MYELODYSPLASTISCHES SYNDROM - DR. VERONIKA PAPP
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
MYELODYSPLASTISCHES SYNDROM DR. VERONIKA PAPP
MDS; PRÄLEUKÄMIEN
• heterogene Gruppe klonaler neoplastischer Erkrankungen der hämatopoetischen Zellen
• qualitativ und quantitativ veränderte Zellproliferation im Knochenmark; Dysplasie in einer oder mehreren
myeloischen Zellreihen
• wesentliches Kriterium: die Zahl der im Knochenmark und/oder im peripheren Blut nachweisbaren Blasten
• Zytopenie des peripheren Blutes (Anämie, Granulozytopenie oder Thrombozytopenie), trotz normo- oder
hyperzellulären Knochenmarks (ineffektive Hämatopoese)
• häufig Übergang in akute oder chronische myeloische Leukämien (präleukämische Syndrome)
• Erkrankungsgipfel nach dem 70. Lebensjahr
• meist unklare Atiologie
• sekundäres myelodysplastisches Syndrom induziert durch Zytostatika, radioaktive Strahlung, Benzol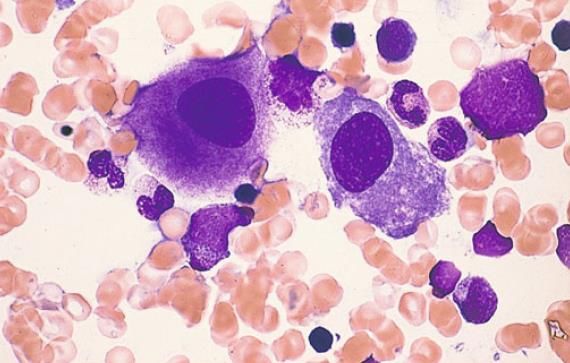
ÄTIOLOGIE Primär (>90%): meistens spontan, d. h. ohne erkennbare Ursache Sekundär (
KLINIK • in 20% der Fälle asymptomatischer Zufallsbefund • im Verlauf durch Zytopenie (80%) – Mono-, Bi- oder Panzytopenie: o unspezifische Anämiesymptome: Müdigkeit, Schwäche o Neutropenie: erhöhte Infektanfälligkeit o Thrombozytopenie: Blutungsneigung • Hepato-/Splenomegalie (30/20%), Lymphome (10%)
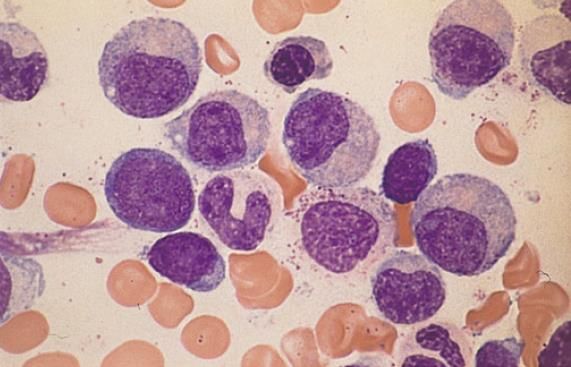
PATHOGENESE
• Erythropoese:
o Anämie durch ineffektive Erythropoese
o Erythroblasten sprechen vermindert auf EPO an
o entstehen megaloblastäre Formen und infolge gestörter Eisenverwertung Sideroblasten
o gesteigerte Bildung von Hb F
o verminderte osmotische Resistenz der Erythrozyten, Pyruvatkinase-Gehalt ist herabgesetzt, veränderte Antigenexpression an der
Zelloberfläche
• Granulozytopoese:
o Neutropenie als Erstsymptom oder im Krankheitsverlauf entwickelt – Infektanfälligkeit!
o Linksverschiebung der Granulozytopoese wegen der Reifungsstörung der weissen Reihe
o diverse Abnormitäten der Neutrophilen:
vergrößerte Granula, Hypogranulation oder fehlende Granula, Hyper- oder Hyposegmentierung des Zellkerns, basophiler Plasmaring in der
Zellperipherie
o Blastenanteil initial 20 % akute myeloische Leukämie!
• Thrombozytopoese:
o Thrombozytopenie bei 50 % der Patienten, bei 5 % als einzige Zytopenie
o im Blutausstrich: neben Riesenplättchen auch granulafreie Exemplare
o im Knochenmark: oft kleine Megakaryozyten mit wenig gelapptem Kern, können auch mehrkernig sein
o Blutungsneigung wegen verminderter Plättchenaggregation und eines Glanzmann-Defektes (verändertes Glykoprotein GPIIb/IIIa)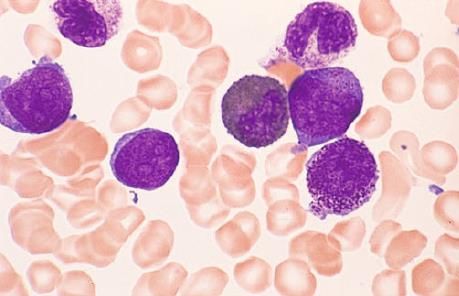
DYSPLASIEZEICHEN
• Im Knochenmark:
o Störungen der Erythropoese (Dyserythropoese): quantitative und qualitative Abnormitäten der kernhaltigen Vorstufen im
Knochenmark und der Erythrozyten im peripheren Blut
o Anteil der Erythropoese im Knochenmark: vermehrt (>50%) oder vermindert (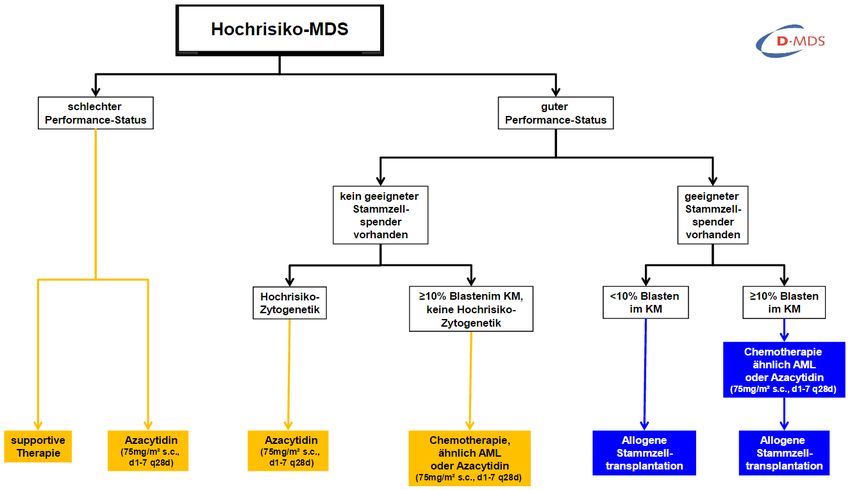
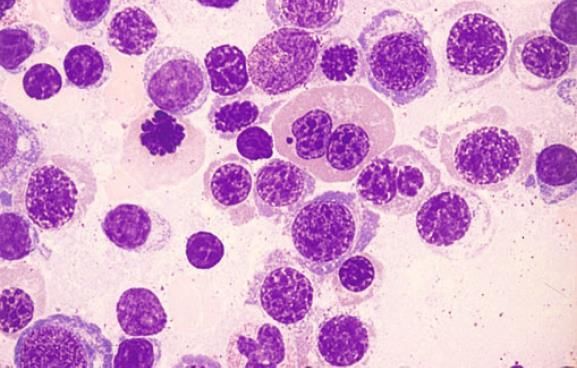
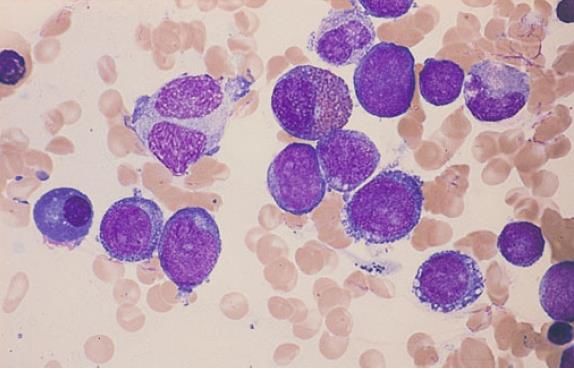
BLASTTYPEN BEI MDS
• Ungranulierte Blasten:
o die Zellen variieren zwischen von normalen Myeloblasten nicht unterscheidbaren bis hin zu
Zellen unterschiedlicher Grösse, die als unklassifizierbar einzustufen sind
o Zytoplasmagranula fehlen immer
o der Kern zeigt gut abgrenzbare Nucleoli und ein aufgelockertes Chromatingerust
• Granulierte Blasten:
o Primärgranula in den Zellen (azurophile Granula)
o das Kern-Zytoplasma-Verhältnis ist geringer
o der Kern ist zentral gelegen
Links 2 Blasten Typ I, rechts 1 Blast Typ II mit deutlichen
Granula, darunter ein Promyelozyt mit sehr groben Primargranula.
Zwischen Blast I und Blast II 1 Eosinophiler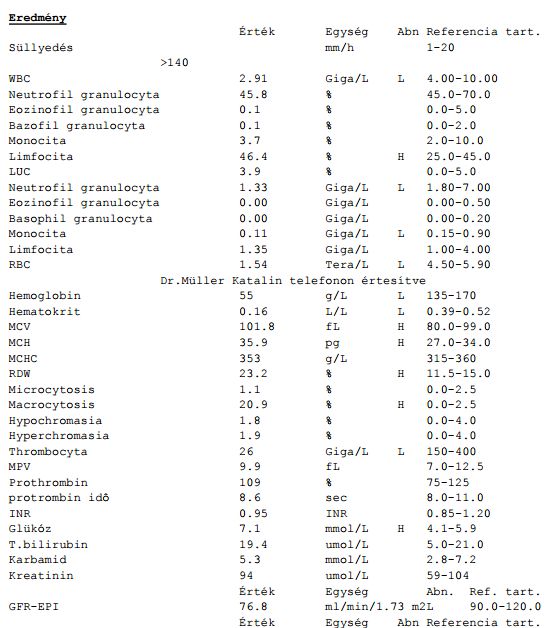
ZYTOGENETISCHE VERÄNDERUNGEN
• das Spektrum ist heterogen
• genetisch ähnelt das MDS der AML und anderen myeloischen Neoplasien
• Translokationen und Inversionen sind selten, vor allem Zugewinnen und Verlusten von genetischem Material
• häufige beobachtete zytogenetische Veränderungen:
o Deletionen im langen Arm der Chromosomen 5, 7 und 20, sowie eine Monosomie 7 und Trisomie 8
• typische Mutationen:
o Mutation der Splice-Maschinerie wie SF3B1, U2AF1 und ZRSF2 (wobei SF3B1-Mutationen beim MDS ganz spezifisch mit dem Vorliegen
von Ringsideroblasten assoziiert sind)
o Mutationen in Genen, die Einfluss auf die epigenetische Regulation nehmen (DNMT3A, ASXL1, EZH2 und TET2)
o JAK2V617F-Mutationen sind mit Thrombozytose assoziiert
o wie bei AML - jedoch mit geringerer Frequenz – : z. B. NRAS, FLT3-ITD, MLL-PTD, NPM1
• Zytogenetik ist ein wichtiger Parameter in verschiedenen Prognose-Scores beim MDS
• Mutationen können als Progressionsfaktor bewertet werden
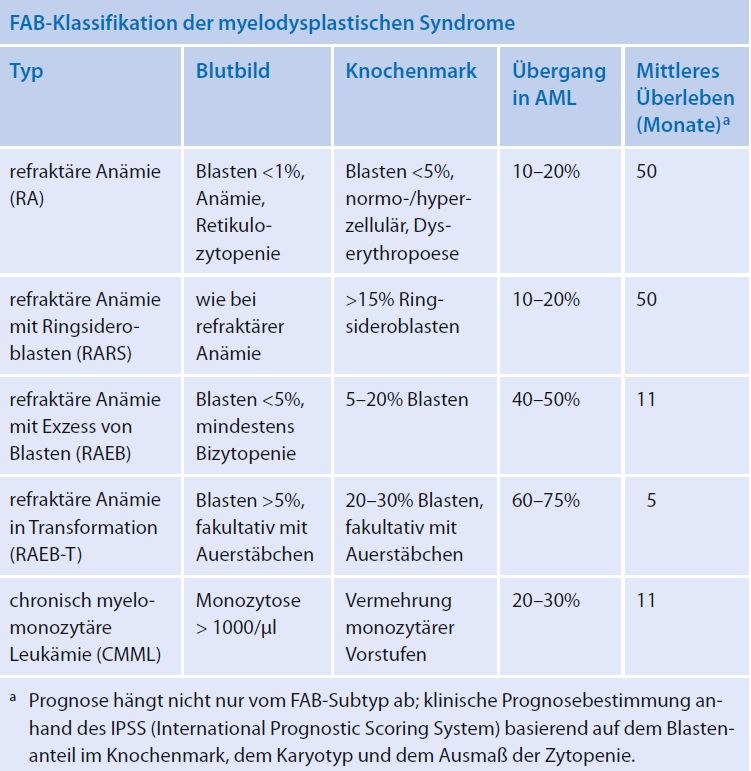
KLASSIFIKATION: FRENCH-AMERICAN-BRITISH COOPERATION GROUP (FAB) UND WORLD HEALTH ORGANIZATION (WHO) http://www.mds-register.de/whoklassifikation/ - RAEB-T mit einem Blastenanteil >20% gilt gemas WHO-Definition als AML - dieWHO-Klassifikation (unterscheidet 8 Untergruppen nach morphologischen und zytogenetischen Kriterien; RAEB-T und CMML sind ausgegliedert) – ermöglicht bessere Aussagen zur Prognose - CMML gehört nach WHO-Kl. zu den Myeloproliferative Neoplasien
1. REFRAKTÄRE ZYTOPENIE MIT
EINLINIENDYSPLASIE (RCUD)
• wird unterteilt in:
o refraktäre Anämie (RA)
o refraktäre Neutropenie (RN)
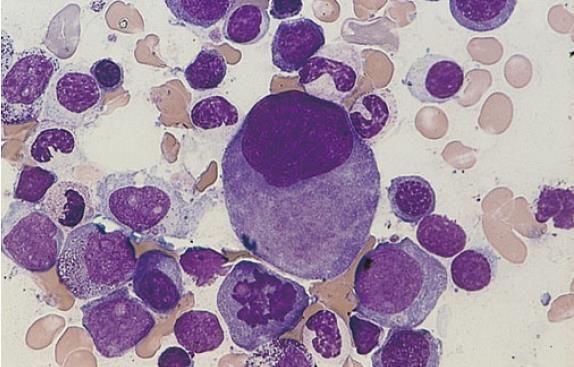
o refraktäre Thrombozytopenie (RT) Myelodysplastische Syndrome (MDS). Erythroblasten mit
megaloblastischen Veränderungen, Mitte oben zweikernige Form.
• Befunde: Refraktäre Anämie (RA)
Löffler, Haferlach. Hämatologische Erkrankungen. 2. Auflage. Springer2. REFRAKTÄRE ANÄMIE MIT
RINGSIDEROBLASTEN (RARS)
• Dyserythropoese, aber keine diagnostisch
verwertbare Veränderungen der Granulozytopoese
und der Megakaryopoese
• Anteil der Blasten im Knochenmark liegt 15% Ringsideroblasten mit ungewöhnlich groben Eisengranula
• extrem hohe Korrelation (ca. 75%) von
Löffler, Haferlach. Hämatologische Erkrankungen. 2. Auflage. Springer
Ringsideroblasten mit SF3B1-Mutationen3. MDS MIT ISOLIERTER DEL(5Q) (5Q-MINUS-SYNDROM) • im peripheren Blut: Anämie mit oft normalen oder erhöhten Thrombozyten • Blastenanteil im Blut und Knochenmark liegt
MDS MIT ISOLIERTER DEL(5Q) (5Q-MINUS-SYNDROM) - 2
2 Megakaryozyten mit rundlichen Kernen und unterschiedlich ausgereiftem Grosser Megakaryozyt mit unsegmentiertem Kern und z. T. noch basophilem
Zytoplasma, ein typischer Befund für dieses Syndrom Zytoplasma. Darunter Erythroblastenmitose und 1 Myeloblast.
Löffler, Haferlach. Hämatologische Erkrankungen. 2. Auflage. Springer4. REFRAKTÄRE ANÄMIE MIT BLASTENVERMEHRUNG 1 (RAEB-1) • im peripheren Blut: Zytopenie,
5. REFRAKTÄRE ANÄMIE MIT BLASTENVERMEHRUNG 2 (RAEB-2) • im peripheren Blut: Zytopenie bei
6. MDS, UNKLASSIFIZIERT (MDS-U) • im peripheren Blut: Zytopenie, wenige Blasten, keine Auer-Stäbchen • im Knochenmark: Dysplasien in einer myeloischen Zellreihe,
7. REFRAKTÄRE ZYTOPENIE MIT MULTILINEAREN DYSPLASIEN (RCMD) • im peripheren Blut: Zytopenie, der Blastenanteil liegt
8. REFRAKTÄRE ANÄMIE IN TRANSFORMATION
(RAEB-T)
• Subtyp RAEB-T wurde von einem pädiatrischen Klassifizierungsvorschlag
beibehalten (bei Erwachsenen nicht mehr verwendet und diese Fälle werden
den AML zugeordnet)
• im Vergleich zu RAEB-1 und -2 findet man im Knochenmark >20% bis zu 30%
Blasten, manchmal auch Auer-Stäbchen
• relativ schneller Übergang in eine AML
Übergang von RAEB-Ta zur AML. Blastenanteil knapp unter 30 %. Gruppe von
Myeloblasten, 1 unauffälliger eosinophiler Myelozyt
Löffler, Haferlach. Hämatologische Erkrankungen. 2. Auflage. Springer9. MDS DES KINDESALTERS
• tritt primär oder sekundär nach kongenitalen oder erworbenen Knochenmarkinsuffizienzsyndromen, sowie
therapiebedingt nach zytotoxischer Therapie
• RARS und 5q-minus-Syndrom sind extrem selten
• isolierte Anämie – wie bei RA des Erwachsenen – ist ungewöhnlich, eher Neutropenie und Thrombopenie
• hypozelluläres Knochenmark ist häufiger, deshalb passen einige Kinder nicht in die Kategorie ≫Low-grade-
MDS≪ - ≫refraktare Zytopenie des Kindesalters≪ (RCC) wurde eingeführt
o von Knochenmarkinsuffizienzerkrankungen - speziell von aplastischer Anamie (AA) und angeborenen Insuffizienzerkrankungen –
ist schwierig abzugrenzen
• RAEB-T wird bei Erwachsenen den AML zugeordnet, im Kindesalter den MDS
o wie bei Erwachsenen sollten Patienten mit t(8;21), inv(16), t(16;16) oder mit t(15;17) unabhängig vom Blastenanteil als AML
eingestuft werdenDIAGNOSTIK
• Anamnese, Klinik
• bei Verdacht auf MDS Knochenmarkaspiration oder Stanzbiopsie zur histologischen Untersuchung
• routinemässige Blutbildwerte:
o im Blutbild: Erythrozytopenie, Bizytopenie, Panzytopenie
o Ferritin, LDH, Vitamin B12, Folsäure, Erythropoetin, Säurehämolysetest
• Hystologie: zur Bestimmung der Zellanzahl, der Fibrose und abnormal lokalisierter unreifen Vorstufen
• Zytologie: Pappenheim-Färbung (Kombination der May-Grünwald-Färbung mit der Giemsa-Färbung), PAS-
Reaktion (Anfärbung von Kohlenhydraten), POX- (peroxidase) Reaktion, Eisenfärbung
• klassische Zytogenetik, die FISH-Technik und molekuläre Verfahren
• Knochenmark: meist erhöhte Zelldichte, Erythropoese und Granulozytopoese zeigen Reifungsstörungen
(Ringsideroblasten, Anisozytose, Poikilozytose bzw. hypogranulierte Granulozyten), evtl. ubersegmentierte,
unterschiedlich grosse Megakaryozyten
• Chromosomenanalyse (50% der Fälle zeigen Abberationen) – 5., 7., 8., 20. Chromosom
• Ausschluss von DifferenzialdiagnosenDIFFERENZIALDIAGNOSE • myeloproliferative Neoplasien – Hystologie, Zytogenetik • Haarzell-Leukämie – Blutbild, Zytologie • akute Leukämien – Blastenanzahl >20% • aplastische Anämie - Hystologie, Zytologie • paroxysmale nächtliche Hämoglobinurie – Flow-Zytometrie • megaloblastäre Anämie – Vitamin B12, Folsäure • nutritiv-toxische oder reaktive Knochenmarkveränderungen – Anamnese • reaktive Knochenmarkveränderungen (Sepsis, AIDS, chronische Infektionen) – Zytologie, Anamnese • Hyperspleniesyndrom – Anamnese, Klinik, Splenomegalie • Immunthrombozytopenie – Zytologie, Klinik • congenitale dyserythropoetische Anämie (selten) – Zytologie, Molekularbiologie
THERAPIE - SYMPTOMATISCHE THERAPIE
• Anämie:
o Erythrozytentransfusionen bei Absinken des Hb unter 8–9 g/dl
o Transfusionen sollten zwecks Zerstörung der T-Zellen bestrahlt werden, um Graft-versus-Host-Reaktionen zu vermeiden
o nach >20 Transfusionen Behandlung mit Desferrioxamin wegen Eisenüberlastung
o in 25 % der Fälle hat rekombinantes Erythropoetin den Transfusionsbedarf deutlich gesenkt (unter 500 U/l
EPO-Wert)
• Neutropenie:
o Wachstumsfaktoren G-CSF und GM-CSF erhöhen die Neutrophilenzahl, verbessern aber die Infektabwehr nicht
(die gebildeten Neutrophilen sind nicht voll funktionstüchtig)
o Breitbandantibiotika bei unklarem Fieber (Haemokultur)
o Pneumococcus-Impfung
• Thrombozytopenie:
o Transfusionen von bestrahlten Thrombozyten bei Blutungen
o bei ungenügender Blutstillung Antifibrinolytika (ε-Aminocapronsäure)
• Vitamine und Anabolika:
o Vitamin B6, Androgene und Anabolika haben bei Erwachsenen keine gesicherte WirksamkeitTHERAPIE - KAUSALE THERAPIE
• Immunsuppression:
o mit MDS assoziierte Autoimmunphänomene - wie rheumatoide Arthritis -sprechen gut auf Prednison an
das Blutbild besert sich
steigende Infektanfälligkeit
o junge Patienten mit niedrigen Plättchenzahlen bessern sich häufig durch Antithymozytenglobulin (ATG)
• Antiangiogenese im Knochenmark:
o Suppression der Neoangiogenese mit lenalidomid (Revlimid) (Thalidomid-Analoge) führte bei 40 % der
Patienten zum Hämoglobinanstieg, besonders bei Patienten mit dem 5q-Syndrom
Transfusionsbedarf nahm ab (beim Therapiebeginn Zytopenie!)
• DNA-Hypomethylierung:
• Beseitigung der mit Inaktivierung verbundenen Hypermethylisierung von Genen durch Hemmung von
Methyltransferasen
• hypomethylisierende Pharmaka: Azacitidin und sein Metabolit Decitabin
• Remissionsrate bis zu 60 %
• Indikation: intermedier und high risk Patienten, bei denen Knochenmarktransplantation kontraindiziert istTHERAPIE - INTENSIVE THERAPIE
• Antileukämische Chemotherapie:
o Remissions-Induktions-Chemotherapie wie bei der akuten Myeloblastenleukämie /Polychemotherapie z. B.
TAD-Regime (Thioguanin, Cytarabin [Ara-C], Daunorubicin), ggf. HAM-Regime (Ara-C, Mitoxantron)/
Indiziert bei PatientenTHERAPIE • ‚Low risk’ MDS-Patienten: o symptomatische Therapie o immunsuppressive Therapie – Antithymozytenglobulin und/oder cyclosporin A o Immunmodulationstherapie - lenalidomid • ‚High risk’ MDS-Patienten: o Azacitidin (DNA-Hypermethylierung) o intensive Polychemotherapie o allogene Stammzelltransplantation
THERAPIESTRUKTUR https://www.onkopedia.com/de/onkopedia/guidelines/myelodysplastische-syndrome-mds/@@view/html/index.html
PROGNOSE • ungünstige Prognose: o Blastenanzahl > 5% o komplexe Chromosomenaberrationen o LDH erhöht o hochgradige Zytopenie o schlechter Allgemeinzustand, Vorerkrankungen o höheres Lebensalter o >60% der Patienten sterben an Komplikationen (Infektionen, Blutungen, AML)
PROGNOSE SCORES:
INTERNATIONALE PROGNOSE-SCORE- IPPS, IPSS-R
Definition des IPSS (International Prognostic Scoring System)
Definition des IPSS-R (International Prognostic Scoring System-Revised)DANKE FÜR IHRE AUFMERKSAMKEIT!
Sie können auch lesen