Expression und Regulation von CD90 (Thy-1) auf makrovaskulären Endothelzellen
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Expression und Regulation von CD90 (Thy-1) auf
makrovaskulären Endothelzellen
Dissertation
zur Erlangung des akademischen Grades
Dr. med.
an der Medizinischen Fakultät
der Universität Leipzig
eingereicht von
Migle Uptaite
16.05.1984 / Vilnius
angefertigt an der
Klinik für Visceral-, Transplantations-, Thorax- und Gefäßchirurgie, Universität Leipzig
Betreuerin: Prof. Dr. rer. nat. Gabriela Aust
Beschluss über die Verleihung des Doktorgrades vom 18.06.2013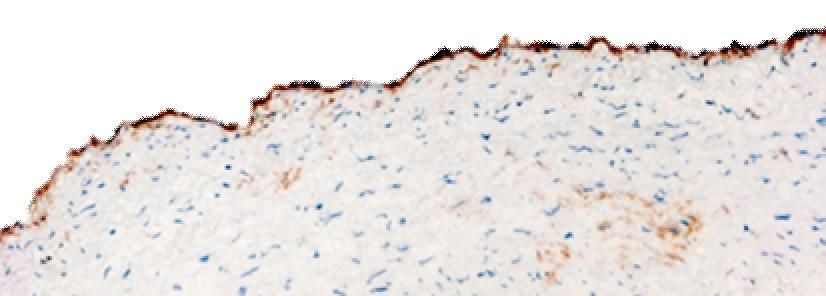
Meiner Tochter Doroteja
1Bibliographische Beschreibung
Migle Uptaite
Expression und Regulation von CD90 (Thy-1) auf makrovaskulären Endothelzellen
Universität Leipzig, Dissertation
59 Seiten, 106 Literaturangaben, 16 Abbildungen, 7 Tabellen
Referat:
CD90 (Thy-1) ist ein Adhäsionsmolekül, das auf aktivierten mikrovaskulären Endothelzellen
(EC) die Bindung von Entzündungszellen über die Interaktion mit dem Integrin m 2 (Mac-1,
CD11b/CD18) vermittelt. Die Expression von CD90 auf mikrovaskulären EC wurde sowohl
in-vitro als auch in-vivo nachgewiesen. Zur Expression von CD90 auf makrovaskulären EC
gibt es nur wenige und sich zum Teil widersprechende in-vitro Daten. In-situ konnte die
Expression von CD90 auf diesen Zellen bisher nicht gezeigt werden.
Die Atherosklerose ist ein stufenweise verlaufendes chronisch-entzündliches Geschehen in
den arteriellen Gefäßen. In der vorliegenden Arbeit wurde in atherosklerotisch-veränderten
Gefäßen die Expression von CD90 auf humanen makrovaskulären EC in-situ demonstriert.
Die Spezifität der immunhistologischen Darstellung von CD90 auf den makrovaskulären EC
atherosklerotischer Gefäße wurde mittels Doppelmarkierung in der Immunfluoreszenz
geklärt. CD90 wurde in jedem Atherosklerosestadium auf EC nachgewiesen. Eine Zunahme
der CD90 Expression in höheren Atherosklerosestadien wurde demonstriert.
Weiterhin sollte mittels Stimulationsversuchen in-vitro geklärt werden, wie die CD90
Expression auf makrovaskulären EC im Rahmen der Atherosklerose reguliert wird. Die
Expression von CD90 konnte in-vitro durch pro-inflammatorische Zytokine, die im Prozess
der Atherosklerose eine wichtige Rolle spielen, tendenziell erhöht werden. Durch die
Stimulation mit den lipid-beladenen Schaumzellen, die zahlreich in atherosklerotischen
Läsionen vorhanden sind, konnte die CD90 Expression nicht erhöht werden. Die Stimulation
mit D-Glukose führte zur tendenziellen Reduktion der CD90 Expression. Die Stimulation mit
CXCL12, einem bedeutsamen Trigger der Mobilisation der endothelialen Vorläuferzellen,
bewirkte einen signifikanten Anstieg der CD90 Expression.
Die Ergebnisse indizieren, dass CD90 eine wichtige Rolle auf makrovaskulären EC in
entzündlichen Prozessen spielt. Möglicherweise ist CD90 an der Migration von Leukozyten
bzw. endothelialen Vorläuferzellen durch das makrovaskuläre Endothel beteiligt.
2INHALTSVERZEICHNIS
ABKÜRZUNGSVERZEICHNIS 4
1. EINLEITUNG 6
1.1. Atherosklerose 6
1.2. Klassifikation der Atherosklerose 6
1.3. Klinische Manifestationen der Atherosklerose 9
1.3.1. Stenose der Arteria carotis interna und Schlaganfall 9
1.4. Pathogenese der Atherosklerose: Leukozyten-EC Interaktion 11
1.4.1. CD90 - ein endotheliales Adhäsionsmolekül 13
1.5. Fragestellung und Zielsetzung 15
2. MATERIAL UND METHODEN 17
2.1. Patientenkollektiv 17
2.1.1. Gewinnung und Aufarbeitung der Gefäße 17
2.1.2. Herstellung von Gefrierschnitten 18
2.1.3. Histomorphologische Beurteilung 18
2.2. Immunohistochemie 19
2.3. Immunofluoreszenz 20
2.4. Zellkultur 21
2.4.1. Präparation der HUVEC 21
2.4.2. Humane EC aus der Koronararterie 21
2.4.3. Stimulation der EC 22
2.4.3.1. Kultur der monozytären Zelllinie THP-1 23
2.4.4. Durchflußzytometrie 24
2.4.5. Statistische Auswertung und Abbildungen 24
3. ERGEBNISSE 25
3.1. Histomorphologische Charakterisierung der Gefäßpräparate 25
3.2. Expression von CD90 auf makrovaskulären EC der atherosklerotischen Gefäße 28
3.3. CD90 Expression auf makrovaskulären EC in vitro 30
3.3.1. Expression von CD90 auf unstimulierten EC 31
3.3.2. Regulation der Expression von CD90 auf EC durch Zytokine 32
3.3.3. Regulation der Expression von CD90 auf EC durch Glukose 35
3.3.4. Auswirkung der Schaumzellen auf die Expression von CD90 auf EC in vitro 36
4. DISKUSSION 38
4.1. CD90 Expression im Rahmen der Atherosklerose 38
4.1.1. Rolle der neutrophilen Granulozyten in der Atherosklerose 39
4.2. CD90 – ein Adhäsionsmolekül für EPC? 41
4.3. CD90 Expression auf mikro- und makrovaskulären EC 42
4.4. Methodische Diskussion der Ergebnisse 44
ZUSAMMENFASSUNG DER ARBEIT 46
LITERATURVERZEICHNIS 49
Erklärung über die eigenständige Abfassung der Arbeit 57
3Abkürzungsverzeichnis
ACE Angiotensin-konvertierendes Enzym
ACI Arteria carotis interna
AHA American Heart Association, engl.
CD Cluster of differentiation engl.
CXCL CX chemokine ligand, engl.
DAB 3,3'-Diaminobenzidin-Gemisch
DAPI 4´,6-Diamidino-2-Phenylindol
EC Endothelzellen
ECBM endothelial cell basal medium, engl.
EEA Eversionsendarteriektomie
EPC endothelial progenitor cells, engl.
FCS fetal calf serum, engl.
Glut-1 Glucose transporter 1, engl.
GPI Glykophosphatidylinositol
HCAEC human coronary artery endothelial cells, engl.
HDMEC human dermal microvascular endothelial cells, engl.
HUVEC human umbilical vein endothelial cells, engl.
ICAM-1 intercellular adhesion molecule 1, engl.
IL-1 Interleukin-1
IL-4 Interleukin-4
IL-6 Interleukin-6
IS inflammation score, engl.
JAM junctional adhesion molecules, engl.
KHK Koronare Herzkrankheit
LDL low-density-lipoprotein, engl.
LFA-1 lymphocyte function associated antigen 1, engl.
Lu-ECAM lung endothelial cell adhesion molecule, engl.
MAC-1 macrophage-1 antigen, engl.
MMP Matrix-Metalloprotease
4pAVK Periphere arterielle Verschlusskrankheit
PBS phosphate buffered saline, engl.
PFA Paraformaldehyd
PMA phorbol 12-myristate 13-acetate, engl.
RANTES regulated and normal T cell expressed and secreted, engl.
RPMI Roswell park memorial institute medium, engl.
TEA Thrombendarteriektomie
THP-1 human acute monocytic leukemia cell line
Thy-1 Thymocyte differentiation antigen 1, engl.
TIA Transitorisch ischämische Attacke
TNF- Tumornekrosefaktor-
VCAM-1 vascular cell adhesion molecule 1, engl.
VEGFR-3 vascular endothelial growth factor receptor 3, engl.
VLA-4 very late antigen 4, engl.
vWF von Willebrand Faktor
51. Einleitung
1.1. Atherosklerose
Atherosklerose und ihre Folgeerkrankungen wie koronare Herzkrankheit (KHK), Schlaganfall
und periphere arterielle Verschlusskrankheit (pAVK) führen in den westlichen
Industriestaaten die Statistik der Todesursachen an [1]. Nach der Definition der World Health
Organisation (WHO) handelt sich bei der Atherosklerose um Veränderungen der Intima
arterieller Blutgefäße. Diese Veränderungen, die herdförmige Ansammlungen bzw.
Ablagerungen von Lipiden, komplexen Kohlehydraten, Blut und Blutbestandteilen sowie von
Kalzium und Bindegewebe umfassen, können variabel kombiniert und mit Veränderungen
der Media der Arterien verbunden sein [2, 3].
Atherosklerose ist eine komplexe multifaktorielle Erkrankung, die sich in der Arterienwand als
Reaktion auf verschiedene Formen von schädlichen Reizen entwickelt und in einer
übermäßigen entzündlichen und fibro-proliferativen Reaktion resultiert [4, 5]. In den 1980er
Jahren wurde erstmals die Hypothese aufgestellt, dass Atherosklerose eine Reaktion auf
eine Läsion ist, die durch Lipide, Makrophagen und in der Gefäßwand akkumulierten glatten
Muskelzellen ausgelöst wird [5]. Diese „Response-to-injury-Hypothese“ ist akzeptiert, da sich
die meisten experimentellen Ergebnisse mit dieser Theorie vereinbaren lassen [6].
1.2. Klassifikation der Atherosklerose
Atherosklerotische Plaques unterliegen in ihrem Fortschreiten zellulären und
morphologischen Veränderungen. Das American Heart Association (AHA) Committee on
Vascular Lesions empfiehlt die morphologische Klassifikation atherosklerotischer Plaques
anhand von verschiedenen Stadien [7]. Diese Stadieneinteilung basiert auf Beobachtungen
der „natürlichen” Entwicklung von Plaques von ihren symptomlosen Frühstadien bis zu
klinisch symptomatischen Läsionen [7]. Der atherosklerotische Prozess wird in sechs
Stadien eingeteilt, die möglicherweise dem natürlichen zeitlichen Verlauf der Erkrankung
entsprechen (Abb. 1).
6Abbildung 1: AHA-Stadien der Plaqueformation nach Stary. Jedes Stadium ist durch Plaques
mit charakteristischer Morphologie gekennzeichnet [8, 9]
Jedes Stadium ist durch Plaques mit charakteristischer Morphologie gekennzeichnet (Tab.
1). Die Klassifikation bietet somit ein Spektrum typischer Morphologien, mit denen klinische
Untersuchungsergebnisse korreliert werden können [7, 8, 10].
7Stadium I: Die Atherosklerose beginnt mit der Entwicklung sogenannter „fatty
Initialläsion streaks“ [6]. Die Endothelzellen (EC) werden z.B. durch Lipoproteine
aktiviert und die extrazellulären Lipide beginnen sich in der Intima
anzureichern [9]. In die Arterienwand werden Monozyten rekrutiert. Sie
differenzieren sich zu Makrophagen und beginnen die Scavenger-
Rezeptoren zu exprimieren, die modifizierte Lipoproteine binden. Durch
Aufnahme der modifizierten Lipoproteine wandeln sich Makrophagen in
lipidbeladene Schaumzellen um [9]. Mikroskopisch sind
Lipideinlagerungen in der Intima mit dem Vorkommen von einzelnen
Schaumzellen verbunden [8].
Stadium II: Die Schaumzellen und andere Gefäßwandzellen sezernieren Zytokine,
Fettstreifenläsion welche die die Leukozytenrekrutierung verstärken und zusätzlich die
Migration und Proliferation der glatten Muskelzellen bedingen [9]. Die
glatten Muskelzellen lagern ebenfalls Lipide ein. Mikroskopisch findet
man eine erhöhte Makrophagenanzahl ohne eingelagerte
Lipidtröpfchen, stratifizierte Schaumzellschichten und glatte
Muskelzellen mit Lipideinlagerungen [8].
Stadium III: Diese Läsion ist durch die extrazellulären Lipidtröpfchen
Präatherom gekennzeichnet. Nekrotische Areale mit abgestorbenen Zellen können
sichtbar werden. Es ist die Übergangsphase zum Atherom [8].
Stadium IV: Die extrazellulären Lipide konfluieren zu einem Lipidkern [8].
Atherom
Stadium V: Zusätzlich zum Lipidkern ist diese Läsion durch einer Ausbildung von
Fibroatherom fibrösen, teilweise verkalkten Bindegewebsschichten gekennzeichnet
[8].
Stadium VI: Die Schaumzellen und andere Gefäßwandzellen sezernieren Matrix-
Läsion mit degradierende Enzyme [9]. Dies führt zum Verlust an
Oberflächendefekt Extrazellularsubstanz und verursacht somit eine Ausdünnung und
Schwächung der fibrotischen Deckplatte [11]. Die Deckplatte kann
dadurch aufbrechen. Wenn die Deckplatte aufbricht, steht der
nekrotische Kern des Plaque im direkten Kontakt mit dem Gefäßlumen,
was zur Thrombenbildung führt [9, 11]. Dieser Typ der
atherosklerotischen Läsion ist deshalb durch Deckplattenaufbrüche,
Fissuren, Ulzerationen mit Einblutungen oder Thrombenbildung
gekennzeichnet [8].
Tabelle 1: AHA-Stadien der Plaqueformation nach Stary [8, 10]
81.3. Klinische Manifestationen der Atherosklerose
Da es sich bei der Atherosklerose nicht um eine lokal begrenzte, sondern um eine
systemische Erkrankung handelt, sind bei Patienten mehrere Gefäßregionen von den
krankhaften Veränderungen betroffen.
Bei einer Manifestation in den epikardialen Koronararterien kommt es als Ausdruck der KHK
zu einer Minderperfusion der Myokardabschnitte, die durch die betroffenen Koronararterien
versorgt werden. Die Folge ist ein lokales Missverhältnis zwischen Sauerstoffbedarf und -
angebot, dass zu regionalen myokardialen Ischämien führt. Die klinischen Manifestationen
reichen von der asymptomatischen Ischämie über die stabile Angina pectoris bis hin zu dem
akuten Koronarsyndrom [6].
Bei einer Manifestation der Atherosklerose in den Becken- und Beinarterien kommt es zu
Durchblutungsstörungen in den Extremitäten. Dies wird als pAVK bezeichnet.
Bei einer Manifestation in extrakraniellen Hirngefäßen kommt es zu zerebralen
Durchblutungsstörungen, denen ein Schlaganfall folgen kann [12, 13].
1.3.1. Stenose der Arteria carotis interna und Schlaganfall
Der Schlaganfall (Apoplex) zählt zu den häufigsten Erkrankungen in Deutschland und ist
weltweit eine der führenden Ursachen von Morbidität und Mortalität [13]. In der deutschen
Todesursachenstatistik belegt er 2006 mit 65.133 Todesällen (7,9%) Platz 4 (Statistisches
Bundesamt 2006). Die prospektive populationsbezogene Oxford Vascular Study [14] zeigte
sogar erstmals in einer westlichen Bevölkerung, dass die Rate der zerebrovaskulären
Erkrankungen die Häufigkeit kardiovaskulärer Erkrankungen übertrifft oder zumindest
erreicht. Heute ist der Schlaganfall die häufigste Ursache dauerhafter Behinderung und in
Industrieländern die teuerste Krankheit überhaupt [13].
Das Gehirn wird aus zwei arteriellen Gefäßen mit Blut versorgt: aus der Arteria vertebralis,
einem Ast der Arteria subclavia, und aus der Arteria carotis interna (ACI). Dabei übernimmt
die ACI etwa 70% des geförderten Gesamtvolumenstroms [15]. Etwa 60% der Schlaganfälle
sind durch Veränderungen der hirnversorgenden Gefäße, insbesondere der ACI, verursacht.
Diesem sogenannten ischämischen Schlaganfall liegt ein Sistieren der Blut- und damit der
Sauerstoffversorgung im Gehirngewebe durch Ablösung von an der Stenose entstandenen
Blutgerinnseln mit konsekutiver Verstopfung nachgeschalteter Hirngefäße zu Grunde. Das
führt zu einem Funktionsverlust und schließlich zum Absterben von Hirngewebe.
Bedingt durch die große Anzahl möglicherweise betroffener Hirnareale gibt es eine Vielzahl
klinischer Erscheinungsformen [13]. Dabei reicht das Spektrum von kurzfristigen
Sehstörungen eines Auges (Amaurosis fugax), über sensomotorische Syndrome mit oder
9ohne kortikale Ausfallerscheinung bis zum malignen Mediainfarkt bei komplettem Verschluss
der ACI [16]. Der zeitliche Krankheitsverlauf ist sehr variabel. Die Symptome können bei der
sogenannten transitorisch ischämischen Attacke (TIA) nur Minuten oder Stunden andauern
oder beim vollendeten Hirninsult dauerhaft bestehen [13]. Nur etwa 30 bis 50% aller
Patienten realisieren Prodromalsymptome vor einem Schlaganfall im Sinne einer TIA [16].
Alle Patienten mit einer ACI-Stenose müssen medikamentös behandelt werden.
Thrombozytenaggregationshemmer und Antihypertensiva stellen die Hauptkomponenten
dieser Therapie dar. Die Therapie mit Statinen, deren Verabreichung in Anlehnung an die
Koronarprävention basiert, wird ebenfalls empfohlen [17].
Bei der operativen Therapie der Stenose der ACI werden zwei unterschiedliche
Operationstechniken durchgeführt. Bei der Thrombendarteriektomie (TEA) wird der
obliterierende Innenschichtzylinder an der Bifurkation der A. carotis communis über eine
Längsarteriotomie ausgeschält. Die Arteriotomie wird mittels autologer Venenpatchplastik
oder Kunststoff verschlossen. Im langjährigen postoperativen Verlauf zeigt diese Methode
eine höhere Komplikationsrate im Vergleich zu der Eversionsendarteriektomie (EEA), d.h. es
kommt öfter zu Rezidivstenosen oder Patchaneurysmen. Bei EEA wird die ACI an ihrem
Abgang aus der A. carotis communis schräg durchtrennt. Die Adventitia und eng anliegende
Teile des Mediaschlauches werden über den Stenosezylinder kranialwärts gestülpt, bis eine
unauffällige intimale Gewebsschicht erreicht wird und hier die arteriosklerotischen
Veränderungen stufenlos auslaufen. Die EEA bietet den Vorteil, dass vor allem eine kürzere
Abklemmzeit und eine physiologische Wiederherstellung der Strombahn durch unmittelbare
Streckung elongierter ACI Abschnitte im Rahmen der Gabelrekonstruktion der A. carotis
durchgeführt werden können. Bei der EEA ist kein Fremdmaterial nötig [18, 19].
Primärpräventiv kann die Endarteriektomie nicht allgemein empfohlen werden, sie ist nur in
Einzelfällen sinnvoll. Meta-Analysen zeigten für Patienten mit bisher asymptomatischen
mittel- bis hochgradigen Stenosen (> 50%) der ACI nur eine leichte Reduktion der
Schlaganfallhäufigkeit nach Endarteriektomie und empfahlen daher keine Operation [20, 21].
Sekundärpräventiv ist eine operative Versorgung der ACI-Stenose bei einem
Stenosierungsgrad ab 70% sinnvoll. Bei einem Stenosegrad von 70-80% beträgt die
Risikoreduktion durch die operative Versorgung einen weiteren Schlaganfall zu erleiden ca.
27%, bei einem Stenosegrad von >80% - sogar 48% [20, 21].
Als Alternative zu der operativen Versorgung der ACI-Stenose kann die transkutane
Angioplastie mit oder ohne Stent derzeit nicht generell empfohlen werden. Indikationen zur
perkutanen interventionellen Therapie der ACI-Stenose mit Stentimplantation sind bei
Patienten mit symptomatischer mindestens 50%iger, oder asymptomatischer mindestens
80%iger ACI-Stenose und beim Vorliegen eines erhöhten Operationsrisikos zu vertreten. Bei
10Patienten unter 75 Jahren mit asymptomatischer, mindestens 70%iger ACI-Stenose ist die
perkutane interventionelle Therapie ebenso möglich. [20-22].
1.4. Pathogenese der Atherosklerose: Leukozyten-EC Interaktion
Diabetes mellitus, Dyslipidämie, arterielle Hypertonie und Übergewicht sind die wichtigsten
Risikofaktoren für die Entwicklung einer Atherosklerose [23]. Sie sind mit einer Erhöhung von
oxidativem Stress und Entzündung im Endothel assoziiert [4]. Eine zentrale Rolle in der
Entwicklung der Atherosklerose spielt die Dyslipoproteinämie, insbesondere der Low-
Density-Lipoproteine (LDL). LDL werden in der Intima der Arterien akkumuliert, unterliegen
dort einer oxidativen Modifikation und induzieren eine Expression von Adhäsionsmolekülen,
wie z.B. ICAM-1 (engl. intercellular adhesion molecule 1) auf EC [24]. Die Hyperglykämie bei
Diabetes mellitus verursacht Veränderungen von Makromolekülen auf den EC, die ebenfalls
zur vermehrten Produktion von pro-inflammatorischen Zytokinen in EC führen [24].
Die vermehrte Expression der endothelialen Adhäsionsmoleküle fördert die Adhäsion von
Leukozyten an EC und schließlich deren Transmigration durch das Endothel. Die
Extravasation erfordert eine ganze Reihe von überlappenden synergistischen Interaktionen
zwischen den Adhäsionsmolekülen auf den Leukozyten und EC [4, 25]. Während der
Extravasation werden das Endothel und die Leukozyten durch Zytokine aktiviert. Diese
Zytokine können spezifisch für bestimmte endotheliale Adhäsionproteine sein, z.B.
Interleukin-4 (IL-4) für VCAM-1 (engl. vascular cell adhesion molecule 1), oder sie können
unspezifisch wirken und die Expression von mehreren endothelialen Adhäsionproteinen
induzieren wie z.B. Tumornekrosefaktor- (TNF- und Interleukin-1 (IL-1) für E-Selektin,
ICAM-1 und VCAM-1 [26].
Die Extravasation von Leukozyten aus dem Blut in das perivaskuläre Gewebe spielt eine
Schlüsselrolle im Prozess der Atherosklerose [25]. Die Extravasation schließt „Erfassen“ der
zirkulierenden Leukozyten am Endothel, das „tethering“ und „rolling“ der Leukozyten entlang
des Endothels, ihre Anhaftung und feste Adhäsion sowie die anschließende Transmigration
ein [25].
Der erste Kontakt des Leukozyten mit der Gefäßwand ist meist ein zufälliges Ereignis, etwa
durch lokale Veränderungen in den Flusseigenschaften des Blutes [26]. Nach dem ersten
Kontakt des Leukozyten an den EC ist ein Rollen (engl. rolling) entlang der Gefäßwand zu
beobachten. Adhäsionsproteine der Selektin-Familie sind vor allem in der Anfangsphase der
Extravasion von Leukozyten beteiligt (Tab. 2) [26, 27].
Eine feste Adhäsion von Leukozyten an das Endothel ist von der Expression von 2
Integrinen wie Mac-1 (engl. macrophage-1 antigen, CD11b/CD18, m 2) oder LFA-1 (engl.
11lymphocyte function associated antigen 1, CD11a/CD18, L 2) auf der Oberfläche von
Leukozyten abhängig. Die 2 Integrine auf der Leukozytenoberfläche interagieren mit
Endothelzellproteinen der Immunglobulin (Ig)-Superfamilie wie z.B. ICAM-1 und VCAM-1 [25,
26, 28]. Eine Bindung der 2 Integrine wie Mac-1 auf der Leukozytenoberfläche an ICAM-1
ist für eine feste Adhäsion und spätere Migration der Leukozyten durch das Endothel
besonders wichtig. 1993 konnte aber gezeigt werden, dass neutrophile Granulozyten von
ICAM-1-null Mäusen trotz Fehlen von ICAM-1 ins entzündete Peritoneum auswandern
können [29]. Diese Beobachtung weist auf die Existenz eines zweiten Liganden, der mit
Mac-1 interagiert, hin [26]. Saalbach et al. zeigten 2004, dass CD90 (Thy-1, engl. Thymocyte
differentiation antigen 1) die Adhäsion der Entzündungszellen an aktivierten mikrovaskulären
EC über die Interaktion mit Mac-1 auf Leukozyten in-vitro vermittelt (Abb. 2) [25, 30]. Diese
über 2 Integrin und CD90 vermittelte Adhäsion von Leukozyten spielt ebenfalls eine
wichtige Rolle bei der Extravasion von Leukozyten in entzündetes Gewebe [25, 30].
Abbildung 2: Feste Adhäsion und transendotheliale Migration der Leukozyten: eine feste
Adhäsion von Leukozyten an das Endothel ist von der Interaktion der 2-Integrine wie VLA-4
(engl. very late antigen 4), LFA-1 oder Mac-1 auf den Leukozyten mit VCAM-1, ICAM-1 oder
CD90 auf den EC abhängig. Die transendotheliale Migration der Leukozyten erfordert
weitere Adhäsionsmoleküle wie CD31 (PECAM-1, engl. platelet endothelial cell adhesion
molecule 1) oder CD99 sowie JAM’s (engl. junctional adhesion molecules) (Modifiziert nach
KEGG PATHWAY Database) [31].
12In den letzten Schritten der Kaskade migrieren die adhärenten Leukozyten zwischen den
Endothelzellverbindungen, und akkumulieren schließlich in der subendothelialen
extrazellulären Matrix, am Ort der entzündlichen oder Immunreaktion. In diesen Schritten
sind u.a. CD31 auf EC sowie Integrine auf Leukozyten beteiligt (Tab. 2) [26].
Migrationsschritt Adhäsionsproteine Adhäsionsproteine In Stimulation der
der Leukozyten der Endothelzellen Adhäsionsproteine der
Endothelzellen involvierte
Zytokine
Tethering und L-Selektin [32, 33] GlyCAM-1 [34], MCP-1, IL-8, RANTES and
Rollen sLex, CD34 MIP-1 [32]
PSGL-1 [35] MAdCAM-1 [33, Thrombin, Histamin [34], IL-
36], PSGL-1 [35] 13 [36], TNF- , IL-1 [33]
P-Selektin [33, 36] TNF- , IL-1[34], IL-13, INF-
E-Selektin [33, 36] [36]
4 1 (VLA-4) [32, VCAM-1 [32, 37] TNF- , IL-1 [34]
37]
Feste Adhäsion 4 1 (VLA-4) [32, VACM-1[32, 35, 37, TNF- , IL-1 [34], IL-4 [36,
35-38] 38] 39] IL-13 [36]
L 2 (LFA-1) [32, ICAM-1 [32, 35, 38] TNF- [34], IL-1, INF- [34,
35, 36, 38] 36], LPS, shear stress [36]
m 2 (MAC-1)[32] ICAM-1 [40], CD90 TNF- [38], IL-1 [41]
[25]
4 7 [35, 36]
9 1 [36] MAD-CAM [35]
Transmigration PECAM-1 [34], PECAM-1 [34, 35] TNF- [40]
CD99 [40]
CD99 [40] JAM-1, JAM-2, IL-1 [36]
JAM-3, CD99 [35,
40]
Tabelle 2: Die Leukozytenmigration schließt mehrere Phasen ein: zunächst „Tethering“ und
„Rollen“, feste Haftung und schließlich transendotheliale Migration. Endotheliale
Adhäsionsproteine sind in jedem Leukozytenmigrationsschritt beteiligt und werden durch
bestimmte Zytokine während der Atheroskleroseentwicklung aktiviert.
1.4.1. CD90 - ein endotheliales Adhäsionsmolekül
13CD90 ist ein relativ kleines Glykoprotein mit einem Molekulargewicht von 25-37 kDA [42],
welches eine einzige Immunoglobulin (Ig)-ähnliche Domäne enthält und über einen
Glykophosphatidylinositol (GPI)-Anker in die Zellmembran integriert ist. CD90 wird im
Menschen auf der Oberfläche von Fibroblasten, Nervenzellen, EC und einer Subpopulation
der CD34+ hämatopoetischen Stammzellen exprimiert [30]. Es spielt eine Rolle bei der
Regulation der Zelladhäsion, des Zellwachstums und der Zelldifferenzierung [43].
Hinsichtlich der Funktion von CD90 auf EC konnte gezeigt werden, dass es die Adhäsion der
Leukozyten an aktivierte mikrovaskulären EC sowie die Adhäsion von Melanomzellen an das
mikrovaskuläre Endothel vermittelt [25, 30].
Auffällig ist, dass zur Expression von CD90 auf EC widersprüchliche Daten publiziert
wurden. Mason et al (1997) demonstrierten die Expression von CD90 auf HDMEC (engl.
human dermal microvascular endothelial cells) sowie HUVEC (engl. human umbilical vein
endothelial cells) in-vitro, die sich durch Stimulation mit TNF- und PMA (engl. phorbol 12-
myristate 13-acetate) verstärken ließ [43]. Im Unterschied dazu konnten Takeuchi et al.
(1997) kein CD90 auf ruhenden dermalen mikrovaskulären EC (DMEC) der Ratte
nachweisen. Die Expression konnte aber durch eine Entzündungsreaktion auf DMEC
induziert werden [44]. Saalbach et al. (2000) fanden keine Expression von CD90 auf
ruhenden HDMEC in-vitro und in-situ. CD90 konnte aber in-vitro nach Stimulation mit
Phorbolester induziert werden. In der entzündlich veränderten Haut bei Psoriasis wurde
CD90 ebenfalls auf mikrovaskulären EC nachgewiesen [45]. Lee et al. (1998) haben die
Expression von CD90 vor und nach Ballondilatation der A. carotis im Rattenmodell
analysiert. Kleine Blutgefäße der Adventitia exprimierten vor der Ballondilatation kein CD90.
Nach der Ballondilatation wurde CD90 auf dem Endothel dieser Mikrogefäße nachgewiesen.
Die makrovaskulären EC der ACI blieben vor und nach der Dilatation CD90-negativ [46].
Ishizu et al. (1997) haben gezeigt, dass isolierte ruhende EC der Vena cava inferior der
Ratte CD90 exprimieren. In-vivo war CD90 jedoch nicht nachweisbar [47].
Insgesamt wurde nur in wenigen Arbeiten die Expression von CD90 auf makrovaskulären EC
analysiert. Die Ergebnisse dieser Untersuchungen sind widersprüchlich (Tab. 3); zudem
wurden überwiegend HUVEC genutzt. HUVEC werden zwar aus einem großen Gefäß, der
Nabelschnurvene, isoliert, sie stammen damit allerdings aus fötalem Gewebe. Sie
unterscheiden sich dadurch von adulten makrovaskulären EC hinsichtlich der Expression
von Oberflächenantigenen und der Stimulierbarkeit mit Zytokinen [43]. Zusammenfassend
lässt sich feststellen, dass die Expression von CD90 auf adulten humanen makrovaskulären
arteriellen EC bisher weder in-vitro noch in-vivo gezeigt wurde.
Autor (Jahr), Mikrovaskuläre EC Makrovaskuläre EC
untersuchte Aktivierte EC Ruhende EC Aktivierte EC Ruhende EC
Spezies
14In vivo
Mason et al CD90+ neue
(1997), Mensch Gefäße in
[43] Plaques
Ishizu et al CD90- EC der
(1997), Ratte V.cava inf.
[47]
Lee et al (1998), CD90+ EC der CD90- EC der CD90- EC der CD90- EC der
Ratte [46] kleinen kleinen A. carotis int. A. carotis int.
adventitiellen adventitiellen
Blutgefäßen der Blutgefäßen der
A. carotis int. A. carotis int.
Saalbach et al CD90+ HDMEC CD90- HDMEC
(2000), Mensch
[48]
Wetzel et al CD90+ HDMEC CD90- HDMEC
(2006), Mensch
[45]
In vitro
Mason et al CD90+ HDMEC CD90+ HDMEC CD90+ HUVEC CD90+ HUVEC
(1996), Mensch
[43]
Takeuchi et al CD90+ DMEC CD90- DMEC
(1997), Ratte
[44]
Ishizu et al CD90+: EC der
(1997), Ratte V. cava inferior
[47]
Saalbach et al CD90+ HDMEC CD90- HDMEC
(1999, 2002),
Mensch [48]
Wetzel et al CD90+ HDMEC CD90- HDMEC CD90- HUVEC CD90- HUVEC
(2006), Mensch CD90- HMEC-1
[45]
Tabelle 3: Die Daten verschiedenen Autoren zur Expression von CD90 auf mikro- und
makrovaskulären EC in-vitro und in-vivo.
1.5. Fragestellung und Zielsetzung
15Das humane CD90, ein membrangebundenes Glykoprotein, wird auf der Oberfläche von
aktivierten mikrovaskulären EC, Fibroblasten, Nervenzellen und einer Subpopulation von
CD34+ hämatopoetischen Stammzellen exprimiert [30]. CD90 fungiert als Adhäsionsmolekül
auf aktivierten mikrovaskulären EC, indem es die Bindung von Leukozyten über die
Interaktion mit dem Integrin m 2 (Mac-1, CD11b/CD18) oder dem Adhäsions-GPCR CD97
an das Endothel vermittelt [49]. Die Expression von Thy-1 auf mikrovaskulären EC wurde
sowohl in-vitro als auch in-vivo u.a. in der Haut [45, 48, 50], in neu einsprossenden Gefäßen
artherosklerotischer Plaques [43] und auf EC der kleinen adventitiellen Blutgefäßen der A.
carotis interna [46] gezeigt.
Die Expression von CD90 auf humanen makrovaskulären EC wurde bisher nur in-vitro
demonstriert, eine CD90 Expression in-vivo konnte bisher nicht gezeigt werden.
Ziel der vorliegenden Arbeit war es:
a) CD90 auf humanen makrovaskulären EC in-vivo nachzuweisen. Neben
Operationspräparaten von Patienten mit einer Stenose der ACI, die entsprechend der
AHA Klassifikation die höchsten Atherosklerosestadien zeigen, wurden
Gefäßtransplantate von Organspendern, die meist nur eine geringe Ausprägung der
Atherosklerose aufwiesen, untersucht. Die Expression von CD90 auf EC wurde zum
Atherosklerosestadium korreliert. Zusätzlich wurde die Spezifität der
immunhistologischen Darstellung von CD90 in der inneren luminalen Zellschicht
atherosklerotischer Gefäße mittels Doppelmarkierung in der Immunfluoreszenz
geklärt.
b) Offen ist, wie die Expression von CD90 auf makrovaskulären EC induziert bzw.
stimuliert wird. Da pro-inflammatorische Zytokine direkt in den atherosklerotischen
Prozess involviert sind, wurde die Expression von CD90 auf makrovaskulären EC in-
vitro nach Stimulation mit pro-inflammatorischen Zytokinen, Chemokinen und
Wachstumsfaktoren untersucht. Weiterhin wurde geprüft, ob Glukose die Expression
von CD90 in-vitro reguliert, da Diabetes mellitus mit einem früheren Auftreten einer
Atherosklerose assoziiert ist [51]. Außerdem wurde die Wirkung von Lipid-beladenen
Schaumzellen, die zahlreich in atherosklerotischen Läsionen vorhanden sind, auf die
Expression von CD90 auf EC analysiert.
162. Material und Methoden
2.1. Patientenkollektiv
In der Arbeit wurden Operationspräparate der ACI und von Arterien weiterer Lokalisationen
untersucht. Alle ACI Proben stammen von Patienten, die in der Klinik für Viszeral-,
Transplantations-, Thorax- und Gefäßchirurgie des Universitätsklinikums Leipzig
endarteriektomiert wurden. Der Eingriff wurde bei symptomatischer oder asymptomatischer
Stenose der ACI durchgeführt. Bei allen Patienten lag eine Einverständniserklärung zur
Verwendung der während der Operation entnommenen Gefäße für Forschungszwecke vor
(Ethikantrag „Nachweis von CD97 in Gefäßen“; Reg.-Nr. 148-2005, genehmigt durch die
Ethik-Kommission an der Medizinischen Fakultät der Universität Leipzig am 15.7.2005). Die
Präparate aus anderen Lokalisationen stammen von Transplantatspendern, von denen keine
weiteren Daten verfügbar waren. Die Zustimmung zur Gefäßentnahme bei diesen Spendern
wurde von der Deutschen Stiftung für Organtransplantation in Abstimmung mit der
Deutschen Gesellschaft für Gewebetransplantation dokumentiert.
Im Rahmen der Studie wurden insgesamt 139 Gefäßpräparate zufällig ausgewählt, davon
waren 72 Präparate der ACI (ACI-Patientengruppe). Die übrigen 67 Präparate wurden aus
verschiedenen Lokalisationen entnommen (Tab. 4).
Arterie Anzahl
Arteria carotis communis 3
Truncus brachiocephalicus 1
Arteria subclavia 4
Arteria mesenterica superior 1
Arteria iliaca communis 1
Arteria iliaca interna 16
Arteria illiaca externa 4
Arteria femoralis communis 8
Arteria profunda femoris 12
Arteria femoralis superficialis 2
Arteria poplitea 15
Tabelle 4: Lokalisation der von Transplantatspendern entnommenen Arterien
2.1.1. Gewinnung und Aufarbeitung der Gefäße
17Die 72 Gefäßpräparate der ACI-Patientengruppe wurden mittels EEA oder TEA gewonnen.
Die EEA ermöglicht, dass die Plaquestruktur über die gesamte Ausdehnung erhalten bleibt.
Die Desobliterate wurden intraoperativ sofort in PBS (engl. phosphate buffered saline) gelegt
und innerhalb von einer Stunde in Tissue Tek® eingebettet, eingefroren und bis zur
Verwendung bei -80°C aufbewahrt. Die ACI-Patientengruppe wurde aufgrund der
Anamnese, der klinischen Untersuchung und ggf. des Befundes aus der kraniellen
Computertomographie in asymptomatische oder symptomatische Patienten eingeteilt.
Ischämiebedingte zerebrale Symptome (Amaurosis fugax, transiente ischämische Attacke
oder vollendeter Schlaganfall) bedeuteten eine Zuordnung zur symptomatischen ACI-
Stenose. Zufällig, z.B. im Rahmen von Routineuntersuchungen bei bestehender pAVK
entdeckte Stenosen wurden als asymptomatisch gewertet.
Aus dem Transplantatspender wurden die Gefäße steril entnommen, in PBS gelegt und
innerhalb von 24 Stunden in Tissue Tek® eingebettet, eingefroren und bis zur Verwendung
bei –80°C aufbewahrt.
2.1.2. Herstellung von Gefrierschnitten
Für die Herstellung von Gefrierschnitten wurden die Gefäße im Kryostaten bei –20°C mit
dem Einbettmaterial Tissue Tek® bedeckt. Es wurden 6 m dicke Schnitte hergestellt, auf
Superfrost® Objektträger aufgezogen und bei Raumtemperatur getrocknet. Die Schnitte
wurden bis zur Verwendung bei –80°C aufbewahrt.
2.1.3. Histomorphologische Beurteilung
Ein Schnitt des Gefäßpräparates wurde mit Hämatoxylin-Eosin gefärbt. In einem weiteren
Schnitt wurde die Elastika mittels van-Gieson-Färbung dargestellt. Durch die Beurteilung
beider Färbungen können die Anteile des Plaques im Gefäß zu lokalisiert und bestimmt
werden [52]. Jedes Gefäßpräparat wurde bezüglich Lipidkerngröße, Plaquehemorrhagie,
Verkalkung, Plaqueruptur und Thrombus auf der Oberfläche beurteilt.
Als ein Lipidkern wurde amorphes Material, das Cholesterinkristalle enthält, definiert. Er
wurde als „groß“ gewertet, wenn er mehr als 50% der Dicke des Plaques einnahm.
Plaquehämorrhagie lag vor, wenn es innerhalb des Plaques eine Zone mit geronnenem Blut
gab, die die Plaque-Architektur zerstörte. Die Einblutungen durften nicht mit frischen
Gewebezerreißungen vergesellschaftet sein, um einen Artefakt durch intraoperativ
verursachte Einblutungen auszuschließen. Eine Plaqueruptur lag vor, wenn es eine klare
Verbindung zwischen den Lipidkern und dem Lumen gab, die mit einem Riss in der fibrösen
18Kappe vergesellschaftet war und die offenbar nicht während der Operation entstanden ist.
Als Thrombus wurde eine organisierte Ansammlung von Fibrin und Erythrozyten an der
Endotheloberfläche des Gefäßes definiert [10]. Als Verkalkung wurde eine entweder
feingranulär-dissiminierte oder grobschollig-fokale Kalkablagerung gewertet [10].
Um die unterschiedlichen Plaquemorphologien zu klassifizieren, wurden die AHA-Stadien
verwendet [7, 8]. Die Plaques der ACI-Patientengruppe wurden zusätzlich in komplizierte
und nicht-komplizierte Plaques (engl. complicated and noncomplicated plaques) eingeteilt.
Diese Klassifikation erfolgte in Anlehnung an die Arbeit von Milei et al (2003) [53]. Als
kompliziert wurden Plaques definiert, die entweder einen Riss, einen Oberflächenthrombus
und/oder eine Einblutung zeigten oder die verkalkt und gleichzeitig ulzeriert waren. Als nicht-
kompliziert wurden die Plaques gewertet, die entweder verkalkt waren und viel fibrotisches
Gewebe und kleine Lipidansammlungen aufwiesen oder die einen großen Lipidkern, aber
keinen Riss, keine Einblutung oder keinen Oberflächenthrombus aufwiesen [53].
2.2. Immunohistochemie
Um verschiedene Zelltypen in den Gefäßen zu differenzieren, wurden die Untersuchungen
durch imunohistochemische Färbungen am Kryostatschnitt ergänzt. Es wurden Färbungen
für CD3, CD68, CD31, von Willebrand Faktor (vWF) und CD90 durchgeführt.
Die Fixierung der Schnitte für die CD3, CD31 und CD90 Färbung erfolgte mit eiskaltem
Aceton, für die CD68 und vWF Färbung mit Methanol bei Raumtemperatur jeweils für 10
Minuten. Anschließend wurden die Schnitte 3 x 5 Minuten mit PBS gespült, die Aktivität der
endogenen Peroxidase 30 Minuten mit 0,3% H2O2 geblockt und die Schnitte für 3 x 5
Minuten mit PBS gespült. Unspezifische Bindungen wurden für 30 Minuten mit Pferdeserum
abgesättigt. Nach Spülen für 3 x 5 Minuten mit PBS wurde der Primärantikörper (Tab. 5)
aufgetragen und über Nacht bei 4°C inkubiert.
Antigen Antikörper Firma Clone Verdünnung Fixierung Identifizierung
CD68 Kaninchen- Dako KP1 1:2000 Methanol Makrophagen
anti-human Cytomation,
Hamburg
CD3 Maus-anti- Dako F7.2.38 1:100 Methanol T-
human Cytomation, Lymphozyten
Hamburg
CD31 Maus-anti- ImmunoTools, MEM- 1:100 Aceton EC
human Friesoythe 05
CD90 Maus-anti- Geschenk von AS02 1:1500 Aceton EC
human A. Saalbach,
Universität
Leipzig
19vWF Kaninchen- Dako - 1:2000 Methanol EC
anti-human Cytomation,
Hamburg
Tabelle 5: In den histologischen Untersuchungen verwendete primäre Antikörper.
Nach der Inkubation mit dem primären Antikörper erfolgte eine dreimalige Spülung der
Schnitte mit PBS. Zur Detektion der des gebundenen Antikörpers wurde der ImmPRESS™
Kit (Vector Laboratories ImmPRESS, Burlingame, CA) genutzt. Der Sekundärantikörper ist
direkt mit Peroxidase gekoppelt, wodurch nur sehr wenig Hintergrundfärbung entsteht. Die
Schnitte wurden mit einem Spezies-spezifischen sekundären ImmPRESS™ Antikörper für
30 Minuten inkubiert, danach für 3 x 5 Minuten mit PBS gespült, mit dem 3,3'-
Diaminobenzidin-Gemisch (DAB, SIGMAFAST™ 3,3‘-Diaminobenzidin Tabletts, Sigma-
Aldrich Chemie GmbH, Steinheim) überschichtet und unter Sichtkontrolle entwickelt. Nach
Spülung mit PBS wurde die Gegenfärbung mit Mayer´s Haemalaun durchgeführt.
Anschließend erfolgten für 5 Minuten die Bläuung der Schnitte unter fließendem
Leitungswasser und schließlich die Eindeckung.
2.3. Immunofluoreszenz
Um zu bestätigen, dass CD90 auf EC exprimiert wird, wurden Doppelimmunofluoreszenz
Färbungen für den vWF und CD90 durchgeführt. Die Schnitte wurden in 4%
Paraformaldehyd (PFA) für 10 Minuten fixiert und anschließend für 3 x 5 min in PBS gespült.
Endogene Autofluoreszenz wurde mit 0,1% Na-Borohydrid in PBS gequencht. Die Schnitte
wurden für 3 x 5 min in PBS gespült. Unspezifische Bindungen wurden mit 5%
Ziegennormalserum für eine Stunde bei Raumtemperatur geblockt. Der erste primäre-
Antikörper (Kaninchen-anti-human vWF; Dakocytomation, Hamburg) wurde mit 5%
Ziegennormalserum verdünnt (Verdünnung 1:2000) und über Nacht bei 4°C auf den
Schnitten inkubiert. Die Schnitte wurden für 3 x 5 min in PBS gespült und mit einem
sekundären Ziege-anti-Kaninchen Alexa-Fluor 546 (Alexa Fluor® 546 F(ab')2 fragment of
goat anti-rabbit IgG (H+L), Katalognummer A-11071, Invitrogen, Karlsruhe) in der
Verdünnung 1:100 für eine Stunde bei Raumtemperatur inkubiert. Nach dreimaliger Spülung
mit PBS wurden die Schnitte mit 5 % Huhn-Normalserum für eine Stunde bei
Raumtemperatur inkubiert. Anschließend wurden die Schnitte mit dem zweiten primären
Antikörper (Maus-anti-human CD90-Antikörper; clone AS02, Geschenk A. Saalbach, Leipzig)
in der Verdünnung 1:400 für 5 Stunden bei Raumtemperatur inkubiert. Die Schnitte wurden 3
x 5 min in PBS gespült und mit dem sekundären Antikörper Huhn-anti-Maus Alexa-Fluor 488
(Alexa Fluor® 488 chicken anti-mouse IgG (H+L), Katalognummer A-21200, Invitrogen,
20Karlsruhe) für eine Stunde bei Raumtemperatur inkubiert. Nach dreimaliger Spülung mit PBS
wurden die Präparate mit 4´,6-Diamidino-2-Phenylindol (DAPI) gefärbt und eingedeckt.
2.4. Zellkultur
2.4.1. Präparation der HUVEC
Die Nabelschnüre wurden gekühlt in PBS gelagert und innerhalb von 48 Stunden EC
präpariert. Noch vorhandenes Blut in der Umbilikalvene wurde durch Spülen mit sterilem
PBS entfernt. Die Venen wurden zum Ablösen der Endothelzellen von der Gefäßwand mit
Collagenase I (100 U/ml, Katalognummer 17018-029, Invitrogen, Karlsruhe) 20 min bei 37°C
intraluminal inkubiert. Die abgelösten EC wurden durch Spülung mit 10 ml PBS in ein
Zentrifugenröhrchen überführt und zentrifugiert (10 min, 284 g). Das Pellet wurde in 6 ml
Endothelzellmedium (ECBM, engl. Endothelial Cell Basal Medium, Katalognummer C-22210,
PromoCell mit SupplementPack, Katalognummer C-39210, PromoCell, Heidelberg)
resuspendiert, auf eine mit 0,2% Gelatine beschichtete Petrischale verteilt und bei 37°C
inkubiert. Die angewachsenen HUVEC wurden täglich kontrolliert. Sie zeigten eine typische
Pflastersteinmorphologie. Bei Erreichen einer Subkonfluenz von ca. 80-90%, in der Regel
nach 3 Tagen, wurden die Zellen gesplittet. Dazu wurde das Medium entfernt und die Zellen
zweimal mit 5 ml PBS gespült, um Zelldebris und nichtadhärente Zellen aus der Zellkultur
vollständig zu entfernen. Im Anschluss wurden die Zellen bei 37°C mit 1 ml einer 0.05 U/ml
Trypsinlösung inkubiert und regelmäßig unter dem Mikroskop kontrolliert, bis sich alle Zellen
vom Boden des Kulturgefäßes gelöst hatten. Die Trypsinreaktion wurde durch Zugabe von
PBS im Überschuss gestoppt und die Zellsuspension bei 242 g, 5 min zentrifugiert. Das
Pellet wurde in 5 ml Endothelzellmedium resuspendiert und in einem neuen, mit Gelatine
beschichteten Zellkulturgefäß ausgesät. Bei frisch angelegten Kulturen wurde der erste
Mediumwechsel ein oder zwei Tage nach der Präparation durchgeführt, bei älteren Kulturen
aller zwei bis drei Tage.
2.4.2. Humane EC aus der Koronararterie
Für die Experimente wurden tiefgefrorene humane EC aus der Koronararterie (HCAEC, engl.
Human Coronary Artery Endothelial Cells; Katalognummer C-12221, PromoCell, Heidelberg)
verwendet. Das Kryoröhrchen wurde in einem Wasserbad (37° C) so lange erwärmt, bis der
Inhalt aufgetaut war. Die resuspendierten Zellen wurden in ein Kulturgefäß mit
vorgewärmtem Medium übergetragen und in einem Inkubator (37°C, 5% CO2) kultiviert. Das
21Medium wurde nach 16 bis 24 Stunden ersetzt. Beim Erreichen einer 70 bis 90% Konfluenz
wurden die Zellen geteilt. Zum Ablösen der Zellen wurde der DetachKit (Katalognummer
C41200/C-41210/C-41220, PromoCell, Heidelberg) entsprechend den Herstellervorschriften
verwendet.
2.4.3. Stimulation der EC
Die Reinheit der Endothelzellkultur wurde mittels Durchflusszytometrie durch Nachweis von
CD31 bestimmt. HUVEC wurden der in der ersten bis dritten Passage verwendet, HCAEC in
der vierten bis fünften Passage. Die Zellen wurden in einer 6-well-Platte mit einer Dichte von
3x105 Zellen/ml Medium ausgesät. Bei einer Subkonfluenz von 80-90% wurden die Zellen
stimuliert. Dazu wurde das Medium entfernt und die Zellen wurden mit PBS gespült. Die EC
wurden mit Zytokinen oder anderen Stimulantien inkubiert, von denen bekannt ist, dass sie
eine wichtige Rolle in der Pathogenese der Atherosklerose spielen (Tab. 6).
Substanz Firma Katalognummer Eingesetzte
(Literatur) Konzentration
IL-1 [54, 55] ImmunoTools, Friesoythe 11349012 20pg/ml
IL-6 [56, 57] ImmunoTools, Friesoythe 11340060 50ng/ml
TNF- [56, 57] ImmunoTools, Friesoythe 11343013 50ng/ml
INF- [55, 57] ImmunoTools, Friesoythe 11343534 50ng/ml
PDGF-BB [9] ImmunoTools, Friesoythe 11343672 50ng/ml
VEGF [58] ImmunoTools, Friesoythe 11343662 50ng/ml
LPS [59, 60] Sigma-Aldrich Chemie L6529 100ng/ml
GmbH, Steinheim
sCD40L [55, 61-63] ImmunoTools, Friesoythe 11343343 0,5 g/ml
CXCL7 [61, 64] ImmunoTools, Friesoythe 11343392 10ng/ml
CXCL12 [61, 63, 65] ImmunoTools, Friesoythe 11343362 50ng/ml
IL-8 [66, 67] ImmunoTools, Friesoythe 11340080 50ng/ml
Histamin [54] Sigma-Aldrich Chemie 53300 100 M
GmbH, Steinheim
Adiponektin [24] Geschenk, Institut für 200 ng/ml
Biochemie, Universität
Leipzig
Leptin [68] Geschenk, Institut für 100 ng/ml
Biochemie, Universität
22Leipzig
D-(+)-Glukose Sigma-Aldrich Chemie G7021 5,5, 10,5, 15,5,
[55, 69, 70] GmbH, Steinheim 20,5 mmol/l
Tabelle 6: In den Experimenten eingesetzte Substanzen, die eine Rolle in der Entstehung
der Atherosklerose spielen.
Die Stimulantien wurden in Endothelzellmedium gelöst. Als Positivkontrolle diente PMA
(Katalognummer P 8139, Sigma-Aldrich Chemie GmbH, Steinheim) in einer Konzentration
von 10 ng/ml. EC mit ECBM und ohne zusätzlichen Stimulantien wurden als Negativkontrolle
mitgeführt. Die Inkubationszeit betrug je nach Versuch 48 bis 96 Stunden.
2.4.3.1. Kultur der monozytären Zelllinie THP-1
Neben Zytokinen und anderen chemischen Substanzen, die zur Entwicklung der
Atherosklerose beitragen, wurde untersucht, ob die Expression von CD90 durch
Schaumzellen (engl. foam cells), die sich in den atherosklerotischen Plaques befinden,
beeinflusst wird.
Schaumzellen wurden durch Behandlung der monozytären THP-1 Zellen (engl. human acute
monocytic leukemia cell line) mit oxidiertem LDL (ox-LDL) gewonnen. Das Kryoröhrchen mit
den THP-1 Zellen (Lot No. 07B012, Sigma-Aldrich, Steinheim) wurde in einem Wasserbad
(37° C) erwärmt und die resuspendierten Zellen in vorgewärmten RPMI 1640 (engl. Roswell
Park Memorial Institute medium, Katalognr.: R8758, Sigma-Aldrich, Steinheim) mit 10% FCS
(engl. fetal calf serum, PromoCell, Katalognr.: C-37355, Heidelberg) in einer Konzentration
von 1×105/ml in die Kulturgefäße ausgesät. Die Zellen wurden nach zwei Tagen geteilt.
Für den Versuch wurden die THP-1 Zellen in einer Konzentration von 1×10 6/ml mit ECBM in
einer 6-well Platte ausgesät. Nach 24 Stunden wurden die Zellen mit 10ng/ml PMA für 24
Stunden stimuliert. Parallel erfolgte die Herstellung des ox-LDL. Zunächst wurde eine 40µM
Cu2SO4-Lösung (Katalognr.: C1297, Sigma-Aldrich, Steinheim) hergestellt. 10µl dieser
Lösung wurde mit 50µg LDL (Katalognr.: 437644, Calbiochem, Darmstadt) für 24 h bei 37°C
inkubiert.
Nach 24 Stunden Stimulation mit PMA wurden die THP-1 Zellen bei 242 g, 20°C, für 5 min
zentrifugiert und anschließend mit 50 µg oxLDL/ml ECBM für 48 Stunden inkubiert, um die
Umwandlung der THP-1 zu Schaumzellen zu induzieren. Als Negativkontrolle diente
Medium, dem nur 20 µl 1 mM Cu2SO4-Lösung/ml ECBM hinzugefügt wurde. Anschließend
wurden die Stimulationsversuche der HUVEC mit den ox-LDL behandelten THP-1 Zellen
sowie mit deren Überständen durchgeführt.
232.4.4. Durchflußzytometrie
Für die Analyse der HUVEC und HCAEC in der Durchflusszytometrie wurden die Zellen
zweimal in PBS gewaschen und bei 242 g für 5 Minuten zentrifugiert. 2 x 105 pelletierte
Zellen wurden mit 30 L des verdünnten CD90, ICAM-1 sowie CD31 Antikörpers
resuspendiert und bei 4°C für 30 min auf Eis inkubiert. Anschließend wurden die Zellen
dreimal mit 100 L PBS gewaschen. Danach wurden die Zellen mit Phycoerythrin-
konjugiertem Ziege-anti-Maus Antikörper (Katalog-Nr.: R0480, Dako, Hamburg), 1:100
verdünnt, für 30 min bei 4°C inkubiert. Anschließend wurden die Zellen dreimal in PBS
gewaschen und mit 100 L 1% Paraformaldehyd (PFA)/PBS fixiert. Im Durchflusszytometer
(FACSCalibur flow cytometer, BD, Heidelberg) wurden jeweils 10000 Zellen gemessen und
mit dem Programm Cell Quest Pro (BD CellQuest Pro™ Software) ausgewertet.
2.4.5. Statistische Auswertung und Abbildungen
Von den histologischen Färbungen wurde je eine repräsentative Abbildung ausgewählt. Die
Daten aus Zellkulturexperimenten sind als Mittelwerte +/- Standardfehler von mindestens
drei unabhängigen Experimenten dargestellt. Die statistische Auswertung wurde mittels
Mann-Whitney-U-Test, Fischer-Test oder Chi-Quadrat-Test (WinSTAT, Version 2012.1, R.
Fitch Software, Bad Krozingen) durchgeführt.
243. Ergebnisse
3.1. Histomorphologische Charakterisierung der Gefäßpräparate
Die histomorphologische Charakterisierung aller Gefäßpräparate erfolgte durch
Begutachtung der mit Hämatoxylin-Eosin bzw. Elastica-van Gieson gefärbten Schnitte. In die
weitere histologische Auswertung wurden nur die Gefäßpräparate einbezogen, in denen die
Endothelzellen vorhanden waren. Das war in 117 von insgesamt 139 Präparaten der Fall.
Weiterhin wurden die Gefäßpräparate nach der AHA-Klassifikation [8] sechs
Atherosklerosestadien zugeordnet (Abb. 3). Am häufigsten gehörten die Gefäße den
Atherosklerosestadien III (n=26), V (n=36) und VI (n=23) an.
Abbildung 3: Alle Gefäßpräparate wurden nach AHA-Klassifikation in sechs
Arteriosklerosestadien zugeordnet.
Die Plaques der ACI-Patientengruppe wurden nach folgenden Kriterien beurteilt: Lipidgehalt
der Verschlusskerne, Blutung im Plaque, sowie Plaqueruptur und Thrombusformation.
Anschließend wurden die verschiedenen Plaquetypen der ACI-Patienten nach
histomorphologischen Kriterien entsprechend Milei et al. [53] in zwei Gruppen unterteilt:
komplizierte Plaques und nicht-komplizierte Plaques (engl. complicated und non-
complicated) (Abb. 4).
25Abbildung 4: Prozentuale Verteilung der komplizierten und nicht-komplizierten Plaques in der
ACI-Patientengruppe pro Arteriosklerosetadium.
Um die Ausprägung der entzündlichen Reaktion in Plaques in der ACI-Patientengruppe
genauer beurteilen zu können, wurden zusätzlich Makrophagen und Lymphozyten mittels
Immunohistologie identifiziert. Alle Gefäßpräparate in der ACI-Patienten Gruppe wurden mit
anti-CD68 und anti-CD3 Antikörpern gefärbt. Anhand der Ausprägung der CD68 und CD3
Expression in den Präparaten wurde ein Entzündungswert (engl. Inflammation Score, IS) in
Anlehnung an die Arbeit von Redgrave et al. 2006 gebildet. Ein Entzündungswert kleiner
oder gleich zwei wurde als nicht relevante entzündliche Infiltration gewertet [10]. Die
Zunahme der Entzündungszellen in der Plaque war im Stadium V vs. Stadium VI statistisch
signifikant (p=0,04) (Abb. 5).
Abbildung 5: Die Plaques der ACI-Patientengruppe wurden in zwei Kategorien unterteilt: mit
IS (engl. Inflammation Score) 1-2 und IS 3-6.
26Anhand der klinischen Angaben zu den Patienten in der ACI-Patientengruppe (Amaurosis
fugax, TIA, oder Schlaganfall bzw. keine Symptome) wurden die Plaques entsprechend der
Publikation von Redgrave et al. [10] in symptomatisch und asymptomatisch klassifiziert.
Insgesamt wurden 31 Plaques als symptomatisch und 22 Plaques als asymptomatisch
gewertet (Abb. 6).
Abbildung 6: Verteilung der symptomatischen und asymptomatischen Plaques pro
Arteriosklerosestadium in der ACI-Patientengruppe.
Die morphologischen Kriterien wurden in Abhängigkeit von klinischen Daten bezüglich der
Symptomatik der ACI-Patienten ausgewertet. Eine Korrelation zwischen der
symptomatischen ACI-Stenose und dem Vorhandensein eines Oberflächenthrombus konnte
nachgewiesen werden (p=0,05). Die symptomatische ACI-Stenose korrelierte mit weiteren
untersuchten histologischen Merkmalen nicht (Tab. 7). Der IS war in symptomatischen
Plaques ebenfalls nicht signifikant erhöht. Die Ergebnisse der histologischen Beurteilung der
Gefäße waren mit Daten in der Literatur vergleichbar [10, 53, 71].
Histologische Ereignisse in Asymptomatisch Symptomatisch p
der ACI Patientengruppe, n n (%) n (%)
Verkalkung, 34 13 (59,1) 21 (67,7) NS
Großer Lipidkern, 3 8 (36,4) 23 (74,2) NS
Plaqueblutung, 19 7 (31,8) 12 (38,7) NS
Plaqueriss, 17 3 (13,6) 14 (45,2) NS
Oberflächenthrombus, 9 1 (4,6) 8 (25,8) 0,05
Komplizierte Plaque, 33 9 (40,9) 24 (77,4) NS
Tabelle 7: Histomorphologische Kriterien und Symptomatik der ACI-Stenose. Anzahl (n),
nicht signifikant (NS).
273.2. Expression von CD90 auf makrovaskulären EC der atherosklerotischen Gefäße
Alle Gefäßpräparate wurden mit einem CD31 Antikörper immunhistologisch gefärbt. CD31
wird in den untersuchten Gefäßen vor allem stark von EC exprimiert. Da CD31, ein
Bestandteil der endothelialen Interzellularverbindungen, auch von Leukozyten exprimiert
werden kann [72, 73], musste sichergestellt werden, dass es sich bei der CD31-positiven
Zellschicht in den Gefäßen tatsächlich um EC handelt. In Serienschnitten wurden
Kontrollfärbungen mit einem vWF Antikörper durchgeführt. Der vWF wird ebenfalls auf EC
exprimiert und ist neben CD31 einer der am häufigsten verwendeten immunhistochemischen
Marker für EC [72, 74, 75]. 117 von 139 Präparaten enthielten eindeutig eine EC-Schicht.
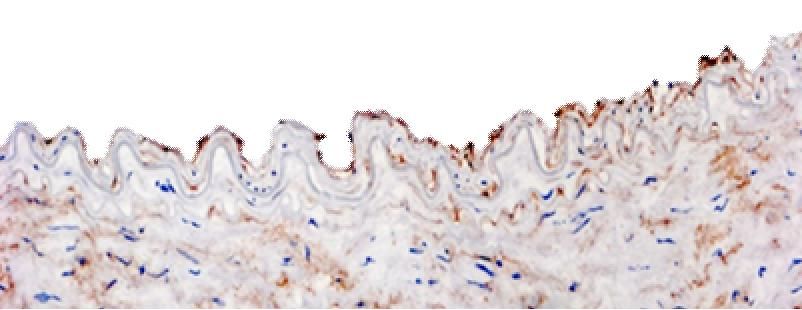
Diese wurden anschließend mit dem anti-CD90 (anti-Thy-1) Antikörper gefärbt.
Es ist bekannt, dass CD90 nicht nur von aktivierten EC, sondern u. a. auch von Fibroblasten
exprimiert wird [30]. Bei der lichtmikroskopischen Beurteilung der CD90-gefärbten
atherosklerotischen Gefäßpräparate konnte man erkennen, dass sich häufig unmittelbar
unter der obersten Schicht, die den EC entspricht, Bindegewebe findet. Hier sind u.a.
Fibroblasten lokalisiert. Sie sind in der histologischen Färbung CD90 positiv.
Die Unterscheidung von CD90-positiven Fibroblasten, die direkt unter den EC liegen, und
tatsächlich CD90-positiven EC war deshalb oft herausfordernd, insbesondere dann, wenn es
sich um fortgeschrittene Atherosklerosestadien handelte. Die Spezifität der
immunhistologischen Darstellung von CD90 in der inneren luminalen Zellschicht
atherosklerotischer Gefäße wurde letztlich mittels Doppelmarkierung von CD90 und vWF,
dem EC Marker [72] in der Immunfluoreszenz geklärt. Dabei konnte man erkennen, dass auf
der obersten Zellschicht die identischen Zellen das CD90 und vWF exprimierten (Abb 7).
A B C
Abbildung 7: Doppelimmunfluoreszenzfärbung mit CD90 (grün) und vWF (rot) einer
atherosklerotischen Arterie. A. CD90 ist auf EC (oberste Zellschicht) und auf Fibroblasten
(tieferliegende Zellschichten) lokalisiert. B. Der vWF ist nur auf EC vorhanden. C. Die
Überlappung der Färbungen (gelb) zeigt, dass vWF und CD90 von den gleichen Zellen auf
der obersten luminal-gerichteten Zellschicht exprimiert wird, die den EC entspricht.
28Sie können auch lesen

















































