AUF ZU NEUEN CHANCEN MIT PADCEVTM - NEU - dccdn.de
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

NEU
BEI VORBEHANDELTEN PATIENTEN MIT LOKAL FORTGESCHRITTENEM
ODER METASTASIERTEM UROTHELKARZINOM*
AUF ZU NEUEN CHANCEN MIT PADCEV
GEZIELTE ANTIKÖRPER-WIRKSTOFF-KONJUGAT THERAPIE (ADC)1
TM
VERLÄNGERTES ÜBERLEBEN VS. CHEMOTHERAPIE**, 1:
Medianes Gesamtüberleben: 12,9 Monate vs. 9,0 Monate
HR: 0,70; 95% KI: 0,56 – 0,89; p = 0,001.
* PADCEV ist als Monotherapie angezeigt zur Behandlung von erwachsenen Patienten mit lokal fortgeschrittenem oder metastasiertem
Urothelkarzinom, die zuvor eine platinhaltige Chemotherapie und einen Programmed Death Receptor-1- oder Programmed Death Ligand-1-
Inhibitor erhalten haben.1
** U
ntersucht wurde Enfortumab Vedotin vs. Chemotherapie nach Wahl des Arztes (Docetaxel, Paclitaxel – beide in dieser Indikation in
Deutschland nicht zugelassen2, 3 – oder Vinflunin).1
20 mg & 30 mg Vial / i. v. Anwendung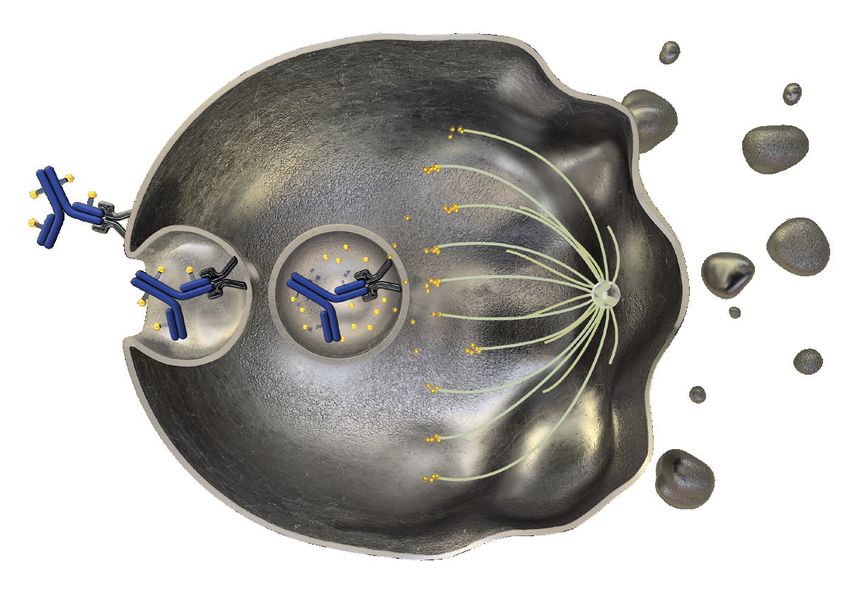
PADCEV – Signifikante Verbesserungen bei Gesamtüberleben
(OS), progressionsfreiem Überleben (PFS) und
Gesamtansprechrate (ORR) im Vergleich zur Chemotherapie*, c, 1, 4
Primärer Endpunkt: OS
100
Senkung des
Medianes OS
Sterberisikos
80
12,9 Monate um
30%
60
(95% KI: 10,6 – 15,2)
OS (%)
40 Medianes OS
20
9,0 Monate
(95% KI: 8,1 – 10,7)
PADCEV vs.
Chemotherapie
HR: 0,70;
PADCEV (n = 301) 95% KI: 0,56 – 0,89;
Chemotherapie (n = 307) p = 0,001*†1
0
0 3 6 9 12 15 18 21 24
Patienten Zeit (Monate)
unter Risiko
PADCEV 301 257 222 130 63 33 7 2 0
Chemotherapie 307 250 198 101 51 29 6 2 0
* mOS-Unterschied von 3,9 Monaten mit PADCEV (mOS = 12,9 Monate) im Vergleich zur Chemotherapie nach Wahl des Arztes (Docetaxel, Paclitaxel – beide in dieser Indikation in Deutschland
nicht zugelassen2, 3 – oder Vinflunin; mOS = 9,0 Monate).1
† einseitiger p-Wert, festgelegte Wirksamkeitsgrenze = 0,00679 (bereinigt nach beobachteten Todesfällen von 301).
Ausgewählte sekundäre Endpunkte
RR
O
41 % Gesamtansprechrate mit PADCEV vs. 18 % Chemotherapie* (p < 0,001a)
41% Schnelles Ansprechen: Im Median 1,87 (1,1 – 5,7) Monate bis zum Ansprechen bei
Patienten mit PADCEV5
tion
uk
ed
38 % Risikoreduktion für Krankheitsprogression oder Tod mit PADCEV vs. Chemotherapie*, b, c
Risikor
38 % Signifikanter medianer PFS Vorteil von PADCEV (5,6 Monate) vs. Chemotherapie (3,7 Monate)*, c
HR: 0,62; 95% KI: 0,51 – 0,75; p < 0,00001b, c
tion
uk
Schmerzred
Anteil der Patienten mit Schmerzreduktion: 52 % mit PADCEV vs. 29 % Chemotherapie*, 6
52% OR: 2,76; 95% KI: 1,81 – 4,2
Basierend auf den EORTC QLQ-C30-Ergebnissen.
* Untersucht wurde Enfortumab Vedotin vs. Chemotherapie nach Wahl des Arztes (Docetaxel, Paclitaxel – beide in dieser Indikation in Deutschland nicht zugelassen2, 3 – oder Vinflunin).1
a Einseitiger p-Wert; festgelegte Wirksamkeitsgrenze = 0,025 (bereinigt nach 100 % Informationsanteil).1
b Einseitiger p-Wert, festgelegte Wirksamkeitsgrenze = 0,02189 (bereinigt nach beobachtetem PFS1 von 432 Ereignissen).1
c Das PFS wurde vom Prüfarzt beurteilt und war definiert als die Zeit vom Tag der Randomisierung bis zum Tag des radiologischen Fortschreitens der Erkrankung (gem. RECIST v1.1)
oder bis zum Tod jeglicher Ursache.Bei welchen Patienten kann ich PADCEV einsetzen?1
Häufige Therapiefolge bei Patienten mit lokal fortgeschrittenem/
metastasiertem Urothelkarzinom
NEU
Platinhaltige PD-(L)1
Chemotherapie Inhibitor
PADCEV
PADCEV zeigt ein handhabbares Verträglichkeitsprofil 1, 4
UNERWÜNSCHTE EREIGNISSE, DIE BEI ≥ 20 % (JEDER GRAD) ODER ≥ 5 % (GRAD ≥ 3) DER PATIENTEN
GEMELDET WURDEN (N = 587)*
PADCEV (N = 296) Chemotherapie (N = 291)
Unerwünschtes Ereignis Alle Grade Grad ≥ 3 Alle Grade Grad ≥ 3
Anzahl Patienten (%) Anzahl Patienten (%)
Alle unerwünschten Ereignisse 278 (93,9) 152 (51,4) 267 (91,8) 145 (49,8)
Alopezie 134 (45,3) 0 106 (36,4) 0
Periphere sensorische Neuropathie† 100 (33,8) 9 (3,0) 62 (21,3) 6 (2,1)
Pruritus 95 (32,1) 4 (1,4) 13 (4,5) 0
Fatigue 92 (31,1) 19 (6,4) 66 (22,7) 13 (4,5)
Verminderter Appetit 91 (30,7) 9 (3,0) 68 (23,4) 5 (1,7)
Diarrhö 72 (24,3) 10 (3,4) 48 (16,5) 5 (1,7)
Dysgeusie 72 (24,3) 0 21 (7,2) 0 (0)
Übelkeit 67 (22,6) 3 (1,0) 63 (21,6) 4 (1,4)
Makulopapulärer Hautausschlag 48 (16,2) 22 (7,4) 5 (1,7) 0 (0)
Anämie 34 (11,5) 8 (2,7) 59 (20,3) 22 (7,6)
Verringerung der Neutrophilen 30 (10,1) 18 (6,1) 49 (16,8) 39 (13,4)
Neutropenie 20 (6,8) 14 (4,7) 24 (8,2) 18 (6,2)
Verringerung weiße Blutzellen 16 (5,4) 4 (1,4) 31 (10,7) 20 (6,9)
Febrile Neutropenie 2 (0,7) 2 (0,7) 16 (5,5) 16 (5,5)
Die Abbruchraten aufgrund von therapiebedingten unerwünschten Ereignissen waren bei PADCEV
und Chemotherapie ähnlich (14 % vs. 11 %). 5
• Unter PADCEV traten hämatologische Nebenwirkungen seltener auf als bei der Chemotherapie.
• Bei PADCEV wurden fünf sogenannte „Nebenwirkungen von besonderem Interesse“ identifiziert – Hautreaktionen,
Hyperglykämie, periphere Neuropathie, Augenerkrankungen, infusionsbezogene Reaktionen (siehe folgende
Seiten). Diese Nebenwirkungen waren meist mild bis moderat und lassen sich teilweise auf den Wirkmechanismus
von PADCEV zurückführen.
Bitte beachten Sie die wichtigen Hinweise und zusätzlichen Informationen zu Häufigkeit und Management
dieser “Nebenwirkungen von besonderem Interesse” in der Fachinformation und dem PADCEV-Dosierungs- und
Anwendungsleitfaden.
* Die Sicherheitspopulation umfasste alle Patienten, die eine beliebige Menge des Studienmedikaments erhielten. Eingeschlossen sind behandlungsassoziierte unerwünschte Ereignisse, die bei
mindestens 20 % der Patienten in einer der Behandlungsgruppen auftraten, oder behandlungsassoziierte unerwünschte Ereignisse des Grades ≥ 3, die bei mindestens 5 % der Patienten in
einer der Behandlungsgruppen auftraten. Als behandlungsassoziierte unerwünschte Ereignisse gelten solche, bei denen eine begründete Möglichkeit besteht, dass sie nach Einschätzung des
Prüfers durch die Studienbehandlung verursacht wurden. Wenn Daten über den Zusammenhang mit der Behandlung fehlten, wurde davon ausgegangen, dass das Ereignis mit der Behandlung
zusammenhing.4
† Insgesamt hatten 113 Patienten (55 im PADCEV-Arm und 58 im Chemotherapie-Arm) eine vorbestehende periphere Neuropathie.4Einleitung der PADCEV-Therapie1
Keine Biomarker-Tests und keine Prämedikation erforderlich
1.25
mg/kg
Individuelle, 30-minütige Infusion 3 Infusionen monatlich
körpergewichtsbasierte (am Tag 1, 8 und 15 eines
Dosierung* 28-tägigen Zyklus)
* Empfohlene Dosierung von PADCEV: 1,25mg/kg Körpergewicht bis maximal 125 mg für Patienten mit einem Gewicht ≥ 100 kg.
Die Therapie mit PADCEV kann bei Patienten mit lokal fortgeschrittenem oder metastasiertem Urothelkarzinom nach
einer platinhaltigen Chemotherapie und einem PD-1- oder PD-L1-Inhibitor initiiert werden.
PADCEV wird nun auch in den europäischen Leitlinien der EAU und der ESMO empfohlen.7, 8
Abkürzungen
ADC = Antibody-Drug-Conjugate (Antikörper-Wirkstoff-Konjugat)
EORTC QLQ-C30 = European Organisation for Research and Treatment of Cancer quality of life questionnaire for cancer patients
Kl = Konfidenzintervall
HR = Hazard Ratio
mOS = medianes Gesamtüberleben
mPFS = medianes progressionsfreies Überleben
OR = Odds Ratio
ORR = Gesamtansprechrate
OS = Gesamtüberleben
PD-1 = programmed death-1
PD-L1 = programmed death-ligand 1
PFS = progressionsfreies Überleben
UC = UrothelkarzinomPADCEV – ADC-Therapie mit gezieltem Wirkansatz
ohne Biomarker-Testung1
Wirkmechanismus von PADCEV
PADCEV bindet an Nectin-4-
1 exprimierende Zellen
Internalisierung des PADCEV-
2 Nectin-4-Komplexes
Freisetzung des zytotoxisch
3 wirkenden MMAE durch Proteolyse 1
Hemmung der zellulären Mikrotubuli
4 durch MMAE
2 3 4 5
5 Zellzyklusarrest und Apoptose
Verbesserte Wirksamkeit im Vergleich zur Chemotherapie**, 1
terberisiko um 30 % gesenkt.1
•S
HR: 0,70; 95% KI: 0,56 – 0,89; p = 0,0011
• Medianes Gesamtüberleben um 3,9 Monate verlängert.1
12,9 Monate vs. 9,0 Monate1
• Mit 41 % mehr als doppelt so hohe Ansprechrate (ORR).1
41 % (n = 177/288); 95% KI: 35,0 % – 46,5 % vs. 18 % (n = 53/296); 95% KI: 13,7 % – 22,8 %; p < 0,0011
Gezielte ADC (Antikörper-Wirkstoff-Konjugat)-Therapie1
• ohne Biomarker-Testung1
• handhabbares Sicherheitsprofil1
•L ebensqualität erhalten***, 6
* PADCEV™ ist als Monotherapie angezeigt zur Behandlung von erwachsenen Patienten mit lokal fortgeschrittenem oder metastasiertem Urothelkarzinom, die zuvor eine platinhaltige Chemotherapie
und einen Programmed Death Receptor-1- oder Programmed Death Ligand-1-Inhibitor erhalten haben.1
** Untersucht wurde Enfortumab Vedotin vs. Chemotherapie nach Wahl des Arztes (Docetaxel, Paclitaxel - beide in dieser Indikation in Deutschland nicht zugelassen2, 3 - oder Vinflunin).1
*** Gemessen in Woche 12 vs. Baseline anhand des EORTC QLQ-C30-Fragebogens.PADCEV Fachinformation
PADCEV Dosierungs- und Anwendungsleitfaden
Referenzen:
1 PADCEV™ Fachinformation, aktueller Stand.
2 Als Beispiel für Docetaxel – Fachinformation Taxotere®, Stand April 2021_2.
3 Als Beispiel für Paclitaxel – Fachinformation Abraxane®, Stand April 2021.
4 Powles T, Rosenberg JE, Sonpavde GP, et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. N Engl J Med. 2021;384(12):1125-1135.
5 Powles T, Rosenberg JE, Sonpavde GP, et al. Enfortumab Vedotin in Previously Treated Advanced UrothelialCarcinoma. N Engl J Med. 2021;384(12):1125-1135. Supplement.
6 Mamtani R et al.; Quality of Life, Functioning, and Symptoms in Patients With Previously Treated Locally Advanced or Metastatic Urothelial Carcinoma From EV-301: A Randomized Phase 3 Trial of Enfortumab Vedotin vs Chemotherapy ; Abstract 4539, ASCO 2021,
June 4-8 Journal of Clinical Oncology / Volume 39, Issue 15_suppl https://ascopubs.org/doi/abs/10.1200/JCO.2021.39.15_suppl.4539.
7 BLADDER CANCER: ESMO CLINICAL PRACTICE GUIDELINE FOR DIAGNOSIS,TREATMENT AND FOLLOW-UP ; Powles T. et al., Annals of Oncology (2021), doi: https://doi.org/10.1016/j.annonc.2021.11.012.
8 Mottet-EAU-EANM-ESUR-ESTRO-SIOG-Guidelines-on-Prostate-Cancer-2022.
Astellas Pharma GmbH | Ridlerstraße 57 | 80339 München 20 mg & 30 mg Vial / i. v. Anwendung
PadcevTM 20 mg / 30 mg Pulver für ein Konzentrat zur Herstellung einer Infusionslösung. Wirkstoff: Enfortumab Vedotin. Zusammensetzung: Eine Durchstechflasche mit Pulver für ein Konzentrat zur Herstellung einer Infusionslösung enthält: Wirkstoff: 20 mg / 30 mg Enfortumab
Vedotin. Nach Rekonstitution enthält 1 ml der Lösung 10 mg Enfortumab Vedotin. Sonstige Bestandteile: Histidin, Histidinhydrochlorid-Monohydrat, Trehalose-Dihydrat (Ph.Eur.), Polysorbat 20. Anwendungsgebiete: Padcev ist als Monotherapie angezeigt zur Behandlung von erwachsenen
Patienten mit lokal fortgeschrittenem oder metastasiertem Urothelkarzinom, die zuvor eine platinhaltige Chemotherapie und einen Programmed Death Receptor-1- oder Programmed Death Ligand-1-Inhibitor erhalten haben (siehe Abschnitt 5.1 „Pharmakodynamische Eigenschaften“ in der
Fachinformation). Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile. Nebenwirkungen: Zusammenfassung des Sicherheitsprofils: Die häufigsten Nebenwirkungen mit Enfortumab Vedotin waren Alopezie (48,8 %), Fatigue (46,8 %), vermin-
derter Appetit (44,9 %), periphere sensorische Neuropathie (38,7 %), Diarrhoe (37,6 %), Übelkeit (36 %), Pruritus (33,4 %), Dysgeusie (29,9 %), Anämie (26,5 %), erniedrigtes Gewicht (23,4 %), makulo-papulöser Ausschlag (22,9 %), trockene Haut (21,6 %), Erbrechen (18,4 %), erhöhte
Aspartataminotransferase (15,3 %), Hyperglykämie (13,1 %), trockenes Auge (12,8 %), erhöhte Alaninaminotransferase (12,1 %) und Ausschlag (10,4 %). Die häufigsten schwerwiegenden Nebenwirkungen waren Diarrhoe (2 %) und Hyperglykämie (2 %). Neun Prozent der Patienten setzten
Enfortumab Vedotin wegen Nebenwirkungen dauerhaft ab; die häufigste Nebenwirkung (≥ 2 %), die zum Absetzen der Dosis führte, war periphere sensorische Neuropathie (4 %). Nebenwirkungen, die zu einer Dosisunterbrechung führten, traten bei 44 % der Patienten auf; die häufigsten
Nebenwirkungen (≥ 2 %), die zu einer Unterbrechung der Behandlung führten, waren periphere sensorische Neuropathie (15 %), Fatigue (7 %), makulo-papulöser Ausschlag (4 %), erhöhte Aspartataminotransferase (4 %), erhöhte Alaninaminotransferase (4 %), Anämie (3 %), Diarrhoe (3 %)
und Hyperglykämie (3 %). Bei 30 % der Patienten war eine Dosisreduktion aufgrund einer Nebenwirkung notwendig; die häufigsten Nebenwirkungen (≥ 2 %), die zu einer Dosisreduktion führten, waren periphere sensorische Neuropathie (10 %), Fatigue (5 %), makulo-papulöser Ausschlag
(4 %) und verminderter Appetit (2 %). Auflistung der Nebenwirkungen (tabellarische Auflistung siehe Tabelle 3 in der Fachinformation): Die Sicherheit von Enfortumab Vedotin als Monotherapie wurde untersucht bei 680 Patienten mit lokal fortgeschrittenem oder metastasiertem Urothelkar-
zinom, die 1,25 mg/kg an den Tagen 1, 8 und 15 eines 28-Tage-Zyklus in klinischen Studien erhielten (siehe Tabelle 3 in der Fachinformation). Die Patienten waren für eine mediane Dauer von 4,7 Monaten (Bereich: 0,3 bis 34,8 Monate) gegenüber Enfortumab Vedotin exponiert. In diesem
Abschnitt sind die während klinischer Studien beobachteten Nebenwirkungen nach MedDRA-Systemorganklasse und darin nach Häufigkeitskategorien aufgeführt. Die Häufigkeitskategorien sind wie folgt definiert: sehr häufig (≥ 1/10); häufig (≥ 1/100, < 1/10); gelegentlich (≥ 1/1.000,
< 1/100); selten (≥ 1/10.000, < 1/1.000); sehr selten (< 1/10.000); nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitsgruppe sind die Nebenwirkungen nach abnehmendem Schweregrad angegeben. Erkrankungen des Blutes und
des Lymphsystems: Sehr häufig: Anämie; Nicht bekannt (basierend auf weltweiten Erfahrungen nach der Markteinführung): Neutropenie, febrile Neutropenie, erniedrigte Neutrophilenzahl. Stoffwechsel- und Ernährungsstörungen: Sehr häufig: Hyperglykämie, verminderter Appetit. Erkran-
kungen des Nervensystems: Sehr häufig: Periphere sensorische Neuropathie, Dysgeusie; Häufig: Periphere Neuropathie, periphere motorische Neuropathie, periphere sensomotorische Neuropathie, Parästhesie, Hypoästhesie, Gangstörung, muskuläre Schwäche; Gelegentlich: Demyelinisierende
Polyneuropathie, Polyneuropathie, Neurotoxizität, motorische Funktionsstörung, Dysästhesie, Muskelatrophie, Neuralgie, Peroneuslähmung, Sinnesempfindungsverlust, brennendes Gefühl auf der Haut, Brennen. Augenerkrankungen: Sehr häufig: Trockenes Auge. Erkrankungen des Gastroin-
testinaltrakts: Sehr häufig: Diarrhoe, Erbrechen, Übelkeit. Erkrankungen der Haut und des Unterhautgewebes: Sehr häufig: Alopezie, Pruritus, Ausschlag, makulo-papulöser Ausschlag, trockene Haut; Häufig: Medikamentenausschlag, Exfoliation der Haut, Konjunktivitis, bullöse Dermatitis,
Blasen, Stomatitis, palmar-plantares Erythrodysästhesie-Syndrom, Ekzem, Erythem, erythematöser Ausschlag, makulöser Ausschlag, papulöser Ausschlag, Ausschlag mit Juckreiz, blasiger Hautausschlag; Gelegentlich: Generalisierte exfoliative Dermatitis, Erythema multiforme, exfoliativer
Hautausschlag, Pemphigoid, makulovesikuläre Hautreaktion, Dermatitis, allergische Dermatitis, Kontaktdermatitis, Intertrigo, Hautreizung, Stauungsdermatitis, Blutblase; Nicht bekannt (basierend auf weltweiten Erfahrungen nach der Markteinführung): Epidermolysis acuta toxica, Stevens-
Johnson-Syndrom, Epidermalnekrose, symmetrisches arzneimittelbedingtes intertriginöses und flexurales Exanthem. Allgemeine Erkrankungen und Beschwerden am Verabreichungsort: Sehr häufig: Fatigue; Häufig: Extravasat an der Infusionsstelle. Untersuchungen: Sehr häufig: Erhöhte
Alaninaminotransferase, erhöhte Aspartataminotransferase, erniedrigtes Gewicht. Beschreibung ausgewählter Nebenwirkungen: Immunogenität: Insgesamt wurden 590 Patienten auf eine Immunogenität gegenüber Enfortumab Vedotin 1,25 mg/kg getestet. Bei 15 Patienten wurde bestätigt,
dass sie zu Beginn der Studie positiv für Anti-Wirkstoff-Antikörper waren, und bei Patienten, die zu Beginn der Studie negativ waren (n = 575), waren insgesamt 16 (2,8 %) nach Beginn der Studie positiv (13 vorübergehend und 3 dauerhaft). Aufgrund der begrenzten Anzahl von Patienten mit
Antikörpern gegen Padcev können keine Rückschlüsse auf einen möglichen Einfluss der Immunogenität auf die Wirksamkeit, Sicherheit oder Pharmakokinetik gezogen werden. Für eine detaillierte Beschreibung der ausgewählten Nebenwirkungen Hautreaktionen, Hyperglykämie, Periphere
Neuropathie und Augenerkrankungen siehe Abschnitt 4.8 „Nebenwirkungen“ in der Fachinformation. Meldung des Verdachts auf Nebenwirkungen: Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von großer Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung
des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung dem Bundesinstitut für Impfstoffe und biomedizinische Arzneimittel, Paul-Ehrlich-Institut, Paul-Ehrlich-Str. 51 59, 63225 Langen,
Tel: +49 6103 77 0, Fax: +49 6103 77 1234, Website: www.pei.de anzuzeigen. Besondere Warnhinweise und Vorsichtsmaßnahmen für die Anwendung: Rückverfolgbarkeit: Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels
und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden. Hautreaktionen: Hautreaktionen werden mit Enfortumab Vedotin als Folge der Bindung von Enfortumab Vedotin an das in der Haut exprimierte Nectin-4 in Verbindung gebracht. Fieber oder
grippeähnliche Symptome können das erste Anzeichen einer schweren Hautreaktion sein. Wenn dies auftritt, sollen die Patienten beobachtet werden. Leichte bis moderate Hautreaktionen, vorwiegend makulo-papulöser Ausschlag, wurden berichtet (siehe Abschnitt „Nebenwirkungen“).
Schwere kutane Nebenwirkungen, einschließlich Stevens-Johnson-Syndrom (SJS) und Epidermolysis acuta toxica (toxische epidermale Nekrolyse, TEN), mit tödlichem Ausgang sind ebenfalls bei Patienten aufgetreten, die mit Enfortumab Vedotin behandelt wurden, vorwiegend während des
ersten Behandlungszyklus. In klinischen Studien betrug die mediane Zeit bis zum Auftreten von schweren Hautreaktionen 0,6 Monate (Bereich: 0,1 bis 6,4). Die Patienten sollen beginnend mit dem ersten Zyklus und während der gesamten Behandlung auf Hautreaktionen überwacht werden.
Bei leichten bis moderaten Hautreaktionen kann eine geeignete Behandlung, wie z. B. topische Kortikosteroide und Antihistaminika, erwogen werden. Bei Verdacht auf SJS oder TEN, oder im Falle von beginnenden bullösen Hautläsionen, unterbrechen Sie die Behandlung sofort und überweisen
Sie an einen Facharzt. Eine histologische Bestätigung, einschließlich der Erwägung von mehreren Biopsien, ist für die Früherkennung entscheidend, da Diagnose und Intervention die Prognose verbessern können. Setzen Sie Padcev bei bestätigtem SJS oder TEN, Grad 4- oder wiederkehrenden
schweren Hautreaktionen dauerhaft ab. Bei sich verschlechternden Grad 2-Hautreaktionen, Grad 2-Hautreaktionen mit Fieber oder bei Grad 3-Hautreaktionen soll die Behandlung unterbrochen werden bis Grad ≤ 1 erreicht ist und eine Überweisung an einen Facharzt in Erwägung gezogen
werden. Die Behandlung soll in der gleichen Dosisstufe wieder aufgenommen oder eine Dosisreduktion um eine Dosisstufe in Betracht gezogen werden (siehe Abschnitt 4.2 „Dosierung und Art der Anwendung“ in der Fachinformation). Hyperglykämie: Hyperglykämie und diabetische Ketoazi-
dose, einschließlich tödlicher Ereignisse, traten bei Patienten mit und ohne vorbestehenden Diabetes mellitus auf, die mit Enfortumab Vedotin behandelt wurden (siehe Abschnitt „Nebenwirkungen“). Hyperglykämie trat häufiger bei Patienten mit vorbestehender Hyperglykämie oder einem
hohen Body-Mass-Index (≥ 30 kg/m2) auf. Patienten mit einem Ausgangs-Hämoglobin A1C (HbA1c) ≥ 8 % wurden von den klinischen Studien ausgeschlossen. Der Glucosespiegel im Blut soll bei Patienten mit Diabetes mellitus oder dem Risiko für Diabetes mellitus oder Hyperglykämie vor
Verabreichung der Dosis und regelmäßig im Behandlungsverlauf je nach klinischen Erfordernissen kontrolliert werden. Wenn die Glucose im Blut auf > 13,9 mmol/l (> 250 mg/dl) erhöht ist, soll Padcev pausiert werden, bis die Glucose im Blut ≤ 13,9 mmol/l (≤ 250 mg/dl) beträgt, und es soll
entsprechend behandelt werden (siehe Abschnitt 4.2 „Dosierung und Art der Anwendung“ in der Fachinformation). Periphere Neuropathie: Periphere Neuropathie, vorwiegend periphere sensorische Neuropathie, ist unter Enfortumab Vedotin einschließlich Reaktionen vom Grad ≥ 3 aufgetreten
(siehe Abschnitt „Nebenwirkungen“). Patienten mit vorbestehender peripherer Neuropathie Grad ≥ 2 wurden von den klinischen Studien ausgeschlossen. Die Patienten sollen auf Symptome einer neuen oder sich verschlimmernden peripheren Neuropathie überwacht werden, da bei diesen
Patienten eine Verzögerung, Dosisreduktion oder ein Absetzen von Enfortumab Vedotin erforderlich sein könnte (siehe Tabelle 1 in der Fachinformation). Padcev soll bei peripherer Neuropathie Grad ≥ 3 dauerhaft abgesetzt werden (siehe Abschnitt 4.2 „Dosierung und Art der Anwendung“ in
der Fachinformation). Augenerkrankungen: Bei Patienten, die mit Enfortumab Vedotin behandelt wurden, traten Augenerkrankungen, vorwiegend trockenes Auge, auf (siehe Abschnitt „Nebenwirkungen“). Die Patienten sollen auf Augenerkrankungen überwacht
werden. Ziehen Sie künstliche Tränenflüssigkeit zur Prophylaxe des trockenen Auges und eine Überweisung zur augenärztlichen Untersuchung in Betracht, wenn die okulären Symptome nicht abklingen oder sich verschlimmern. Extravasat an der Infusionsstelle:
Haut- und Weichteilverletzungen wurden nach der Verabreichung von Enfortumab Vedotin beobachtet, wenn ein Extravasat auftrat (siehe Abschnitt „Nebenwirkungen“). Stellen Sie einen guten venösen Zugang sicher, bevor Sie mit Padcev beginnen, und achten
Sie während der Verabreichung auf ein mögliches Extravasat an der Infusionsstelle. Wenn ein Extravasat auftritt, stoppen Sie die Infusion und achten Sie auf Nebenwirkungen. Embryo-fetale Toxizität und Empfängnisverhütung: Schwangere Frauen sollen über das
mögliche Risiko für einen Fetus informiert werden (siehe Abschnitte 4.6 „Fertilität, Schwangerschaft und Stillzeit“ und 5.3 „Präklinische Daten zur Sicherheit“ in der Fachinformation). Frauen im fortpflanzungsfähigen Alter soll geraten werden, innerhalb von 7 Tagen
vor Beginn der Behandlung mit Enfortumab Vedotin einen Schwangerschaftstest durchzuführen und während der Behandlung und für mindestens 12 Monate nach Beendigung der Behandlung eine wirksame Verhütungsmethode anzuwenden. Männern, die mit
Enfortumab Vedotin behandelt werden, wird empfohlen, während der Behandlung und für bis zu 9 Monate nach der letzten Dosis von Padcev kein Kind zu zeugen. Warnhinweise: Arzneimittel für Kinder unzugänglich aufbewahren. Verschreibungspflichtig.
Weitere Einzelheiten enthalten die Fach- und Gebrauchsinformation. Pharmazeutischer Unternehmer: Astellas Pharma Europe B.V., Sylviusweg 62, 2333 BE Leiden, Niederlande; Deutsche Vertretung des pharmazeutischen Unternehmers: Astellas Pharma
GmbH, Ridlerstraße 57, 80339 München. Stand: April 2022.
AST21360 | ENF_2021_0011_DE | Erstellt: Apr 2022 Mehr zu PADCEVSie können auch lesen