PICS: Statistische Analyse von CHIP-seq Daten
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
PICS: Statistische Analyse von CHIP-seq
Daten
Ausarbeitung zum Seminar “Biomedical Informatics” im WS 2012/2013
Sandra Uhlenbrock
10. Dezember 2012
1 Einleitung
Diese Ausarbeitung basiert auf “PICS: Probabilistic Inference for ChIP-seq” von Zhang
et al. (Biometrics 2011) und befasst sich mit einer statistischen Methode zur Analyse von
großen ChIP-seq Datenmengen. Alle verwendeten Abbildungen sind der oben genannten
Veröffentlichung entnommen.
Ziel des Verfahrens ist es, aus den gegebenen ChIP-seq Daten die Bindungsstellen der
Proteine zu schätzen. Bevor ich näher auf die statistischen Methoden eingehe, werde ich
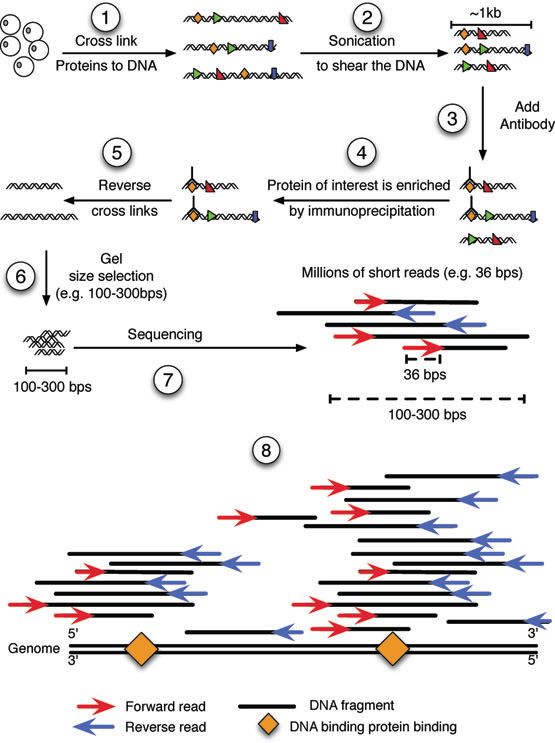
zunächst kurz beschreiben, wie die gegebenen ChIP-seq Daten entstehen beziehungs-
weise woraus sie sich zusammensetzen. Dieses Verfahren ist ebenfalls in Abbildung 1
dargestellt.
In einem ersten Schritt werden die Proteine mit der DNA vernetzt und man erhält Chro-
matin. Mit Hilfe von Ultraschall wird dann in einem zweiten Schritt die DNA mit den
gebundenen Proteinen aus den Zellen gelöst und in Fragmente geteilt. Diese Fragmente
haben eine durchschnittliche Länge von etwa 1000 Basenpaaren (bp). Als nächstes wer-
den die interessanten Proteine, welche je nach Experiment verschieden sein können, durch
die Hinzugabe von passenden Antikörpern markiert und mittels Immunpräzipitation iso-
liert. Im nächsten Schritt wird die DNA wieder vom Protein getrennt. Danach werden
die Fragmente ausgewählt, die eine passende Größe haben. In diesem Fall sind das et-
wa 100 bis 300 bp. Die so gewonnenen DNA Fragmente werden nun sequenziert. Dabei
entstehen Millionen von kurzen reads (z.B. je 36 bp), die entweder den Anfang oder das
Ende eines Fragments darstellen (vorwärts bzw. rückwärts reads). Mit Hilfe dieser reads
werden die Fragmente nun den Stellen im Genom zugeordnet. Auf Grund von sich wie-
derholenden Abfolgen von Basenpaaren, kann es sein, dass ein read sich nicht eindeutig
einer Stelle im Genom zuordnen lässt. In diesem Fall wird der read verworfen. Dies führt
dazu, dass es im Genom Bereiche gibt, in denen keine reads zugeordnet werden können.
1Abbildung 1: Ein ChIP-seq Experiment.
2In der später folgenden Analyse wird versucht, die reads in diesen Bereichen mit Hilfe
der Werte aus benachbarten Bereichen zu approximieren.
Datensätze, die wie oben beschrieben entstanden sind, können nun dazu benutzt werden
die Bindungsstellen der interessanten Proteine zu schätzen. Jedes DNA Fragment des
Datensatzes, bis auf Ausnahmen die durch Fehler in der Immunpräzipitation oder der
Zuordnung entstanden sind, enthält eine Bindungsstelle. Wenn man nun die vorwärts
und rückwärts reads geeignet modelliert, kann man die Bindungsstelle als Mittelwert zwi-
schen der mittleren Position der vorwärts reads und der mittleren Position der rückwärts
reads schätzen. Wie man dies umsetzt werde ich in den folgenden Abschnitten genauer
erläutern.
2 Das Modell
Da ChIP-seq Datensätze häufig Regionen enthalten, in denen keine oder kaum reads
beobachtet werden, teilt man die Daten zunächst in sogenannte Kandidatenregionen
auf. Jede solche Kandidatenregion soll dabei eine minimale Anzahl von vorwärts und
von rückwärts reads enthalten, damit eine Analyse der Region durch PICS sinnvoll ist.
Außerdem sollen die Regionen disjunkt voneinander sein, das heißt sich nicht überlappen,
damit man später jede solche Region einzeln analysieren kann.
Um diese Kandidatenregionen zu bekommen, schiebt man ein Fenster mit einer geeig-
neten Breite in einer geeigneten Schrittgröße über den Datensatz und zählt die Anzahl
der vorwärts reads in der linken Fensterhälfte und die Anzahl der rückwärts reads in der
rechten Fensterhälfte. Nun behält man nur die Fenster, die mindestens einen vorwärts
und einen rückwärts read beinhalten, und fügt sie bei Überlappungen zusammen. Wenn
man nun noch die Zusammengefügten Fenster löscht, die weniger als zwei vorwärts be-
ziehungsweise rückwärts reads beinhalten, erhält man die gewünschten Kandidatenre-
gionen.
2.1 Das Modell für eine einzelne Bindungsstelle
Nehmen wir nun einmal an, dass eine gegebene Kandidatenregion, die nun analysiert
werden soll, nur eine einzelne Bindungsstelle aufweist. In der gegebenen Region bezeichne
fi beziehungsweise rj die Position des i-ten vorwärts beziehungsweise des j-ten rückwärts
reads. Die Anzahl der vorwärts reads in der gegebenen Kandidatenregion bezeichnen wir
mit nf und die Anzahl der rückwärts reads mit nr . Nun modellieren wir die Positionen
der reads mit Hilfe einer studentschen t-Verteilung mit vier Freiheitsgraden wie folgt:
δ δ
fi ∼ t4 (µ − , σf2 ) und rj ∼ t4 (µ + , σr2 ).
2 2
Hierbei bezeichnet µ die Bindungsstelle und δ den Abstand zwischen den Maxima der
Dichten der vorwärts beziehungsweise rückwärts reads. Dieser Abstand ist offensichtlich
zugleich die mittlere Länge der gegebenen DNA Fragmente, deren reads für diese Bin-
dungsstelle betrachtet wurden. σf2 und σr2 , die Varianzen der studentschen t-Verteilungen,
3parameters σf k and σ
Figure 2a shows a candidate region with one binding event,
along with the corresponding PICS parameter estimates. 3.3 Prior Distribution
Typically, the library
3.2 Modeling Multiple Binding Events mation available for t
We use mixture models to address the possibility that the use a Bayesian appro
sets
simulieren die of forwardin and
Varianzen reverse beziehungsweise
den Anfangs- reads within aEndpositionen
candidate region by allowing the δk ’s f
der gegebenen
were generated
DNA Fragmente. by der
Die Dichte multiple closelyt-Verteilung
studentschen spaced binding
ähnelt events. Weder mon
der Dichte Nor- prior fragment le
modelallerdings
malverteilung, the forward
sind and reverse(“Tails”)
die Flanken read positions
etwas mehrusing t-mixture
betont. put
Damit ist diea common prior d
in diesemdistributions:
Fall gut geeignet, da sie Ausreißer besser modelliert. In Abbildung 2.1 istusdie
to incorporate prio
Schätzung von PICS für eine Region mit einer Bindungsstelle mit den vorkommenden
Parametern dargestellt.
(a) One binding event (b)
PICS
Kernel density
0.010
Reads Density
Mappability profile Mappability profile
0.005
1 f
1 r
0.000
> > >> >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
>>
>>>>
>>
>>>>>>>>> >
< < <Typically, the library construction process makes prior infor-
mation available for the length of the DNA fragments. We can
possibility that the use a Bayesian approach to take advantage of this information
n a candidate region by allowing the δk ’s for all binding sites to derive from a com-
d binding events. We mon prior fragment length distribution. Similarly, we can also
tions using t-mixture put a common prior distribution on σf2 k and σr2 k , which allows
us to incorporate
Wahrscheinlichkeiten prioreininformation
dafür, dass about
gegebenes DNA the variability
Fragment of the
vorwärts beziehungsweise
rückwärts gelesen wird gleich sind. In Abbildung 2.2 ist die Schätzung von PICS für eine
Region mit zwei Bindungsstellen mit den vorkommenden Parametern dargestellt.
vent (b) Two binding events
Mappability profile
1 r
>2.4 Modellierung von verworfenen reads
Wie oben schon erwähnt, werden reads, die keiner eindeutigen Stelle im Genom zugeord-
net werden können, vor der Analyse verworfen. Die führt dazu, dass in einigen Bereichen
des Genoms keine reads in den Datensätzen auftreten, obwohl eigentlich welche vorhan-
den sein müssten. Um dieses Problem zu lösen, teilt man jede Kandidatenregion zunächst
in verschiedene Bereiche ein, je nachdem wie gut reads in dem jeweiligen Bereich zuge-
ordnet werden können. Wenn man eine konkrete read-Länge vorgibt, wird dazu jedem
Basenpaar in der Region eine sogenannte Zuordnungsrate zugeordnet, die schätzt wie
viel Prozent der reads, die diese Stelle enthalten, auch eindeutig dieser Stelle zugeordnet
werden können.
Der Einfachheit halber zerlegt man die Kandidatenregion in disjunkte Intervalle, wobei
alle Basenpaare in einem solchen Intervall entweder einen score SL = 0 oder > 0 haben.
Wenn I die Kandidatenregion bezeichnet, schreibt man I = l=0 Il , wobei I0 die Verei-
nigung aller Intervalle mit hoher Zuordnungsrate ist und Il das l-te Intervall bezeichnet,
in dem keine Zuordnung stattfinden kann.
Bezeichnet man nun mit fli beziehungsweise rlj die Positionen der vorwärts beziehungs-
weise rückwärts reads im Intervall Il , dann sind dies für l > 0 unbeobachtete Zufalls-
variablen. Diese werden durch eingeschränkte t-Mischverteilungen modelliert. Das heißt
fli und rli sind so verteilt wie in Abschnitt 2.2, jedoch eingeschränkt auf das Intervall Il .
3 Analyse des Modells
Nachdem nun das Modell für die Daten aufgestellt wurde, muss dieses geeignet analysiert
werden, um die konkreten Bindungsstellen schätzen zu können. Zunächst werden dazu
2
mit Mitteln der Statistik die unbekannten Parameter des Modells, wk , µk , δk , σf2k und σrk
für alle k = 1, . . . , K, geschätzt. Da dieses Verfahren im Detail mathematisch aufwändig
ist und nicht Schwerpunkt dieser Ausarbeitung sein soll, werde ich darauf an dieser Stelle
nicht weiter eingehen.
Als nächstes stellt sich die Frage, wie man aus dem Modell nun die konkreten Bindungs-
stellen extrahiert. Insbesondere muss man dazu bewerten, welche “peaks” im Modell
nun tatsächlich ausschlaggebend genug für eine Bindungsstelle sind, beziehungsweise
mit welcher Sicherheit man dort eine Bindungsstelle im Genom vorfinden wird.
3.1 Wahl der Anzahl der Bindungsstellen in einer Kandidatenregion
Bisher haben wir die Modelle unter der Annahme betrachtet, dass bekannt ist wie viele
Bindungsstellen sich in einer gegebenen Kanditatenregion befinden. Diese Anzahl wurde
mit K bezeichnet. Nun ist diese Annahme in der Praxis im Allgemeinen unbekannt und
muss ebenfalls geschätzt werden.
Dies geschieht mittels des sogenannten Bayesschen Informationskriteriums (BIC). Dazu
stellt man das Modell für jede Kandidatenregion mit verschiedenen Werten von K auf,
in diesem Fall für K = 1, . . . , 15. Dann wählt man das Modell aus, welches den höchsten
6BIC-Wert hat. Dabei berechnet sich die BIC-Wert hier wie folgt:
BIC = 2Q(Θ = Θ̂|Θ̂) − (5K − 1) ln(nf 0 + nr0 ).
Hierbei bezeichnet Θ̂ die geschätzen Werte für die unbekannten Parameter Θ und Q die
log-Likelihoodfunktion für diese Parameter. Insbesondere sieht man aber, dass Modelle
mit vielen Bindungsstellen (das heißt großem K) in Abhängigkeit von der Anzahl der
gesamten reads im Bereich mit hoher Zuordnungsrate “bestraft” werden. Dies ist notwen-
dig, da ansonsten zu häufig Modelle mit vielen nah aneinanderliegenden Bindungsstellen
gewählt werden, da diese sonst einen hohen BIC-Wert erhalten, obwohl dies in der Praxis
nicht so häufig vorkommt.
3.2 Zusammenlegen und Filtern der peaks
Da die Parameterschätzungen in einem statistischen Modell durchgeführt wurden, kann
man für die interessanten Parameter µ und δ Standardfehler µ̂ und δ̂ berechnen. Mit
Hilfe dieser Standardfehler kann man dann zum Beispiel Konfidenzintervalle bestimmen
oder auch die Güte der Schätzung bewerten.
Durch die Bewertung der Modelle mit Hilfe des BIC-Wertes, kann es passieren, dass
zum Beispiel ein Modell mit zwei sehr nah aneinander liegenden geschätzten Bindungs-
stellen gegenüber einem Modell mit nur einer geschätzten Bindungsstelle beim gleichen
zu Grunde liegenden Datensatz bevorzugt wird. Um diesem entgegen zu wirken, werden
peaks, die nah beieinander liegen, zu einem einzelnen peak zusammengefügt.
In einem zweiten Schritt werden mit Hilfe der Standardfehler die Bindungsstellen aus-
sortiert, für die ungewöhnliche Parameter geschätzt wurden oder deren Schätzung nur
sehr ungenau möglich war.
3.3 Bewertung der geschätzten Bindungsstellen
Um herauszufinden welche peaks tatsächlich ausschlaggebend genug für eine Bindungs-
stelle sind, wird jeder möglichen Bindungsstelle ein score zugeordnet. Dazu definieren wir
FChIP beziehungsweise RChIP als die Zahl der aufgetretenen vorwärts beziehungsweise
rückwärts reads im ChIP Datensatz, deren Position innerhalb eines 90% Konfidenz-
intervalles der Dichtefunktion der vorwärts beziehungsweise rückwärts reads liegt. Die
Summe dieser beiden Größen OChIP = FChIP + RChIP bezeichnen wir als den score
einer Bindungsstelle. Dieser score ist also die geschätzte Zahl der DNA Fragmente, die
zu dieser Bindungsstelle gehören, wenn man Ausreißer nicht berücksichtigt.
Wenn zusätzlich ein Kontrolldatensatz vorhanden ist, kann man den score relativ zum
Kontrolldatensatz ausdrücken. Dazu definiert man zunächst Fcont , Rcont und Ocont für
den Kontrolldatensatz analog zu den obigen Größen. Das heißt Fcont ist dann zum Bei-
spiel die Anzahl der vorwärts reads des Kontrolldatensatzes, deren Position innerhalb
des 90% Konfidenzintervalles der aus den ChIP Daten geschätzten Dichtefunktion der
vorwärts reads liegt. Mit diesen Größen kann man dann den score S(b) einer geschätzten
7Bindungsstelle an Position b relativ zu den Kontrolldaten wie folgt definieren.
Ncont OChIP
S(b) = ·
NChIP Ocont + 1
Hierbei bezeichnet NChIP beziehungsweise Ncont die Gesamtanzahl der beobachteten
reads in den ChIP Daten (beziehungsweise im Kontrolldatensatz). Die Normierung mit
dem Quotienten der beiden Gesamtanzahlen dient dazu, um mögliche Unterschiede in
der Gesamtzahl der reads auszugleichen.
4 Anwendungen
PICS wurde in der vorliegenden Veröffentlichung auf zwei realen Datensätzen getestet
und dabei mit drei bestehenden Systemen verglichen. Ich werde in diesem Abschnitt
nicht im Detail auf die Ergebnisse dieses Vergleiches eingehen, sondern vielmehr die
verschiedenen Vergleichsmethoden erläutern.
4.1 Die False Discovery Rate
Wenn ein Kontrolldatensatz gegeben ist, kann man den Anteil der falsch positiven Bin-
dungsstellen in der sogenannten False Discovery Rate (FDR) ausdrücken. Dazu führt
man die Analyse noch einmal mit vertauschten Rollen von ChIP Daten und Kontroll-
daten durch. Das heißt man berechnet sogenannte “null-scores” S0 (b) als scores der
Kontrolldaten mit den ChIP Daten zur Kontrolle. Damit bestimmt man die FDR, als
Quotient der Anzahl der falsch positiven und der insgesamt gefundenen Bindungsstel-
len. Genauer ist die FDR eine Funktion, abhängig von einem Randwert q und wie folgt
definiert.
#{b : S0 (b) > q}
F DR(q) =
#{b : S(b) > q}
Wenn man die Analyse von PICS mit dem Ergebnis anderer Systeme mit Hilfe der
FDR vergleicht, erhält man für beide Testdatensätze unterschiedliche Ergebnisse. Daher
scheint ein Vergleich mittels der FDR in diesem Fall keine hohe Aussagekraft zu haben.
4.2 Vergleiche mittels Motiven
Eine weitere Möglichkeit des Vergleichs verschiedener Analysesysteme bieten sogenannte
Motive. Dazu versucht man biologisch bekannte Motive in den geschätzten Bindungsstel-
len zu finden und bewertet die Ergebnisse nach zwei Kriterien. Das erste ist der Anteil
der geschätzten Bindungsstellen, welche tatsächlich in einem biologisch zu erwartenden
Motiv enthalten sind. Das zweite Kriterium ist der Abstand zwischen der geschätzten
Bindungsstelle und dem Ort des am nächsten gelegenen Motives.
Beim Vergleich mit den Systemen MACS, USeq, QuEST und cisGenome war PICS im
Bezug auf den Anteil der Bindungsstellen in Motiven am besten. Der Abstand zwischen
den Bindungsstellen und den Motiven war bei PICS und bei MACS am geringsten.
84.3 Vergleiche mittels simulierter Daten
Anstelle eines Vergleiches auf experimentellen Daten, kann man die verschiedenen Syste-
me auch mit Hilfe von simulierten Daten vergleichen. Dies hat unter anderem den Vorteil,
dass man Parameter des Modells auf verschiedene Weisen bewusst falsch spezifizieren
kann, um zu sehen, wie die Analysesysteme dies bewältigen.
Auch bei einem Vergleich mittels simulierter Daten hat das PICS System in verschiede-
nen betrachteten Szenarien sehr gute Ergebnisse geliefert.
5 Fazit
PICS ist ein System zur Analyse von ChIP-seq Daten, welches auf einem probabilis-
tischen Ansatz basiert und Bindungsstellen von Proteinen schätzt. Ein Nachteil dieses
Ansatzes ist, dass das System, zum Beispiel durch die aufwendigen Mischverteilungen,
rechenintensiv ist. Vorteile dieses Systems sind, dass a-priori-Verteilungen für die Länge
der DNA Fragmente δ benutzt werden können und dass reads, die auf Grund von wie-
derholenden Folgen von Basenpaaren verworfen wurden, approximiert werden.
Eine objektive Bewertung der Ergebnisse stellt sich als schwierig heraus, da eine solche
Bewertung immer von der gewählten Bewertungsmethode und den Testdaten abhängt.
Es lässt sich aber festhalten, dass der hier gewählte probabilitische Ansatz für die un-
tersuchten Testdatensätze sehr gute Ergebnisse liefert und daher auf jeden Fall konkur-
renzfähig zu bereits bestehenden anderen Ansätzen ist.
Eine mögliche Weiterentwicklung des Systems wäre die zusätzliche Betrachtung von
sogenannten paired-end reads. Da man aus diesen Daten die Länge der DNA Fragmente
direkt erhält, würde dies das Modell für die Analyse vereinfachen. Der Grund wieso PICS
dies bisher nicht unterstützt ist, dass die Gewinnung von paired-end reads biologisch
deutlich aufwendiger ist, als die Gewinnung der hier betrachteten single-end reads, und
daher in der Praxis kaum verwendet wird.
Eine weitere mögliche Erweiterung des PICS Systems besteht darin mehr a-priori-Informationen
zu benutzen, um die Ergebnisse zu verbessern. So könnte man zum Beispiel eine a-priori-
Verteilung für die Position der Bindungsstellen µ benutzen, die man aus den zuvor be-
rechneten Motiven bestimmt.
Außerdem ist es möglich das PICS System auf andere Fragestellungen anzupassen. So
könnte man zum Beispiel anstatt nach der konkreten Position einer Bindungsstelle in
einem gegebenen Datensatz auch danach fragen, in welchen Bereichen sich die Bindungs-
stellen in zwei gegebenen Datensätzen unterscheiden. Um diese Frage effizient beantwor-
ten zu können, müsste man die Modelle, die in PICS benutzt werden, erweitern. Eine
Region, in der sich die Bindungsstellen in den Datensätzen unterscheiden, wird dann
dadurch gekennzeichnet, dass in einem Datensatz prozentual deutlich mehr reads auf
diese Region entfallen als in einem anderen Datensatz. Der probabilitische Ansatz von
PICS scheint auch für solche Fragestellungen gut geeignet zu sein, daher halte ich es
für eine sinnvolle Idee, PICS auf solche Fragestellungen zu erweitern. Man könnte dann
9sogar noch weiter gehen und Fragen untersuchen, die sich auf ganze Gruppen von Da-
tensätzen beziehen. Zum Beispiel kann man fragen in welchen Regionen ein bestimmtes
Protein in einer Gruppe von Datensätzen bindet, aber nicht in einer anderen Gruppe
von Datensätzen.
Alles in allem scheint der probabilitische Ansatz zur Analyse von ChIP-seq Daten sehr
vielversprechend zu sein und das PICS System ist eine erste Implementierung dieses
Ansatzes, die schon viele Schwierigkeiten, wie zum Beispiel verworfene reads, beachtet.
Durch Weiterentwicklungen kann man versuchen das System noch leistungsfähiger zu
machen und auch auf andere Anwendungsgebiete auszuweiten.
10Sie können auch lesen

















































