche Volumen Zellanzahl Füllmenge von Medium Trypsin T25 25 cm2 50 ml 5x105 24ml 7ml Methodik der Zellkultivierung nach Flaschengröße mit Beschreibung der Grenzwerte für Flaschenteilung und der Füllmengen der Reagenzien 3.2.1 Auftauprozess Eingefrorene Zellen wurden in kurzer Zeit im 37 °C Wasserbad aufgetaut und 12 ml FB-Medium zugegeben. Daraufhin wurden die Zellen in eine T75 Zellkulturflasche überführt und bei 37 °C und 5% CO2 mit angefeuchteter Luft in einem Brutschrank kultiviert. 3.2.2 Mediumwechsel Als Medium wurde humanes FB-Medium (RPMI 1640 ergänzt mit 10% fetalem Kälberserum, 0.87X MEMNon-essential amino acids, 0.87mM Sodium Orthopyruvat, 1.75mM Glutamin, 87U/ml Penicillin, 87µg/ml Streptomycin) verwendet, welches ein oder zwei Mal pro Woche, abhängig von Proliferation und Medium-Umsatz der zu kultivierenden Zellen, gewechselt wurde. Als Mediumwechsel wurden 2/3 des Mediums abgenommen und mit frischem Medium aufgefüllt. 3.2.3 Passagieren der Zellen Bei 90% Konfluenz der Zellen oder nach durch Auszählung festgestellter Überschreitung der Zellzahl in der o.g. Tabelle wurden die Zellen abgelöst und passagiert. Dafür wurde das benutzte Medium abgenommen und zwischengelagert. Die Flasche wurde einmalig mit PBS gespült, da das im FB-Medium beinhaltete FCS die Trypsinaktivität inhibiert [75]. Trypsin/EDTA Lsg. wurde hinzugegeben bis der Flaschenboden gleichmäßig benetzt war. Daraufhin wurde die Flasche so kurz wie möglich (maximal 5 min), aufgrund der Zelltoxizität des Trypsins erneut bei 37 °C im Brutschrank kultiviert, bis, kontrolliert
3 Material und Methoden 29 mittels Lichtmikroskop, sich alle Zellen abgelöst hatten. Für eine komplette Ablösung der Zellen war unter Umständen ein leichtes Beklopfen der Flasche notwendig. Das Trypsin/EDTA Lsg. Zellgemisch wurde mit 8 ml frischem FB-Medium verdünnt und bei 1200 rpm für 6 min zentrifugiert, was zur Bildung eines Zellpellets führte. Nach Abnahme des Überstands wurde das Zellpellet mit einem Teil zwischengelagertem benutztem Medium und zwei Teilen frischem Medium aufgelöst und auf eine nächstgrößere Zellkulturflasche überführt. Abbildung 8: Passagierschema nach Auftauen von einer Cryovial 3.2.4 Cryokonservierung In folgenden Versuchen wurden ausschließlich Zellen der Passage 4-8 verwendet. Eine Cryokonservierung wurde nötig, falls Zellreihen unterschiedlich schnell proliferierten, um diese zu konservieren und zu einem späteren Zeitpunkt wiederzuverwenden.
3 Material und Methoden 30 Abbildung 9: Passagierschema mit Zwischenfrieren Das Lösen der Zellen aus den Zellkulturflaschen erfolgte wie unter 3.2.3 beschrieben. Es erfolgte eine Zentrifugation über 6 min mit 1200 rpm. Nach Abnahme des Überstandes wurde das Zellpellet mit 1800 ml FCS verdünnt, in ein Cryovial überführt und mit 200ml DMSO versetzt. Das fibroblastenhaltige Cryovial mit 10% DMSO in FCS Lsg. wurde, aufgrund der Zytotoxizität von DMSO, sofort bei -80 °C gefroren und am Folgetag in flüssigen Stickstoff umgelagert [76]. 3.2.5 Zellzählung Zur Bestimmung der Zellkonzentration wurde eine Zählkammer nach Neubauer benutzt. Hierbei wurde ein Teil der zu bestimmenden Lösung abgenommen und mit Trypanblau verdünnt. Dies erlaubt eine Differenzierung zwischen lebenden und toten Zellen, welche sich blau anfärben lassen [77]. Dann wurden alle ungefärbten, vitalen Zellen in allen 4 Quadranten der Zählkammer ausgezählt. Jeder der 4 Quadranten hat ein Volumen von 0,1 µl (Fläche von 1 mm2 mit einer Kammerhöhe von 0,1 mm). Mit folgender Formel lässt sich somit die Gesamtzahl der vitalen Zellen bestimmen: ℎ äℎ 4 = × ü × 10 4
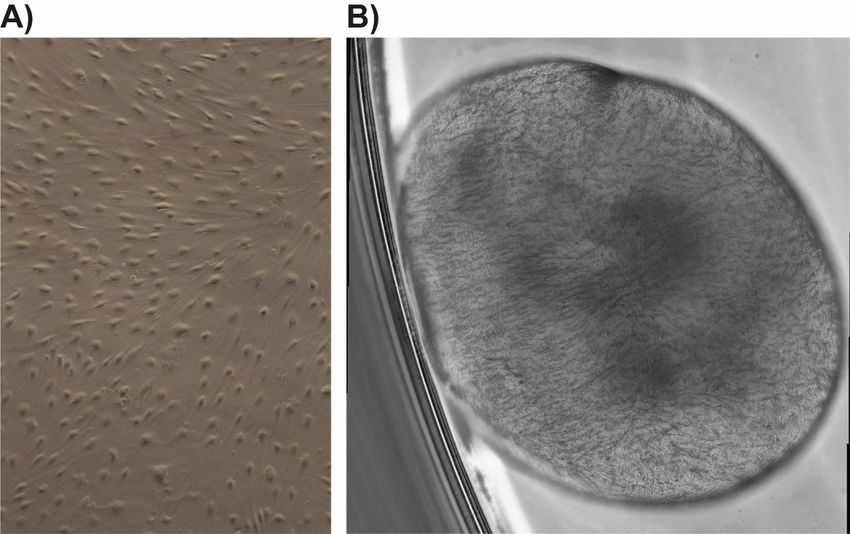
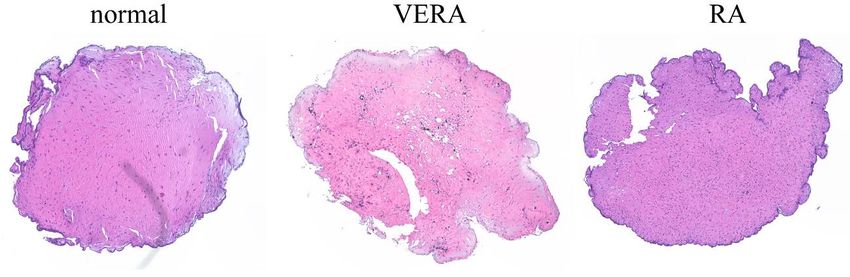
3 Material und Methoden 31 3.3 Experimente 3.3.1 Micromass Die Micromass ist eine 3D-Zellkultur, bestehend aus einem Gerüst (von Mäusen stammende Extrazellulärmatrix), welche mit Fibroblasten besiedelt wird [78]. Im folgenden Versuch wird ein Synovium imitiert. Kultivierte Fibroblasten, die aus menschlichen Synovialmembranen isoliert wurden, behalten ihre Eigenschaft, dünne Synovium-ähnliche Zellschichten zu bilden auch in vitro [72]. Die Zellreihen wurden bis in Passage 4-8 kultiviert und hiernach abgelöst (siehe 3.2.3). Nach Auszählen der Zellen (siehe 3.2.5) wurden 1,04 x 106 Zellen bei 1500 rpm für 8 min abzentrifugiert. Das so gewonnene Zellpellet wurde in 130 µl Matrigel (auf Eis gelagert) aufgenommen und als Triplikat in eine mit PolyHEMA beschichtete 12-Well-Platte pipettiert. Danach erfolgte die Kultivierung der mit Zelltröpfchen bestückten Well-Platten für 45 min bei 37 °C im Brutschrank. Anschließend wurden 2 ml Micromass-Medium (DMEM/F12 ergänzt mit 10% fetalem Kälberserum, 1% Penicillin/ Streptomycin, 0,2% ITS Supplement (500x), 0,1 mmol/L Ascorbinsäure) zu jedem Tropfen hinzugegeben. Das Medium wurde alle 2 Tage ausgetauscht. Die angehenden Micromasses wurden über 21 Tage kultiviert, bis sich eine vollständige 3D-Zellkultur gebildet hatte. Für die Auswertungen wurden die Micromasses mit HBS/Ca [1 mM] gewaschen und über Nacht mit 2 ml 2% Paraformaldehyd in HBS/Ca [1 mM] gelagert. Am Folgetag wurde die Fixierungslösung gegen 2 ml 70% Ethanol ausgetauscht.
3 Material und Methoden 32 Abbildung 10: Micromassentwicklung im zeitlichen Verlauf. Draufsicht auf den Rand der Micromass mit Zusammenziehen bei Zunahme der Sphärizität (nach 1h, 38h und 64h). Unteres Bild entsprechend einer kompletten Micromass nach Ablösen vom Well-Boden und freiem Schwimmen im Medium (nach 20 d Kultivierung). 3.3.1.1 PolyHEMA Beschichtung Zum Beschichten wurde 1g Poly-2-hydroxyethylmethacrylate in 100 ml 98% Ethanol auf einer heißen Platte unter ständigem Rühren aufgelöst. Davon wurde jeweils 1 ml in die Vertiefungen einer 12-Well-Platte gegeben und bei 40 °C gelagert bis das Ethanol vollständig verdampft war. Als Folge dieses Vorgehens blieb eine PolyHEMA Beschichtung zurück. 3.3.1.2 Analyse der Micromasses Die in 70% Ethanol gelagerten 3D-Zellkulturen wurden zur Analyse in Paraffin gebettet. Per Zitadelle wurden 1-2 µm dicke Schnitte gefertigt, welche dann gefärbt
3 Material und Methoden 33 werden konnten (siehe 3.3.2). Mittels Osteomeasure-Software wurde die Schichtdicke, per Adobe Photoshop Zirkularität und Interkonnektivität bestimmt. Für die Interkonnektivität wurde die durchschnittliche Anzahl der Zellen, welche in Aggregaten liegen, berechnet. Die Zirkularität wurde über folgende Formel bestimmt: ä ℎ = 4 × ( 2 ) 3.3.1.3 Durchflusszytometrie für Cytokinbestimmung Es wurden die Cytokin- und Chemokin- Konzentrationen im Micromass-Medium gemessen, welches alle 2 Tage ausgetauscht wurde. Es stand ein LegendplexTM Panel der Firma Biolegend zur Verfügung. Die Bestimmung erfolgt über fluoreszenz- markierte Antikörper, welche spezifische Cytokine binden. Daraufhin wurde ein Biotin-gekoppelter Antikörper zusammen mit Streptavidin-PE hinzugegeben, was zu einer roten Fluoreszenz führt. Dieses Signal konnte mittels Durchflusszytometrie quantifiziert werden. Die Durchführung erfolgte durch eine Verdünnung von 25 µl jeder Probe mit 25 µl Bead mix (1:50 verdünnt mit Assay Pufferlösung) sowie 25 µl Detektionsantikörper (1:10 verdünnt mit Assay Pufferlösung) und 25 µl Assay Pufferlösung. Die entstehende Lösung wurde mit 600 rpm für 2 h bei Raumtemperatur und in Dunkelheit geschüttelt, bevor 25 µl Streptavidin-PE (1:10 verdünnt mit Assay Pufferlösung) hinzugegeben wurden. Daraufhin wurden die Proben ein weiteres Mal mit 600 rpm für 2 h bei Raumtemperatur in Dunkelheit geschüttelt. Nach der folgenden Zentrifugation (5 min bei 1000xg) wurde der Überstand abgenommen. Die Proben wurden mit 200 µl Wasch-Pufferlösung gewaschen und ein weiteres Mal zentrifugiert (5 min bei 1000xg). Die fertigen Proben wurden auf gleichmäßige 300 µl aufgefüllt und mittels Durchflusszytometrie gemessen. Die Auswertung erfolgte mit dem Kaluza Programm Version 1.5. 3.3.2 Rasterelektronenmikroskopie Zur Beurteilung der SFB-Morphologie wurden Aufnahmen mit einem Tescan Lyra 3 Rasterelektronenmikroskop mit 5kV Beschleunigungsspannung gemacht (Zusammenarbeit mit Lasse Kling, Max Planck Institute for the Science of Light, Erlangen). Dazu wurden 22 mm x 22 mm runde Deckgläser mit Fibronektin beschichtet (500µl bei einer Konzentration von 50µg/ml in PBS bei 4 °C über Nacht). Anschließend konnten
Sie können auch lesen

















































