Expression von Tumorstammzellmarkern in Vorläuferläsionen des murinen Pankreaskarzinommodells
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Universitätsklinikum Ulm
Zentrum für Innere Medizin
Klinik für Innere Medizin Ι
Ärztlicher Direktor:
Prof. Dr. Thomas Seufferlein
Expression von Tumorstammzellmarkern in
Vorläuferläsionen des murinen
Pankreaskarzinommodells
Dissertation zur Erlangung des Doktorgrades der Medizin der
Medizinischen Fakultät der Universität Ulm
Eingereicht von
Susanne Bayer
geboren in Regensburg
2020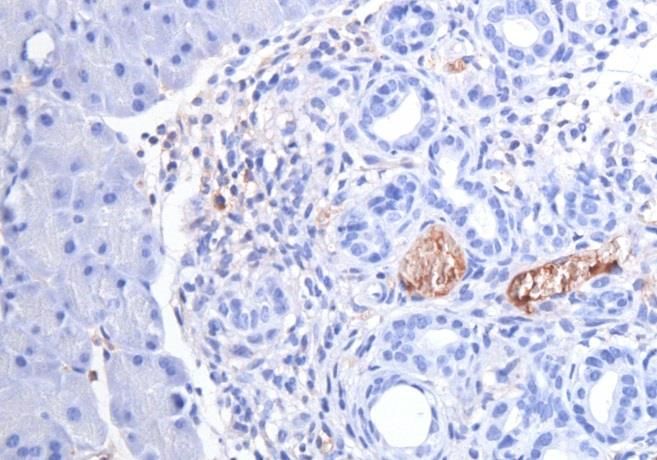
Amtierender Dekan: Prof. Dr. T. Seufferlein 1. Berichterstatter: Prof. Dr. M. Wagner 2. Berichterstatter: PD Dr. B. Baumann Tag der Promotion: 16.07.2021
INHALTSVERZEICHNIS
Abkürzungsverzeichnis ..................................................................................................................................... III
1 Einleitung .......................................................................................................................................................... 1
1.1 Das Pankreaskarzinom...................................................................................................................... 1
1.1.1 Epidemiologie ...................................................................................................1
1.1.2 Risikofaktoren ...................................................................................................1
1.1.3 Pathologie .........................................................................................................3
1.1.4 Vorläuferläsionen ..............................................................................................3
1.1.5 Molekulargenetik ..............................................................................................5
1.2 Die Tumorstammzellhypothese .................................................................................................... 6
1.2.1 Modelle der Tumorentstehung ..........................................................................6
1.2.2 Eigenschaften der Tumorstammzellen...............................................................7
1.2.3 Ursprung der Tumorstammzellen ......................................................................7
1.2.4 Identifikation von Tumorstammzellen ...............................................................8
1.2.5 Bedeutung für die Prognose und Therapie ......................................................12
1.3 Pankreaskarzinom-Modelle ......................................................................................................... 13
1.3.1 Zusätzlicher Verlust von ATM beim KC-Mausmodell ........................................15
1.4 Zielsetzung ........................................................................................................................................... 16
2 Material und Methoden ........................................................................................................................... 17
2.1 Material .................................................................................................................................................. 17
2.1.1 Laborgeräte .....................................................................................................17
2.1.2 Chemikalien ....................................................................................................18
2.1.2 Puffer ..............................................................................................................18
2.1.3 Verbrauchsmaterial .........................................................................................19
2.1.4 Primer .............................................................................................................19
2.1.5 Primärantikörper .............................................................................................19
2.1.6 Sekundärantikörper .........................................................................................19
2.2 Methoden .............................................................................................................................................. 20
2.2.1 Mauslinien ......................................................................................................20
2.2.2 Genotypisierung ..............................................................................................20
2.2.3 Immunhistochemische Färbung ......................................................................22
2.2.4 Auszählungsverfahren .....................................................................................24
I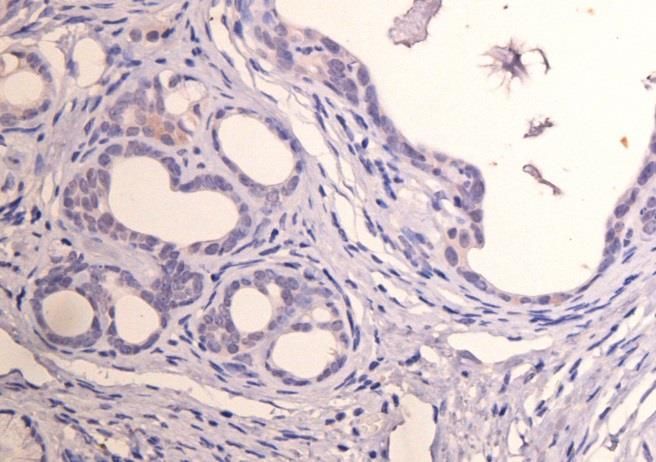
Inhaltsverzeichnis
2.2.5 Statistische Auswertung ..................................................................................24
3 Ergebnisse ..................................................................................................................................................... 25
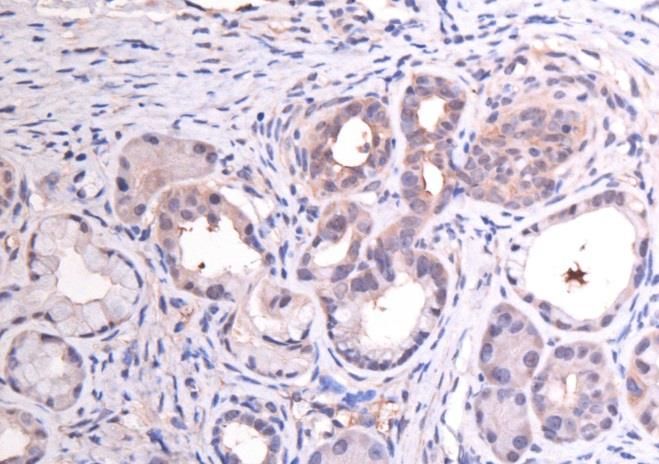
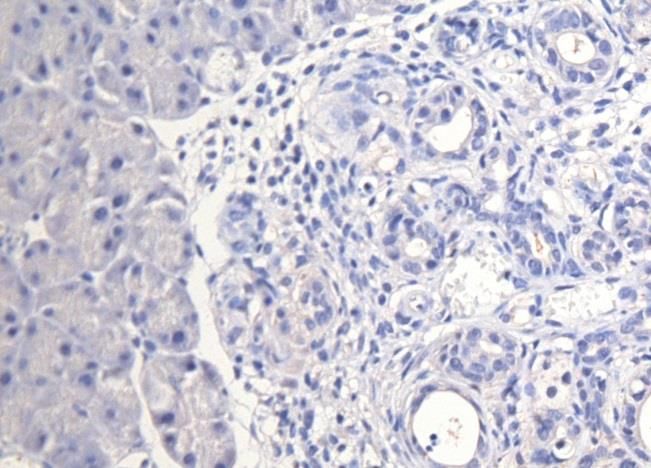
3.1 Etablierung der Färbeprotokolle............................................................................................... 25
3.2 CD24 ........................................................................................................................................................ 25
3.3 CD44 ........................................................................................................................................................ 29
3.4 CD133 ..................................................................................................................................................... 32
3.5 C-Met ....................................................................................................................................................... 35
3.6 EpCam ..................................................................................................................................................... 38
3.7 CXCR4 ..................................................................................................................................................... 41
3.8 Vergleich der Oberflächenmarker ............................................................................................ 44
3.8.1 Genotyp ATM +/+ ............................................................................................44
3.8.2 Genotyp ATM +/-.............................................................................................45
3.8.3 Genotyp ATM -/- .............................................................................................46
3.8.4 Gesamtüberblick .............................................................................................47
3.8.5 Vergleich der Effektstärken .............................................................................48
3.9 Abhängigkeit vom Alter ................................................................................................................. 49
4 Diskussion ..................................................................................................................................................... 51
4.1.1 CD24 ...............................................................................................................51
4.1.2 CD44 ...............................................................................................................52
4.1.3 CD133 .............................................................................................................52
4.1.4 C-mEt ..............................................................................................................53
4.1.5 EpCAM ............................................................................................................54
4.1.6 CXCR4..............................................................................................................55
4.1.7 Vergleich der Oberflächenmarker....................................................................55
4.2 Abhängigkeit vom Alter ................................................................................................................. 57
5 Zusammenfassung ..................................................................................................................................... 58
6 Literaturverzeichnis ................................................................................................................................. 59
7 Danksagung ................................................................................................................................................... 71
8 Lebenslauf...................................................................................................................................................... 72
II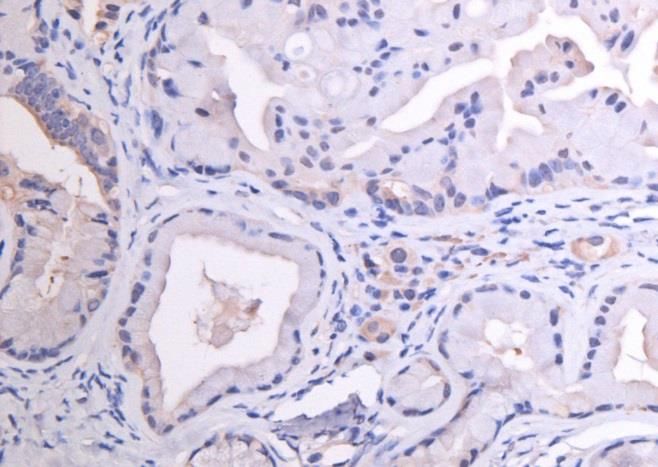
Abkürzungsverzeichnis
ABKÜRZUNGSVERZEICHNIS
ABC ATP binding cassette
ABC-Methode Avidin-Biotin-Komplex-Methode
ACVR1B Activin receptor type-1B
AIB1 Amplified-in-breast cancer 1 (= NCOA3)
AKC Genotyp p48Cre+//LSL-KrasG12D+//ATMfl/fl
ALDH Aldehyd-Dehydrogenase
AML Akute myeloische Leukämie
ANTXR Anthrax toxin receptor 1
APC Adenomatöse Polyposis coli
ATM Ataxia Teleangiectasia Mutated
Bcl-2 B-cell lymphoma 2
BRCA Breast Cancer Gene
CA Carbohydrate-Antigen
CAM Cell Adhesion Molecule
CD Cluster of differentiation
CDKN Cyclin-dependent kinase inhibitor
CIS Carcinoma in situ
c-Met Mesenchymal-epithelial transition factor
c-Myc Avian myelocytomatosis virus oncogene cellular homolog
CSC Cancer stem cell
CXCL12 CXC-Motiv-Chemokinligand 12
CXCR4 CXC-Motiv-Chemokinrezeptor 4
DNA Deoxyribonucleic acid
dNTP Desoxyribonukleosidtriphosphat
DPC4 Deleted in Pancreatic Cancer 4 (= SMAD4)
EGFR Epidermal Growth Factor Receptor
EMT Epitheliale-Mesenchymale-Transition
EpCAM Epithelial cell adhesion molecule
EZM Extrazelluläre Matrix
FAMMMPC- Syndrom familiäres atypisches multiples Muttermal- und Melanom-
Pankreaskarzinom-Syndrom
FDR first-degree relative
G1-S Gap1-Synthese
GAP GTPase-activating protein
GEF Guanine nucleotide exchange factors
GI-Trakt Gastrointestinal-Trakt
GDP Guanosindiphosphat
GSEA Gene set enrichment analysis
GTP Guanosintriphosphat
HBOC-Syndrom Hereditary Breast and Ovarian Cancer Syndrome
Her2 Human epidermal growth factor receptor 2
III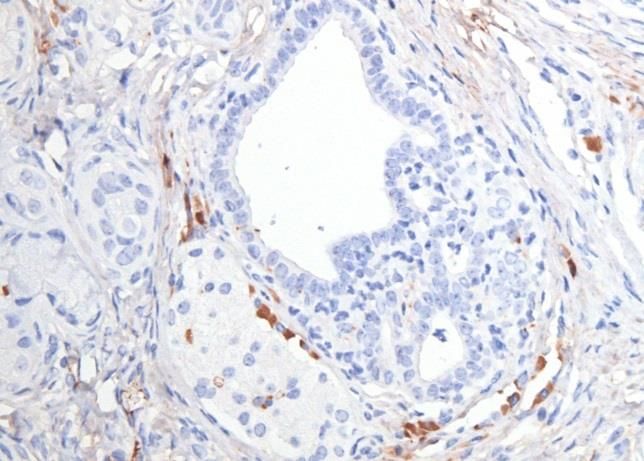
Abkürzungsverzeichnis
HES Hairy/Enhancer of Split
HEY Hairy/enhancer-of-split related with YRPW motif-like protein
HGF Hepatocyte growth factor
HIV Humanes Immundefizienz-Virus
IL Interleukin
INK4A Inhibitor of cyclin-dependent kinase type 4
IPMN Intraduktale papillär-muzinöse Neoplasie
KC Genotyp p48Cre+//LSL-KrasG12D+
KRAS Kirsten rat sarcoma viral oncogene homolog
LRP Low density lipoprotein (LDL) receptor-related protein
LKB1 Liver kinase B1 (= STK11)
MAPK Mitogen-activated protein
MCN Muzinös zystische Neoplasie
MDR Multi-drug resistance
Mg Magnesium
MKK4 Mitogen-Activated Protein Kinase Kinase 4
MMP Matrix-Metalloproteinase
MUC Mucin
MYB Myeloblastosis
NCOA3 Nuclear receptor coactivator 3 (= AIB1)
NOD Non-Obese Diabetic
PanIN Pankreatische Intraepitheliale Neoplasie
PDAC Pancreatic ductal adenocarcinoma
Pdx1 Pancreatic and duodenal homebox 1
PI3K Phosphoinositid-3-Kinase
PML Promyelozytenleukämie (AML M3)
Ptf1a Pancreas-specific transcription factor 1a
PCR Polymerase-Kettenreaktion
RAF Rapidly accelerated fibrosarcoma
RARα Retinoic acid receptor α
RNA Ribonucleic acid
Rpm Revolutions per minute
SCID Severe combined immunodeficiency
SDF-1 Stromal-derived-factor 1
SHH Sonic Hedgehog Pathway
SMAD4 Sma Mothers against decapentaplegic 4 (= DPC4)
STK11 Serin Threonin Kinase 11 (= LKB1)
TBST Tris-Buffered Saline Tween
TGFBR1/2 Transforming growth factor beta receptor 1/2
TP53 Tumor Protein p53
TPA Tetradecanoyl-phorbol-acetat1
VEGF Vascular endothelial growth factor
Wnt Wingless Int 1
IV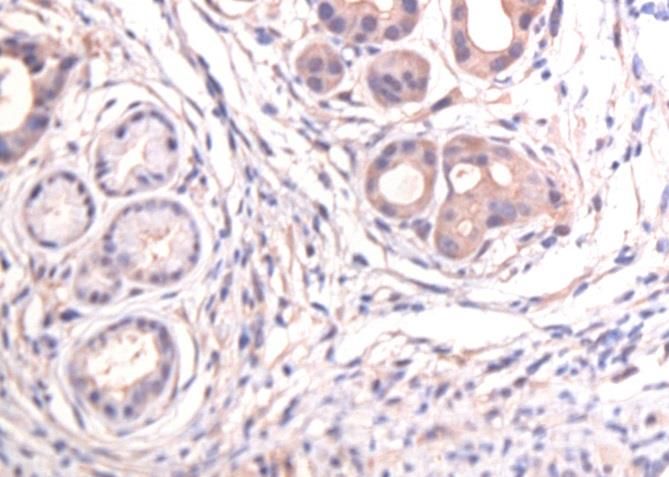
Einleitung
1 EINLEITUNG
1.1 DAS PANKREASKARZINOM
1.1.1 EPIDEMIOLOGIE
In Deutschland erkrankten im Jahr 2012 ca. 478.000 Menschen an Krebs. Hiervon
erhielten etwa 3,3% der betroffenen Männer bzw. 3,8% der Frauen die Diagnose
Bauchspeicheldrüsenkrebs. Das Pankreaskarzinom weist unter allen Krebsarten mit
einer 5-Jahresüberlebensrate von nur ca. 8% die niedrigste Überlebensrate auf und
steht an vierter Stelle der Krebstodesursachen. Aufgrund des demographischen Wandels
steigt die absolute Zahl der Neuerkrankungs- und Sterbefälle in den letzten Jahren
kontinuierlich an (Robert Koch-Institut (Hrsg.) und die Gesellschaft der
epidemiologischen Krebsregister in Deutschland e.V. (Hrsg.) 2015).
Tabelle 1: Übersicht über die wichtigsten epidemiologischen Maßzahlen des
Pankreaskarzinoms in Deutschland (modifiziert nach Robert Koch-Institut (Hrsg.) und die
Gesellschaft der epidemiologischen Krebsregister in Deutschland e.V. (Hrsg.) 2015)
2012
Männer Frauen
Neuerkrankungen 8250 8480
standardisierte Erkrankungsrate1,2 14 10,6
Sterbefälle 7936 8184
standardisierte Sterberate1,2 13,1 9,6
5-Jahres-Prävalenz 7800 8100
relative 5-Jahres-Überlebensrate (2011-2012) 6% 8%
1
je 100.000 Personen, 2altersstandardisiert nach Europastandard
1.1.2 RISIKOFAKTOREN
Das Risiko an einem Pankreaskarzinom zu erkranken steigt mit zunehmendem
Lebensalter. Nur ca. 10% der betroffenen Patienten sind jünger als 50 Jahre (Lowenfels
und Maisonneuve 2006). Einen wichtigen Risikofaktor stellt das Tabakrauchen dar,
welches für 20-25% aller Erkrankungen verantwortlich gemacht wird (Maisonneuve
und Lowenfels 2010). Hierbei verhält sich das Risiko direkt proportional zu Dauer und
Intensität des Rauchens (Silverman et al. 1994). Weiterhin sind Übergewicht oder
Adipositas im jungen Erwachsenenalter (Li et al. 2009) sowie ein hoher Alkoholkonsum
mit einem erhöhten Erkrankungsrisiko assoziiert (Lucenteforte et al. 2012). Zu den
Risikofaktoren zählt weiterhin der Diabetes mellitus, welcher das Risiko an einem
Pankreaskarzinom zu erkranken, um bis zu 80% erhöht (Huxley et al. 2005). Ein
bedeutender prädestinierender Faktor ist außerdem die chronische Pankreatitis, meist
verursacht durch erhöhten Alkoholkonsum, aber auch im Rahmen einer hereditären
Pankreatitis (Lowenfels und Maisonneuve 2006).
1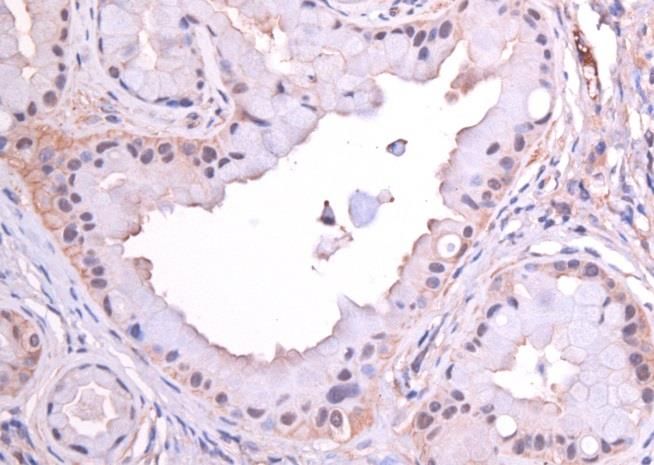
Einleitung
Insgesamt haben ca. 10% der Patienten eine positive Familienanamnese (Klein et al.
2014). Ist beispielsweise ein Verwandter 1. Grades vom Pankreaskarzinom betroffen, so
erhöht sich das individuelle Erkrankungsrisiko um das 2,3-fache (Amundadottir et al.
2004). Generell können hier drei verschiedene Formen unterschieden werden: erstens
Patienten mit einer hereditären Pankreatitis, zweitens eine familiäre Häufung des
Pankreaskarzinoms mit unbekanntem genetischen Hintergrund und drittens spezifische
bekannte Mutationen im Rahmen eines hereditären Tumorsyndroms (Riemann J. F. et al.
2007). Die meisten dieser für das Pankreaskarzinom bekannten Mutationen sind auch
mit anderen Krebsarten assoziiert. Oft gibt es für die diversen Mutationen ein
spezifisches histologisches Erscheinungsbild (Maitra und Hruban 2008). Die häufigsten
bekannten genetisch vererbbaren Mutationen sind in der folgenden Tabelle
zusammengefasst:
Tabelle 2: Übersicht über die mit dem Pankreaskarzinom (PDAC) assoziierten
Keimbahnmutationen (modifiziert nach Becker et al. 2014)
Risikofaktor Gen Erhöhung andere assoziierte Malignome
des PDAC-
Risikos
HBOC-Syndrom BRCA1, BRCA2, 2-3,5 Brust, Ovarien, Prostata
PALB2
Lynch-Syndrom MLH1, MSH2, 8,6 Kolon, Endometrium, Ovarien, Magen,
MSH6, PMS2, Dünndarm, Harnwege, Gehirn, Talgdrüsen
EPCAM
Familiäre APC 4,5-6 Kolon, Duodenum, Schilddrüse, Gehirn,
adenomatöse Papillenkarzinom, Hepatoblastom,
Polyposis Desmoid-Tumor
Peutz-Jeghers- STK11/LKB1 132 Ösophagus, Magen, Dünndarm, Kolon,
Syndrom Lunge, Brust, Uterus, Ovarien
FAMMMPC- P16INK4A/CDKN 47 Melanom
Syndrom 2A
Hereditäre PRSS1, SPINK1 69
Pankreatitis
Cystische CFTR 3,5
Fibrose
Ataxia ATM erhöht Leukämie, Lymphom
telangiectasia
Non-0 1,3
Blutgruppe
Familiäres unbekannt 9 (1 FDR), 32
Pankreas- (3 FDRs)
karzinom
2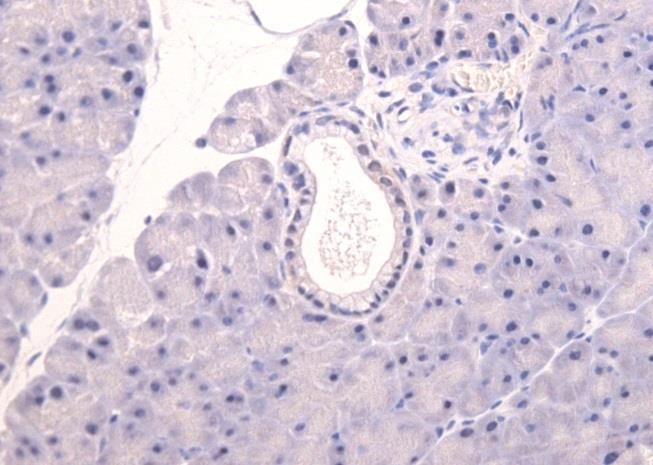
Einleitung
1.1.3 PATHOLOGIE
Maligne Neoplasien des Pankreas können aus dem endokrinen oder exokrinen Anteil
der Bauchspeicheldrüse hervorgehen. Das duktale Adenokarzinom (PDAC) ist mit über
90% die häufigste Krebserkrankung des Pankreas (Hackeng et al. 2016). Es tritt mit
einer Häufigkeit von ca. 60% im Bereich des Caput pancreatis auf, gefolgt vom Corpus
pancreatis mit 15% und der Cauda pancreatis mit 5%. Bei den verbleibenden 20%
handelt es sich um eine diffuse Infiltration der gesamten Bauchspeicheldrüse.
Makroskopisch imponiert das PDAC als schlecht abgrenzbarer, harter Tumor mit
sternförmigen Ausläufern. Mikroskopisch zeigt sich ein infiltrierendes, drüsenbildendes
neoplastisches Epithel mit ausgeprägter demoplastischer Reaktion, also Bildung von
kollagenreichem Bindegewebe (Kumar et al. 2013).
Abbildung 1: Infiltrierendes Pankreaskarzinom (PDAC) mit unregelmäßiger Drüsenstruktur
und desmoplastischem Stroma (Maitra und Hruban 2008)
1.1.4 VORLÄUFERLÄSIONEN
In den meisten Organen entwickeln sich solide Tumoren über eine Reihe von
prämalignen Vorstufen, welche im Laufe der Zeit sukzessiv typische genetische
Mutationen akkumulieren. Ein Karzinom entsteht erst dann, wenn durch die Aktivierung
von Onkogenen und Inaktivierung von Tumorsuppressorgenen die Wachstumskontrolle
einer Zelle auf mehreren Signalwegen beeinträchtigt wird und die Zelle unkontrolliert
proliferieren kann (Vogelstein B. und Kinzler K. W. 1993).
Auch für das Pankreaskarzinom kann ein solches Tumorprogressionsmodell mit
Einschränkungen dargestellt werden. Die Klassifikation der pankreatischen
intraepithelialen Neoplasien (PanIN) teilt Vorläuferläsionen nach morphologischen
Gesichtspunkten in drei Klassen ein (Hruban et al. 2001). PanINs bestehen aus Muzin-
produzierenden, zylindrisch bis kuboidalen Zellen mit variierenden Atypien und
betreffen in der Regel kleine Pankreasgänge mit einem Durchmesser von < 5mm
(Hruban et al. 2004). Dabei werden flache PanIN-1A-Läsionen, papilläre PanIN-1B-
Läsionen, PanIN-2-Läsionen mit mittelgradiger Dysplasie und PanIN-3-Läsionen als
Carcinoma in situ (CIS) mit schwergradiger Dysplasie voneinander unterschieden
(Hruban et al. 2001).
Zu den in frühen Stadien auftretenden genetischen Veränderungen zählen die
Überexpression von Her2-neu (Day et al. 1996) und die aktivierenden Mutationen im
KRAS-Onkogen (Moskaluk et al. 1997) (siehe Abb. 2). Weiterhin zeigt sich bereits
3
Einleitung
frühzeitig eine signifikante Verkürzung der Telomere, welche im Verlauf der
Karzinogenese immer weiter voranschreitet (Matsuda et al. 2015). Inaktivierende
Mutationen des Tumorsuppressorgens p16INK4a/CDKN2A findet man häufig in den
höhergradigen PanIN-2 Läsionen (Moskaluk et al. 1997; Yamamoto et al. 2000). Relativ
spät in der Karzinogenese treten Mutationen der Tumorsuppressorgene p53
(DiGiuseppe et al. 1994), BRCA2 (Goggins et al. 2000) und DPC4/SMAD4 (Wilentz et al.
2000) auf.
Abbildung 2: Tumorprogressionsmodell des Pankreaskarzinoms (Hruban et al. 2000):
Übergang des histologisch unauffälligen Pankreasgangs in eine flache (PanIN-1 A), papilläre
(PanIN-1 B), mittelgradig dysplastische (PanIN-2) und hochgradig dysplastische (PanIN-3)
Gangläsion mit den im entsprechenden Stadium typischerweise neu auftretenden genetischen
Alterationen
Neben den PanINs existieren auch zystische Vorläuferläsionen aus denen ein invasives
Pankreaskarzinom hervorgehen kann (Zamboni et al. 2013). Hierbei werden zwei
verschiedene Ausprägungen voneinander unterschieden:
1.) Die Intraduktale papillär-muzinöse Neoplasie (IPMN) ist eine
muzinproduzierende Neoplasie des Pankreas, die im Pankreashauptgang oder
dessen Seitenästen auftritt. Sie hat meist eine papilläre Architektur (Hruban et al.
2004).
2.) Die Muzinös zystische Neoplasie (MCN) tritt meist bei perimenopausalen
Frauen auf und ist im Gegensatz zur IPMN nicht mit dem Hauptgang assoziiert
(Adsay 2008).
Die klinische Relevanz der genannten Vorläuferläsionen liegt darin, dass diese im
Schnitt 3-5 Jahre früher als das Karzinom auftreten und es somit ein potenzielles
zeitliches Fenster zur Therapie gibt, bevor sich ein Karzinom entwickelt (Maitra und
Hruban 2008).
4Einleitung
1.1.5 MOLEKULARGENETIK
KRAS und andere Onkogene
Die häufigste Mutation des Pankreaskarzinoms ist mit einer Inzidenz von ca. 90% die
aktivierende Mutation des KRAS-Proto-Onkogens (Hruban et al. 1993). Betroffen ist fast
immer das Codon 12 (Nagata et al. 1990). KRAS kodiert für ein kleines G-Protein, das
zwischen zwei Zuständen wechseln kann: aktiv im GTP-gebundenen Zustand und inaktiv
im GDP-gebundenen Zustand. Der Übergang zur aktiven Form wird über GEFs
vermittelt, welche den Austausch von GDP mit GTP fördern. Der Wechsel in die inaktive
Form wird hingegen über GAPs eingeleitet. Diese beschleunigen die Hydrolyse von GTP
zu GDP durch die intrinsische GTPase. Durch aktivierende Mutationen kann die
Funktion der intrinsischen GTPase beeinträchtigt und die Interaktion zwischen KRAS
und den GAPs gestört werden. Daraus resultiert eine permanente Aktivierung von KRAS
und damit einhergehend auch der nachfolgenden Signalwege (Pylayeva-Gupta et al.
2011). Diese haben eine Bedeutung für die Proliferation (Feramisco et al. 1984), das
Zellüberleben (Cox und Der 2003), den Zellmetabolismus (Johannessen et al. 2005), das
Remodelling des Mikroenvironments z.B. durch Förderung der Angiogenese (Rak et al.
1995), die Evasion des Immunsystems (Maudsley et al. 1991; Clark et al. 2009) und die
Metastasierung (Hoogwater et al. 2010). Zu den von KRAS ausgehenden Kaskaden
gehören unter anderem die Signalwege RAF-MAPK, PI3K-AKT und RaIGDS. Weitere für
das Pankreaskarzinom bedeutende Onkogene sind c-Myc, MYB, AIB1/NCOA3 und EGFR
(Maitra und Hruban 2008).
Tumorsuppressorgene
Das mit ca. 85% am häufigsten inaktivierte Tumorsuppressorgen des
Pankreaskarzinoms ist das Gen p16INK4a/CDKN2A auf Chromosom 9. Das Protein gehört
zur CDK-Inhibitor-Familie und hemmt das Fortschreiten des Zellzyklus am G1-S-
Checkpoint (Caldas et al. 1994).
Bei etwa 70% der Pankreaskarzinome tritt eine Mutation des TP53-
Tumorsuppressorgens auf (Scarpa et al. 1993). Wird die DNA einer Zelle geschädigt, so
wird p53 aktiviert und der Arrest des Zellzyklus, die Seneszenz oder die Apoptose der
Zelle wird eingeleitet. Aufgrund der zentralen Rolle bei der Regulation von
Zellwachstum und Zellteilung wird p53 auch als „Wächter des Genoms“ bezeichnet
(Lane 1992). Bei einer Mutation von p53 kommt es nicht nur zu einem Verlust der
tumorsuppressiven Eigenschaften, sondern auch zu einem Zugewinn neuer
pathologischer Funktionen („gain of function“). So konnte gezeigt werden, dass durch die
Transfektion von mutiertem p53 in p53-negative Tumoren die Tumorigenität der
betroffenen Zellen gesteigert wird (Dittmer et al. 1993).
Das Tumorsuppressorgen DPC4 auf Chromosom 18q21.1, welches auch als SMAD4
bezeichnet wird, ist bei etwa der Hälfte aller Pankreaskarzinome inaktiviert (Schutte et
al. 1996). Es gehört zur Gruppe der Smad-Gene und ist am TFG-beta-Signalweg beteiligt,
welcher eine wichtige Rolle bei der Suppression des epithelialen Zellwachstums spielt.
Ein Ausfall von DPC4 führt zu einer Störung dieses Signalwegs und dessen hemmenden
Einfluss auf die Zellproliferation (Miyaki und Kuroki 2003).
5Einleitung
Seltener beteiligte Tumorsuppressorgene sind LKB1/STK11, TGFBR1, TGFBR2, ACVR1B
und MKK4. Letzteres ist meist insbesondere in Metastasen mutiert und hat demnach
wahrscheinlich einen hemmenden Einfluss auf die Metastasierung (Maitra und Hruban
2008).
1.2 DIE TUMORSTAMMZELLHYPOTHESE
1.2.1 MODELLE DER TUMORENTSTEHUNG
Hinsichtlich der Entstehung eines Tumors werden im Wesentlichen zwei verschiedene
Modelle voneinander unterschieden: das stochastische und das hierarchische
Tumormodell (siehe Abb. 3). Diese erklären die Tumorinitiierung, die heterogene
Tumorstruktur und die Eigenschaften der malignen Tumorprogression auf der Basis
verschiedener Zelltypen (Vochem et al. 2014).
Das stochastischen Tumormodell postuliert, dass grundsätzlich jede Tumorzelle das
Potenzial dazu hat, Tumorwachstum zu initiieren und voranzutreiben (Acker 2005). Ein
Tumor besteht demnach aus einer grundsätzlich homogenen Zellpopulation, welche
durch eine jeweils individuelle Kombination aus endogenen und exogenen Faktoren eine
heterogene Tumormasse generiert. Zu den intrinsischen Faktoren zählen genetische
Mutationen sowie die Anwesenheit bestimmter Transkriptionsfaktoren und
Translationsmechanismen. Das Mikromilieu, Zell-Zell-Interaktionen und
Zytokinkonzentrationen spielen hingegen auf der extrinschen Seite eine wichtige Rolle
(Bomken et al. 2010). Diese Faktoren führen zu einer Selektion der am besten
angepassten Zellen, welche im weiteren Verlauf das weitere Tumorwachstum fördern
(Garvalov und Acker 2011). In Anlehnung an Charles Darwins Evolutionstheorie wird
dieses Modell daher auch als das „klonale Evolutionsmodell“ bezeichnet (Greaves und
Maley 2012).
Im Gegensatz dazu steht das hierarchische Tumormodell. Dieses unterteilt die Zellen
eines Tumors analog zur hierarchischen Organisation normaler Gewebe in zwei
Subpopulationen (Acker 2005). Nur eine begrenzte Untergruppe von Zellen, die
sogenannten Tumorstammzellen, besitzt die Fähigkeit zur Selbsterneuerung und ein
entsprechendes Differenzierungspotenzial, um das Tumorwachstum zu generieren und
aufrecht zu erhalten. Differenzierte Tumorzellen, welche den Großteil der Tumormasse
bilden, sind hingegen nicht fähig, sich selbst zu erneuern und haben die Fähigkeit zur
Tumorinitiierung verloren (Bomken et al. 2010).
6Einleitung
Abbildung 3: Stochastisches (a) und hierarchisches (b) Tumormodell (Reya et al. 2001):
Während im stochastischen Tumormodell generell jede Zelle das Potenzial zur Initiierung von
Tumorwachstum besitzt, wird diese Fähigkeit im hierarchischen Modell auf die Subpopulation
der Tumorstammzellen eingegrenzt.
1.2.2 EIGENSCHAFTEN DER TUMORSTAMMZELLEN
Tumorstammzellen sind eine kleine Gruppe von Zellen innerhalb eines Tumors, welche
Stammzell-ähnliche Eigenschaften besitzen. So besitzen Tumorstammzellen einerseits
die Fähigkeit zur Selbsterneuerung und andererseits die Möglichkeit in alle heterogenen
Tumorzellen zu differenzieren, welche die Tumormasse bilden. Tumorstammzellen,
oder auch CSCs, können sich asymmetrisch in eine neue Tumorstammzelle und eine
besser differenzierte Vorläuferzelle oder aber symmetrisch in zwei Tumorstammzellen
oder zwei Vorläuferzellen teilen. Weiterhin können die Tumorstammzellen darüber
definiert werden, dass sie dazu fähig sind, die Bildung eines kontinuierlich wachsenden
Tumors zu wiederholen. Deshalb werden sie auch als tumorinitiierende oder
tumorigene Zellen bezeichnet. Eine tumorinitiierende Zelle kann also bei einer
Transplantation in ein Xenotransplantat-Modell einen Tumor auslösen. Bisher ist nur
eine einzige sichere Möglichkeit bekannt, wie man eine CSC nachweisen kann, nämlich
sie zu transplantieren und zu beobachten, ob sie einen neuen Tumor generieren kann
(Clarke et al. 2006).
1.2.3 URSPRUNG DER TUMORSTAMMZELLEN
Bezüglich der Herkunft der Tumorstammzellen existieren derzeit verschiedene
Hypothesen. Eine Theorie besteht darin, dass die Tumorstammzelle aus einer
herkömmlichen Stammzelle hervorgeht, welche durch Mutationen zu einer
Tumorstammzelle wird. Normale Stammzellen besitzen bereits die Fähigkeit zur
Selbsterneuerung und benötigen daher relativ wenige Mutationen, um zur
Tumorstammzelle zu entarten. Außerdem haben Stammzellen aufgrund ihrer
lebenslänglichen Existenz ausreichend Zeit neue Mutationen zu akkumulieren (Passegué
et al. 2003). Beispielsweise waren in Experimenten mit immundefizienten NOD/SCID-
Mäusen nur [CD34+CD38-]-Zellen, was dem Phänotyp normaler hämatopoetischer
Stammzellen entspricht, fähig, bei Transplantation eine akute myeloische Leukämie
(AML) auszulösen (Bonnet und Dick 1997).
7Einleitung
Die andere Möglichkeit besteht darin, dass die Tumorstammzelle auch aus einer
Vorläuferzelle oder einer bereits differenzierten reifen Zelle entstehen kann, welche
durch Mutationen Stammzelleigenschaften erwirbt (Passegué et al. 2003). So konnte für
den M3-Subtyp der AML konnte gezeigt werden, dass das für diesen Leukämie-Subtyp
typische PML-RARα-Gen nur in [CD34-CD38+]-Zellpopulationen, aber nicht in den
[CD34+CD38-]-Stammzellen gegenwärtig ist (Turhan et al. 1995).
1.2.4 IDENTIFIKATION VON TUMORSTAMMZELLEN
Erste Hinweise auf die Existenz von Tumorstammzellen ergaben sich bereits in den
1960er Jahren. So konnte die Arbeitsgruppe um Robert Bruce zeigen, dass nur ca. 1-4%
aller Lymphomzellen dazu in der Lage sind, in vitro Kolonien zu bilden und bei
Transplantation in eine murine Milz ein Karzinom zu generieren (Bruce und van der
Gaag 1963).
Etwa dreißig Jahre später gelang es dann John Dick und seinen Kollegen erstmals
Tumorstammzellen zu identifizieren und zu isolieren. Dazu wurden AML-Zellen nach
ihren exprimierten Oberflächenmarkern sortiert und in NOD/SCID-Mäuse
transplantiert. Nur die Gruppe der [CD34+CD38-]-Leukämiezellen war dazu in der Lage
bei einer Transplantation eine AML zu induzieren, wohingegen sowohl die [CD34-]- als
auch die [CD38+]-Zellen diese Eigenschaft nicht besaßen. (Lapidot et al. 1994).
Später haben dann Al Hajj et al. zum ersten Mal Stammzellen aus einem soliden Tumor,
nämlich aus Brustkrebsgewebe, isoliert. Bei der Transplantation humaner
Mammakarzinomzellen in immunkompromittierte Mäuse konnte nur ein sehr
begrenzter Teil der Zellen ein neues Karzinom generieren. Diese tumorigenen Zellen
konnten anhand der exprimierten Oberflächenmarker von den nicht-tumorigenen Zellen
unterschieden werden. So konnten bereits weniger als hundert [CD44+CD24-]-Mamma-
CA-Zellen nach Transplantation in den Mäusen einen Tumor hervorrufen, während
selbst zehntausende Zellen mit anderem Phänotyp keine tumorigenen Fähigkeiten
besaßen (Al-Hajj et al. 2003).
Mittlerweile ist es gelungen, für fast alle soliden Tumoren CSCs zu identifizieren,
darunter unter anderem das Glioblastom, das Kolonkarzinom, Lungenkrebs und auch
das Pankreaskarzinom (Bao et al. 2010) (siehe Tab. 3).
Erste Experimente zur Isolation von pankreatischen Tumorstammzellen wurden von Li
et al. durchgeführt. Hierzu wurden humane Pankreaskarzinomzellen in
immunkompromittierte NOD/SCID-Mäuse transplantiert. Eine Subpopulation von
[CD44+CD24+EpCAM+]-Zellen zeigte ein besonders tumorigenes Potenzial und in ca.
50% der Fälle konnte durch die orthotope Transplantation von nur 100 solcher
Tumorzellen ein Karzinom im Xenograft-Modell induziert werden. Zellen mit diesem
Phänotyp zeigten Stammzell-Eigenschaften, wie die Fähigkeit zur Selbsterneuerung und
die Fähigkeit differenzierte Tochterzellen zu bilden (Li et al. 2007).
Weitere Fortschritte bei der Identifikation potenzieller Tumorstammzellen konnten
Patrick Hermann und Kollegen bei ihren Forschungen über Zellen mit [CD133+]- und
[CD133+CXCR4+]-Phänotyp erzielen. Bereits 500 [CD133+]-Pankreaskarzinomzellen
waren dazu in der Lage nach orthotoper Transplantation in immunkompromittierte
Mäuse einen Tumor zu induzieren, während von den [CD133-]-Zellen selbst mehr als
8Einleitung
106 Zellen keinen Tumor generieren konnten. Weiterhin konnte gezeigt werden, dass
nach Aussortierung der [CD133+CXCR4+]-Tumorzellen aus dem Pool der mutmaßlichen
[CD133+]-CSCs zwar weiterhin die Tumorbildung im Xenograft-Modell induziert
werden konnte, dieser Tumor jedoch die Fähigkeit zur Metastasierung verloren hatte. Es
wird also postuliert, dass die Tumorstammzellen selbst eine in sich heterogene Gruppe
sind und ausschließlich die Subpopulation der [CD133+CXCR4+]-CSCs für die
Metastasierung verantwortlich ist. (Hermann et al. 2007).
Schließlich konnten Rasheed et al. zeigen, dass [ALDH+]-Pankreaskarzinomzellen
sowohl in vitro als auch in vivo ein statistisch signifikant höheres tumorigenes Potenzial
besitzen als [ALDH-]- Zellen. Zellen mit diesem Phänotyp finden sich besonders häufig in
Metastasen und scheinen daher eine wichtige Rolle bei der Metastasierung des
Pankreaskarzinoms zu spielen. Auch weisen Patienten mit [ALDH+]-Zellen im
Primärtumor nach Tumorresektion in frühen Krankheitsstadien ein vergleichsweise
schlechteres Gesamtüberleben auf (Rasheed et al. 2010).
Die nachfolgende Tabelle (Tab. 3) bietet einen Überblick über die bisher bekannten
Oberflächenmarker, welche zur Isolierung und Identifikation von Tumorstammzellen
genutzt wurden.
Tabelle 3: Oberflächenmarker von Tumorstammzellen geordnet nach Tumorentität
(modifiziert nach Yang et al. 2015)
Tumorentität Marker Referenz
Pankreaskarzinom CD133, CD44, CD24, CXCR4, (Hermann et al. 2007; Li et al. 2011;
c-Met, ALDH1, ABCG2, EpCAM Wang et al. 2009; Salnikov et al. 2009;
Wei et al. 2011)
Mammakarzinom CD44, ANTXR1, ALDH1, CXCR4 (Aktas et al. 2009; Kasimir-Bauer et al.
2012; Balic et al. 2006; Reuben et al.
2011; Abraham et al. 2005; Chen et al.
2013a; Krohn et al. 2009)
Kolonkarzinom CD133, CD44, CD44v6, CXCR4, (Pang et al. 2010; Zhang et al. 2012;
CD26 Todaro et al. 2014)
Magenkarzinom CD44 (Li et al. 2014)
Glioblastom CD133, MMP-13 (Singh et al. 2004; Inoue et al. 2010)
Lungenkarzinom CXCR4, ABCG2, CD133; ALDH1 (Ho et al. 2007; Nian et al. 2011)
Osteosarkom CD133 (Tirino et al. 2011; Tirino et al. 2013)
Retinoblastom ABCG2 (Seigel et al. 2005)
Kopf-Hals- c-Met (Sun und Wang 2011)
Karzinome
Ovarialkarzinom CD133 (Baba et al. 2009)
In dieser Arbeit wurde insbesondere Augenmerk auf die Expression der
Oberflächenmarker CD44, CD24, CXCR4, EpCAM, c-Met und CD133 gelegt. Auf diese
Marker soll im nachfolgenden Abschnitt näher eingegangen werden.
CD44 ist ein multifunktionales transmembranäres Glykoprotein, das als spezifischer
Rezeptor für Hyaluronsäure agiert und Kontakte zwischen Zellen und zur
extrazellulären Matrix (EZM) vermittelt. Bei den meisten epithelialen Tumoren findet
9Einleitung
sich eine Überexpression des Adhäsionsmoleküls CD44. Die Interaktion zwischen CD44
und Hyaluronsäure fördert unter anderem den EGFR-Signalweg. Dadurch kommt es zu
einem verstärkten Tumorwachstum, Migration von Tumorzellen und Resistenz
gegenüber Chemotherapeutika (Thapa und Wilson 2016). Durch alternatives Splicing
sowie N-Glykosylierung bzw. O-Glykosylierung entstehen verschiedene Unterformen.
Die kleinste Isoform, CD44s, besitzt keine zusätzlichen Exons und wird in den meisten
normalen Zellen gefunden. Im Gegensatz dazu werden einige andere Isoformen
typischerweise von Tumorzellen oder im Rahmen einer Entzündung exprimiert (Misra
et al. 2015).
CD24 ist ein kleines Oberflächenprotein mit GPI-Anker, welches, ebenso wie CD44,
Funktionen im Bereich der Zell-Zell- und Zell-Matrix-Interaktion besitzt. Viele
verschiedene Tumorentitäten, darunter auch das Pankreaskarzinom, sind mit einer
Überexpression von CD24 assoziiert. CD24 scheint dort die Proliferation der
Tumorzellen und die Metastasierung zu fördern. Durch variable Glykosylierungen erhält
das Molekül unterschiedliche Funktionen, von denen viele derzeit noch unverstanden
sind (Jaggupilli und Elkord 2012).
CXCR4 ist ein G-Protein-gekoppelter Rezeptor der zur Gruppe der Chemokinrezeptoren
zählt. Durch Bindung des Liganden CXCL12/SDF-1 an den transmembranären Rezeptor
wird eine intrazelluläre Signalkaskade gestartet. Dies führt über multiple Signalwege zu
Chemotaxis, einem erhöhtem intrazellulärem Calciumspiegel, Zelladhäsion, -überleben
und –proliferation sowie zur Transkription bestimmter Gene. CXCR4 spielt eine wichtige
Rolle bei der Migration von Stammzellen zu ihren CXCL12-exprimierenden Zielorten,
sowie bei der Wundheilung und Angiogenese (Chatterjee et al. 2014). Die
Überexpression von CXCR4 verleiht den Tumorzellen ein aggressiveres Verhalten und
fördert Tumorwachstum, Metastasierung und Vaskularisierung (Darash-Yahana et al.
2004). Bei einigen Tumorentitäten, darunter unter anderem auch das
Pankreaskarzinom, ist die Expression des Chemokinrezeptors CXCR4 mit einer
signifikant schlechteren Überlebensrate und einem kürzeren progressionsfreien
Überleben assoziiert (Zhao et al. 2015). Ursprünglich wurde CXCR4 durch seine
Beteiligung beim Eindringen des HI-Virus in seine Zielzelle entdeckt (Bleul et al. 1996).
EpCAM (epitheliales Zelladhäsionsmolekül) ist ein transmembranäres Glykoprotein, das
von verschiedenen Epithelzellen exprimiert wird und Zell-Zell-Kontakte sowie Zell-
Matrix-Kontakte vermittelt. Weiterhin spielt EpCAM, ähnlich wie andere CAMs, eine
Rolle bei zellulären Prozessen wie der Signaltransduktion, Zellmigration, Proliferation
und Differenzierung. Im Vergleich zu Epithelien gesunden Gewebes zeigt sich eine
deutliche Überexpression bei schnell proliferierenden Tumoren mit epithelialem
Ursprung (Trzpis et al. 2007).
C-Met gehört zu den Rezeptortyrosinkinasen und wird vom c-Met Protoonkogen
kodiert. Stimuliert durch den Liganden HGF werden verschiedene Signalwege aktiviert,
welche über eine veränderte Genexpression invasives Wachstum ermöglichen. Dieses
Programm des invasiven Wachstums verläuft in mehreren Schritten: die Zelle verlässt
zunächst ihre ursprüngliche Umgebung, migriert dann zu ihrem neuen Standort und
lässt sich dort nieder. An ihrem neuen Standort baut die Zelle neue tubuläre Strukturen
auf und kann gegebenenfalls proliferieren. Während des kompletten Prozesses ist die
10Einleitung
Zelle vor Apoptose geschützt. Unter physiologischen Bedingungen besitzt dieses
Programm eine wichtige Bedeutung für die Embryogenese und Organentwicklung
sowie die Wundheilung. C-Met spielt jedoch auch eine wichtige Rolle bei der
Tumorprogression. Durch die pathologische Aktivierung in Tumorzellen wird diesen der
Übertritt physiologischer Barrieren ermöglicht und die neoplastischen Zellen erreichen
die Zirkulation, wo sie geschützt vor Apoptose auch ohne Zellverankerung überleben
können. Nach Extravasation können die Zellen neue Umgebungen besiedeln und durch
Proliferation Metastasen bilden. C-Met wird in vielen soliden Tumoren überexprimiert
und ist mit einer schlechteren Prognose assoziiert. Keimbahnmutationen von c-Met sind
verantwortlich für das hereditäre papilläre Nierenzellkarzinom (Gentile et al. 2008).
CD133, auch Prominin-1 genannt, ist ein transmembranäres Glykoprotein (Miraglia et
al. 1997). Es zählt zu den am besten etablierten Tumorstammzellmarkern und die
Expression korreliert bei vielen verschiedenen Tumorentitäten mit einer schlechteren
Prognose (Chen et al. 2013b; Canis et al. 2012). [CD133+]-Zellpopulationen zeigen ein
besonders hohes Potenzial an Tumorstammzelleigenschaften wie Tumorigenität,
Selbsterneuerung und Metastasierung (Hermann et al. 2007). Weiterhin ist die
Überexpression von CD133 auch mit einer verstärkten Invasivität der Tumorzellen
sowie einer vermehrten Expression von Genen assoziiert, welche die epithelial-
mesenchymale Transition (EMT) fördern (Nomura et al. 2015). CD133 ist beteiligt an
der Glucose- und Transferrin-Aufnahme, Autophagie, Membran-Membran-Interaktion
und der Funktion von Matrixmetalloproteinasen. In [CD133+]-Zellen sind die Il-8,
mTOR, PI3K und MAPK Signalwege besonders aktiv (Li 2013). Kommt es durch
chemische, physikalische oder mutagene Einflüsse zu Zellschäden, wird CD133
hochreguliert. So führt zum Beispiel Hypoxie, mitochondriale Dysfunktion oder
Depletion von mitochondrialer DNA zu einer Hochregulation von CD133 (Griguer et al.
2008). [CD133+]-Zellen sind besonders resistent gegenüber Chemo- und Radiotherapie
(Zhang et al. 2010; Piao et al. 2012).
Jedoch muss bedacht werden, dass bisher kein Oberflächenmarker bekannt ist, der nur
von Tumorstammzellen exprimiert wird. Die Mehrheit der Zellen, die diesen Marker
exprimieren, sind keine Stammzellen. Auch hängt das Expressionsmuster von
Oberflächenmarkern vom jeweiligen Organ ab und Erkenntnisse dürfen nicht auf andere
Organe übertragen werden. Weiterhin könnte die Expression der Stammzellmarker von
der Umgebung der Zelle abhängen. Auch deshalb wird nach neuen Wegen zur
Identifikation von Tumorstammzellen gesucht (Clarke et al. 2006).
Eine vielversprechende Möglichkeit sind hierbei Signalwege, die bei Tumorstammzellen
im Vergleich zu anderen Zellen überexprimiert werden und eine wichtige Rolle für die
Stammzelleigenschaften der CSCs zu spielen scheinen (Takebe et al. 2015). Drei wichtige
solcher Signalwege sollen im Folgenden näher erläutert werden.
1.) Sonic Hedgehog Signalweg (SHH): Der Hedgehog-Signalweg nimmt eine
wichtige Funktion bei der Embryonalentwicklung, der Reparatur von normalem
Gewebe und bei der epithelial-mesenchymalen Transition ein (Beachy et al.
2010). Durch die Bindung der drei Hedgehog Ligandenproteine wird die
Expression von HH Zielgenen eingeleitet. Darunter befinden sich Gene, die für
11Einleitung
Cycline, c-Myc, Bcl-2 und Snail kodieren, welche die zelluläre Differenzierung,
Proliferation und das Zellüberleben regulieren (Amakye et al. 2013).
Pankreaskarzinomzellen zeigten im Vergleich zu normalen Pankreaszellen eine
4,1-fach erhöhte Expression des SHH Pathways, [CD24+CD44+ESA+]-
Pankreaskarzinomzellen sogar eine 46,3-fache Erhöhung. Dies weist darauf hin,
dass SHH in pankreatischen Tumorstammzellen deutlich hochreguliert wird (Li
et al. 2007). Weiterhin konnte gezeigt werden, dass durch IPI-269609, einem
Inhibitor des SHH-Signalwegs, insbesondere die Anzahl der [ALDH+]-
Pankreaskarzinomzellen reduziert werden kann und sowohl Tumorwachstum als
auch Metastasierung gehemmt werden (Feldmann et al. 2008).
2.) Wnt Pathway: Binden Wnt-Liganden an den Rezeptorkomplex aus dem G-
Protein gekoppelten Rezeptor Frizzled und dem Ko-Rezeptor LRP 5/6 wird die
Transkription bestimmter Zielgene induziert (Logan und Nusse 2004). Unter den
zahlreichen bisher bekannten Zielgenen befinden sich unter anderem auch das
Onkogen c-Myc (He et al. 1998) und Cyclin D1 (Morin 1999).
In Vorläuferzellen oder Stammzellen trägt der streng regulierte Wnt-Signalweg
zu deren Selbsterneuerungsfähigkeit bei. Wird der Wnt-Pathway jedoch in Folge
von Mutationen konstitutiv aktiviert, so kann dies zum tumorigenen Potenzial
von Tumorstammzellen beitragen (Reya und Clevers 2005). Weiterhin spielt Wnt
eine wichtige Rolle für das Überleben von CSCs. Die Ausschaltung des β-Catenin-
Gens führt beispielsweise bei Hauttumoren zu einem kompletten Verlust der
identifizierten Tumorstammzellen und einer vollständigen Tumorregression
(Malanchi et al. 2008).
3.) Notch Pathway: Die Interaktion zwischen einem der vier Notch-Rezeptoren und
einem der fünf verschiedenen Liganden zweier benachbarter Zellen führt zur
Transkription von Zielgenen. Zu diesen Zielgenen zählen Gene, wie zum Beispiel
Gene aus der HES- oder HEY-Familie, c-Myc und Cyclin D1, die eine wichtige Rolle
bei der Zelldifferenzierung und dem Zellüberleben spielen (Espinoza et al. 2013).
In Synergie mit KRAS wird durch die Hochregulation des Notch-Signalwegs die
Bildung und Progression von PanINs gefördert (La O et al. 2008). Auch beim
Übergang von PanINs in PDACs spielt Notch eine wichtige Rolle (Plentz et al.
2009).
1.2.5 BEDEUTUNG FÜR DIE PROGNOSE UND THERAPIE
Faktoren wie ein lokal fortgeschrittenes Tumorstadium bei Erstdiagnose, die frühzeitige
Metastasierung, Resistenz gegenüber Chemo- und Radiotherapie sowie die hohe
Rezidivrate sind entscheidend für die schlechte Prognose des Pankreaskarzinoms.
Tumorstammzellen scheinen eine wichtige Bedeutung für diese Faktoren und somit für
das Überleben der Patienten zu haben (Fitzgerald und McCubrey 2014).
Die Expression von Tumorstammzellmarkern wie CD133, CD24, CD44 und ALDH
korreliert mit einer verminderten Überlebensrate der Patienten (Maeda et al. 2008;
Ohara et al. 2013; Rasheed und Matsui 2012).
Mehr als 50% der Pankreaskarzinom-Patienten weisen zum Zeitpunkt der Diagnose
Fernmetastasen auf. Bei dieser Metastasierung scheinen die Tumorstammzellen eine
12Einleitung
zentrale Rolle zu spielen (Bao et al. 2010). [CD133+]-Tumorstammzellen können in zwei
Gruppen unterteilt werden: eine unbewegliche, sesshafte und eine migrierende, die
Metastasen bilden kann. Es wurde gezeigt, dass [CD133+CXCR4+]-Zellen im Gegensatz
zu [CD133+CXCR4-]-Zellen die Fähigkeit der Metastasierung besitzen. Dies deutet
darauf hin, dass CXCR4 eine wichtige Funktion für der Metastasierung besitzt (Hermann
et al. 2007). Die am häufigsten von der Metastasierung betroffenen Organe wie Leber,
Lunge, Lymphknoten und Knochenmark weisen besonders hohe Konzentrationen an
SDF-1 auf, dem spezifischen Liganden für den Chemokinrezeptor CXCR4 (Kucia et al.
2005).
Weiterhin scheinen Tumorstammzellen besonders resistent gegenüber Chemo- und
Radiotherapie zu sein. Resistenzmechanismen wie eine effektive DNA-Reparatur,
erhöhte Toleranz gegenüber DNA-Schäden, Umgehung der Apoptose, niedrige
Mitoseraten, MDR-ABC-Transporter und ALDH-Dehydrogenasen spielen dabei eine
Rolle (Fitzgerald und McCubrey 2014).
Tumorstammzellen erscheinen also als ein vielversprechendes Ziel für die Entwicklung
neuer Therapiestrategien. Konventionelle Therapien, welche die Tumorstammzellen
nicht angreifen, reduzieren zwar zunächst die große Anzahl an normalen Tumorzellen,
jedoch bleiben die tumorigenen Zellen unangetastet und es kann zum Rezidiv kommen.
Ziel muss es also sein, Therapiestrategien zu entwickeln, die sowohl die Masse an
Tumorzellen angreifen, die den Großteil des Tumors bildet, als auch die tumorigenen
Tumorstammzellen (Fitzgerald und McCubrey 2014).
1.3 PANKREASKARZINOM-MODELLE
In den letzten Jahrzehnten konnten große Fortschritte dabei erzielt werden, die
Schlüsseleigenschaften des duktalen Pankreaskarzinoms bei Tieren nachzustellen. Dabei
können verschiedene Modelle unterschieden werden (Murtaugh 2014).
Beim ersten transgenen Mausmodell wurden Mäuse mit KRAS-Mutation und den
Promotoren gangspezifisches Zytokeratin-19 (KRT19) und azinusspezifische Elastase-1
(Ela1) verwendet. Interessanterweise haben die gangspezifischen KRT19-Modelle
jedoch nur eine leichte Entzündung aufgewiesen, die azinusspezifischen Ela1-Modelle
jedoch prämaligne Vorläuferläsionen mit einem gemischt azinär-duktalen
Erscheinungsbild (Brembeck et al. 2003; Grippo et al. 2003).
Problematisch ist, dass es bei einer einfachen Überexpression von KRAS Seneszenz
hervorgerufen wird (Serrano et al. 1997). Deshalb müssen mutierte KRAS-Allele
hergestellt werden. Hierbei wurden zwei verschiedene mutierte Allele entwickelt: im
KRAS Exon 2 wurde das Gly-12 Codon entweder durch Aspartat (G12D) oder Valin
(G12V) ersetzt. Dies sind auch die häufigsten Mutationen im humanen
Pankreaskarzinom. Die G12D und G12V Modelle verhalten sich größtenteils identisch
(Guerra et al. 2003; Tuveson et al. 2004). Vor das mutierte Allel wurde ein Stopp-Codon
eingeführt, sodass das mutierte KRAS nicht abgelesen wird. Das Stopp-Codon wird von
loxP-Stellen flankiert und dadurch für die Cre-Rekombinase markiert. Wenn das Cre-
Rekombinase Enzym in einer Zelle vorhanden ist, wird das Stopp-Codon
herausgeschnitten und KRAS kann somit aktiv werden. Bei den meisten Modellen wird
Cre in den multipotenten embryonalen Vorläuferzellen des frühen embryonalen
13Einleitung
Pankreas unter Kontrolle von Pdx1 oder Ptf1a (=Ptf1-p48) exprimiert. Pdx1 ist ein
Protein, das für die frühe Entwicklung des Pankreas wichtig ist. Sowohl differenzierte
exokrine als auch endokrine Zellen des reifen Pankreas stammen von Pdx1-positiven
Zellen ab. Ptf1-p48 ist ein Marker des frühen Pankreas und persistiert in den reifen
Azinuszellen. Bei der Geburt erscheinen die Bauchspeicheldrüsen zunächst normal,
entwickeln jedoch innerhalb von 1-2 Monate Vorläuferläsionen, welche den PanIN-1 des
humanen Pankreaskarzinoms ähneln. Mit der Zeit werden die PanINs immer häufiger
und dysplastischer und ähneln den humanen PanIN-2 und PanIN-3. Nach einigen
Monaten entwickelt ein Teil der Mäuse aufgrund weiterer somatischer Mutationen ein
invasives und metastasierendes Pankreaskarzinom. Dies gilt sowohl für das [Pdx1-
Cre;LSL-Kras-] als auch für das [Ptf1/p48-Cre;LSL-Kras-]-Modell, kurz KC-Modell
(Hingorani et al. 2005).
Abbildung 4: KRAS-Mausmodell (Murtaugh 2014): Das von lox-P flankierte Stopp-Codon wird
in den Pankreaszellen durch die Cre-Rekombinase ausgeschnitten und so das mutierte KRAS
exprimiert.
Dieser Vorgang kann erheblich beschleunigt werden, wenn zusätzlich
Tumorsuppressorgene wie p16INK4a/CDKN2A, Tp53, Smad4 und Brca2 mutiert sind. In
diesem Falle entwickelt sich innerhalb von wenigen Wochen nach der Geburt ein
invasives und metastasierendes Pankreaskarzinom (Aguirre et al. 2003; Bardeesy et al.
2006a; Bardeesy et al. 2006b; Skoulidis et al. 2010).
14Einleitung
Abbildung 5: Azinär-duktale Metaplasie (Murtaugh 2014): Aus der azinären Ursprungszelle
entsteht über prämaligne Vorläuferläsionen ein invasives Pankreaskarzinom (PDAC) mit
duktalem Erscheinungsbild.
1.3.1 ZUSÄTZLICHER VERLUST VON ATM BEIM KC-MAUSMODELL
ATM (= Ataxia telangiectasia mutated), eine Serin-Threonin-Kinase aus der Gruppe der
PI3-Kinasen, erlangte ihre Bedeutung ursprünglich durch ihre Rolle bei der DNA-
Reparatur, insbesondere der Reparatur von DNA-Doppelstrangbrüchen (Bakkenist und
Kastan 2003).
Patienten mit homozygoter Mutation des ATM-Gens leiden am Krankheitsbild der Ataxia
teleangiectatica (= Louis-Bar-Syndrom), welches unter anderem mit einer progressiven
Neurodegeneration, Teleangiektasien, Immundefizienz, einem erhöhten Risiko für
maligne Neoplasien und einer erhöhten genomischen Instabilität einhergeht. Auch
leiden die Patienten gehäuft an Stoffwechselerkankungen wie Diabetes mellitus oder
Infertilität und zeigen eine erhöhte Empfindlichkeit gegenüber ionisierender Strahlung
(Shiloh und Ziv 2013). Die Inzidenz dieser autosomal-rezessiv vererbbaren Krankheit
beträgt 1/40.000 bis 1/100.000 und ca. 1% der Bevölkerung sind heterozygote Träger
des mutierten ATM-Gens (Fitzgerald et al. 1997). Neben der Funktion bei der DNA-
Reparatur hat ATM aber noch eine Vielzahl weiterer Funktionen, wie zum Beispiel in der
Aufrechterhaltung der Zellhomöostase, der Regulation der Umbauvorgänge des
Chromatins, im Bereich des Zellmetabolismus und des oxidativen Stresses. Weiterhin
hat ATM-Defizienz Auswirkungen auf die Erneuerung von hämatopoetischen
Stammzellen, steigert die Angiogenese und führt zur schnelleren Alterung
telomerdefizienter Mäuse (Shiloh und Ziv 2013).
Sowohl beim sporadischen als auch beim familiären Pankreaskarzinom lassen sich hohe
Mutationsraten im ATM-Gen nachweisen (Biankin et al. 2012; Roberts et al. 2012). Eine
Analyse der ATM-Expression konnte zeigen, dass eine fehlende ATM-Expression im
Vergleich zu einer generell vorhandenen ATM-Expression mit einer verkürzten
Überlebenszeit einhergeht (Kamphues et al. 2015).
Gentechnisch veränderte Mäuse mit zusätzlicher ATM-Defizienz (AKC-Mäuse) zeigen im
Vergleich zum herkömmlichen [Ptf1/p48-Cre; LSL-Kras-]-Modell (KC-Mäuse) eine
veränderte Regulation von 2472 Genen, darunter viele Gene, welche bekannter Weise
eine Rolle beim duktalen Pankreasadenokarzinom spielen (Russell et al. 2015).
Es konnte gezeigt werden, dass die Depletion von ATM verstärkt zur Entwicklung von
neoplastischen Vorläuferläsionen im Pankreas führt. So kommt es verstärkt zur azinär-
duktalen Metaplasie und zur Ausbildung von PanINs der Grade I-III (Russell et al. 2015).
15Einleitung
Ein weiterer Punkt ist die verstärkte epitheliale mesenchymale Transition. Diese führt
unter anderem zu einer Akkumulation von Wachstumsfaktoren, die das
Mikroenvironment des Tumors beeinflussen und das Tumorwachstum fördern. Eine
wichtige Rolle spielt hierbei der durch Depletion von ATM hyperaktive BMP-4-Pathway,
welcher die Transition unterstützt und die Invasivität des Tumors steigert (Russell et al.
2015).
Außerdem konnte gezeigt werden, dass ATM-Defizienz zu einer Überexpression von
stammzellassoziierten Genen führt und stammzellassoziierte Oberflächenmarker wie
CD133 und CXCR4 überexprimiert werden (Russell et al. 2015).
Schließlich beeinflusst ATM auch das Überleben der Mäuse: so verkürzt sich das
durchschnittliche Überleben von KC-Mäusen von 55 Wochen auf nur noch 36 Wochen
bei homozygoten bzw. 45 Wochen bei heterozygoten AKC-Mäusen. Weiterhin
entwickeln AKC-Mäuse auch früher hochproliferative Tumoren und haben eine höhere
Anzahl an Lebermetastasen (Russell et al. 2015).
Stellt man das AKC-Mausmodell dem humanen PDAC gegenüber, so fällt auf, dass es
einige Ähnlichkeiten zu aggressiveren Subtypen des humanen PDACs besitzt und eine
inverse Korrelation zwischen der Expression von ATM-Proteinen und dem WHO-
Grading besteht. Weiterhin zeigen Tumoren mit niedrigerem ATM-Level häufiger
Lymphknotenmetastasen. Auch in humanen Karzinomen zeigte eine geringe Expression
von ATM eine epitheliale mesenchymale Transition und somit einen infiltrativeren
Phänotyp an (Russell et al. 2015).
1.4 ZIELSETZUNG
In dieser Arbeit soll die Verteilung von potenziellen Tumorstammzellen in
Vorläuferläsionen des murinen Pankreaskarzinommodells (KC-Modell) in Abhängigkeit
von einer zusätzlichen ATM-Defizienz untersucht werden. Dabei sollen die
Tumorstammzellen über die Expression der spezifischen Oberflächenmarker CD133,
CD24, CD44, CXCR4, EpCAM und c-Met identifiziert werden. Diese Marker können
mittels immunhistochemischer Färbungen nachgewiesen werden. Anhand der
Verteilung soll insbesondere ermittelt werden, ob Vorläuferläsionen mit einer
zusätzlichen ATM-Defizienz, für die eine stärker ausgeprägte Tumorigenität zu
postulieren ist, mehr Tumorstammzellen gefunden werden können. Dies könnte die
Bedeutung der Tumorstammzellen für die Entstehung und die schlechte Prognose des
Pankreaskarzinoms untermauern. Auch sollen die einzelnen Oberflächenmarker
miteinander verglichen werden, um möglicherweise eine Aussage zu ihrer Eignung als
Tumorstammzellmarker zu treffen.
16Sie können auch lesen

















































