Identifizierung von CNVs (copy number variations) - Antje Krause TFH Wildau
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

Identifizierung von CNVs
(copy number variations)
Antje Krause
TFH Wildau
akrause@tfh-wildau.de
08.02.2007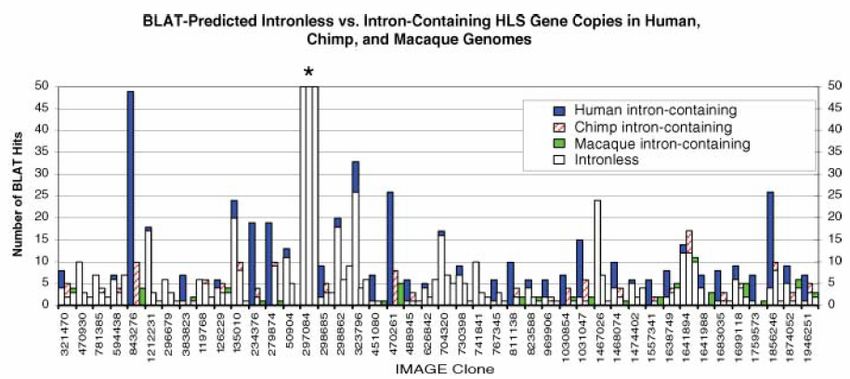
http://www.eva.mpg.de/genetics/images/chimp_human.jpg
• Sequenzierung des menschlichen Genoms (2001)
• Vergleiche zeigen, dass Genome zu 99,9% identisch sind und
sich nur in einzelnen Nukleotiden, sogenannten SNPs (Single
Nucleotide Polymorphisms) unterscheiden
• Sequenzierung des Schimpansen-Genoms (2005)
• Vergleich des menschlichen Genoms und des Schimpansen-
Genoms zeigt, dass sie zu 98,8% identisch sind
• Was macht den Menschen zum Menschen?
• Was macht den Schimpansen zum Schimpansen?
08.02.2007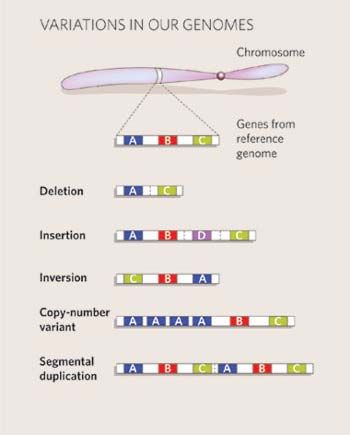
Bisherige Sichtweise auf “das” Genom
• Menschen unterscheiden sich durch kleine Variationen im Genom
(SNPs):
Person X: ...CGCTAGGATAGCTCTCTAGGATCGCCTCGATAGAGA...
Person Y: ...CGCTAGGATAGCTCTCTTGGATCGCCTCGATAGAGA...
davon gibt es ca. 10 Mio in der gesamten Menschheit
• Daneben gibt es große Veränderungen im Karyotyp:
z.B. Trisomie21
≥ 3Mb
im Mikroskop
sichtbar
08.02.2007
http://www.bio-pro.de/de/region/ulm/magazin/01155/index.html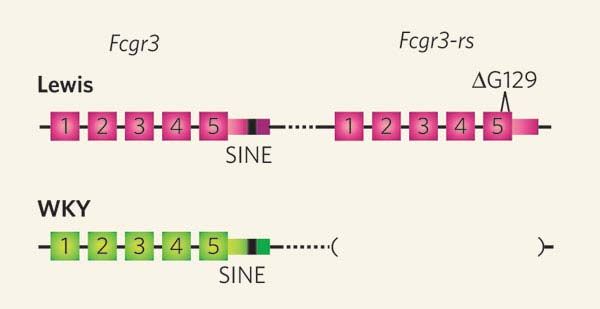
Gibt es noch andere Unterschiede neben SNPs?
• Bereits bekannt: in krankhaft veränderten Zellen (z.B. Tumor-
zellen) kommen einzelne Genregionen häufiger vor als in
gesunden Zellen
• Im Vergleich des menschlichen Genoms mit den Genomen
verschiedener Menschenaffen (Interspezies-Vergleiche) zeigt
sich:
– dass einzelne Gene in unterschiedlicher Kopienzahl vorliegen
können
– dass auch größere Regionen fehlen können, umorganisiert,
umgekehrt bzw. vervielfacht sein können
• Vergleiche zwischen den Genomen verschiedener (gesunder)
Menschen (Intraspezies-Vergleiche) zeigen ähnliche Ergebnisse
08.02.2007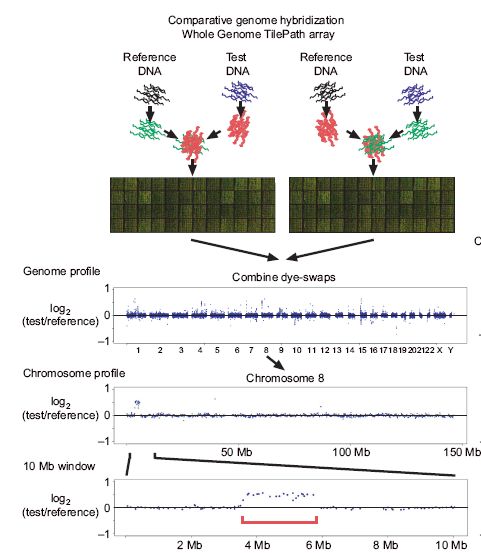
Neue Sichtweise auf “das” Genom • Es gibt sehr kleinen Unterschiede auf Nukleotidebene (
08.02.2007 E. Check, "Patchwork people", Nature 437, 1084-1086, 2005
Begriffsklärung
• Copy Number Variant (CNV):
– DNA-Segment mit einer Länge > 1kb
– kommt in variabler Anzahl vor in Bezug auf ein Referenz-
genom
– beinhaltet sowohl Löschungen (Deletionen) als auch
Duplikationen (Inversionen werden jedoch nicht dazuge-
rechnet)
– schließt somatische Umorganisationen aus, z.B. in Tumoren
• Copy Number Polymorphismus (CNP):
– wenn > 1% der Population Träger einer Variante ist
• Large-Scale Copy-Number Variation (LCV):
– CNVs, die größere Regionen umfassen (>100kb)
08.02.2007Welche Folgen hat das?
• Kann dramatischen Einfluß auf Stoffwechsel haben:
– Löschen einer DNA-Region kann zum Fehlen essentieller Gene
führen
– Extrakopien eines Gens können zur Überproduktion eines
Proteins führen
– Verschieben einer DNA-Region kann Gen-Regulation
durcheinanderbringen
• Variationen erhöhen eventuell Anfälligkeit für Krankheiten, sind
aber häufig nicht Ursache
• häufig sind jedoch Gene betroffen, die mit der Interaktion mit
der Umwelt in Verbindung gebracht werden, also z.B. Immun-
reaktion, Abbau von Medikamenten und Giften, Abwehr von
Pathogenen, Entzündungen
08.02.2007Beispiel: Copy Number Variation
E.Gonzalez et al., The influence of CCL3L1 gene-containing segmental duplications
on HIV-1/AIDS susceptibility. Science, 307(5714):1434-40, 2005.
• CCL3L1: HIV-1–suppressive
Chemokin
• Protein der Immunabwehr
• geringe Kopienzahl führt zu
erhöhter Infektionsanfällig-
keit
• höhere Kopienzahl führt bei
HIV-Infektion zu verzöger-
tem Ausbruch von AIDS
08.02.2007Beispiel: Inversion
H.Stefansson et al., A common inversion under selection in Europeans.
Nature Genetics, 37(2):129-37, 2005.
• Chr. 17: 900kb Region (H1) , die in unterschiedlicher
Orientierung vorkommen kann
• Inversion (H2) vor ca. 3 Mio Jahren entstanden
• betrifft ca. 20% der Europäer, wenige Afrikaner, (bisher) bei
keinen Ostasiaten gefunden
• Untersuchung in Island: Trägerinnen der Inversion haben mehr
Kinder ⇒ positive Selektion!
H1-Klone
H2-Klone
08.02.2007Krankheiten
• Hämophilie A (Bluterkrankheit): Inversion auf X-Chromosom
• Prader-Willi-Syndrom: Deletion auf Chromosom 15
• Nierenentzündung (Glomerulonephritis): CNV im Gen FCGr3
beeinflußt Anfälligkeit für Nierenentzündung, die zu Nieren-
versagen führen kann (zunächst in Ratten untersucht)
T.J.Aitman et al., Copy number polymorphism in Fcgr3 predisposes to
glomerulonephritis in rats and humans. Nature, 439(7078):851-5, 2006 08.02.2007Identifizierung von CNVs
z.B. HapMap, ENSEMBL
(öffentliche) Datenbanken
Durchsuchen von z.B. z.B. Experimenteller
Sequenzdaten am BLAT, Array CGH, Genom-Vergleich
Computer BLAST SNP Array im Labor
Experimentelle z.B. z.B.
FISH, Analyse am
Validierung SW-ARRAY,
PCR Computer
im Labor CBS
(öffentliche) Datenbanken
z.B. Database of Genomic Variation
08.02.2007Ziele und Hauptakteure
• Identifikation von CNVs in offenbar gesunden Individuen in
verschiedenen Populationen
• Entwicklung neuer Methoden zur genomweiten CNV-
Identifikation
• Vernetzung mit Daten aus anderen Datenbanken, Projekten,
Literatur etc.
• “Copy Number Variation Project”
am Sanger Center, UK
• “Database of Genomic Variants”
am TCAG (The Center for Applied Genomics), Canada
• “International HapMap Project”
• “Wellcome Trust Case Control Consortium” (WTCCC)
08.02.2007Identifizierung von CNVs basierend auf Sequenzdaten
M.C.Popesco et al., Human lineage-specific amplification, selection, and neuronal
expression of DUF1220 domains. Science, 313(5791):1304-7, 2006
• Vergleich Mensch, Schimpanse, Makake (als Outgroup)
• Start mit 134 cDNA-Sequenzen von HLS-Genen (Human Lineage
Specific)
• Durchführung von BLAT-Sequenzsuchen gegen das menschliche
Genom und gegen Draft-Sequenzen von Schimpanse und
Makake (Rhesusaffe)
• 86,4% der 134 Gene zeigten eine erhöhte Copy Number im
menschlichen Genom
• BLAT: BLAST-like Alignment Tool, entwickelt an der UCSC als
Teil des Genome Browsers zum schnellen Durchsuchen des
menschlichen Genoms
08.02.2007Identifizierung von CNVs basierend auf Sequenzdaten
• Gen MGC8902: 49 Kopien im Menschen, 10 im Schimpansen,
4 im Makaken (Rhesusaffe)
• enthält 6 DUF1220-Domänen (Funktion unbekannt)
• BLAT-Suche gegen andere Spezies liefert nur Säugetier-
sequenzen, insbesondere von Primaten
• stark exprimiert in Hirnregionen, die mit höheren kognitiven
Funktionen assoziiert werden
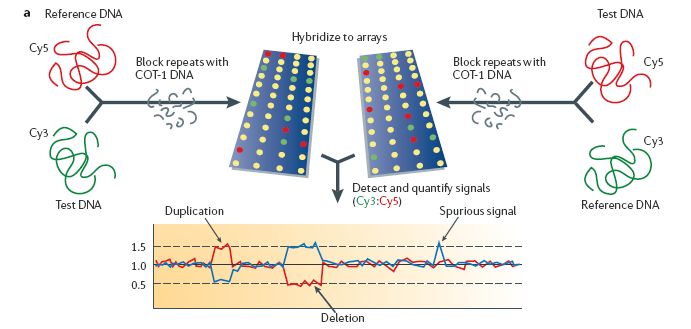
08.02.2007Identifizierung von CNVs basierend auf CGH
• Array-CGH (Comparative Genomics Hybridization)
• basiert auf der Annahme, dass die Copy Number proportional zur
Hybridisierungsintensität ist
• eine Veränderung im Intensitätsverhältnis deutet also auf eine
Vervielfältigung bzw. Deletion hin
• Array sollte dazu möglichst die euchromatischen Regionen des
Genoms abdecken
• aufgebrachte Proben (Test und Referenz) sind mit Farbstoff
markiert
• Repeats sind maskiert
• Euchromatin: weniger dicht gepackter Teil des Genoms, der die
meisten Gene enthält (Gegenteil von Heterochromatin)
08.02.2007Array-CGH (Comparative Genomics Hybridization)
L.Feuk et al., Structural variation in the human genome.
Nature Reviews Genetics, 7(2):85-97, 2006
08.02.2007Array CGH
Test/Referenz
Test: GATTACGGA
Referenz: GATTACGGA
GAT TAC GGA
Test: GATGGA
Referenz: GATTACGGA
GAT TAC GGA
Test: GATTACTACGGA
Referenz: GATTACGGA
GAT TAC GGA
08.02.2007Array CGH
• Vorteile:
– hohe Auflösung
– schnell
– geringe Kosten (hoffentlich in Zukunft)
• Nachteile:
– es gibt noch keine Arrays, die das komplette Genom
abdecken
– experimentelle Artefakte
08.02.2007R.Redon et al., Global variation in copy
number in the human genome. Nature,
444(7118):444-54, 2006
• 26.574 Klone auf Chip
• decken 93,7% der
euchromatischen
Regionen des mensch-
lichen Genoms ab
• Experimente für 82
Individuen (rechts
Vergleich zweier
männlicher Genome)
• log2 (Test/Referenz)
gegen Genom
(Chromosomen)
auftragen
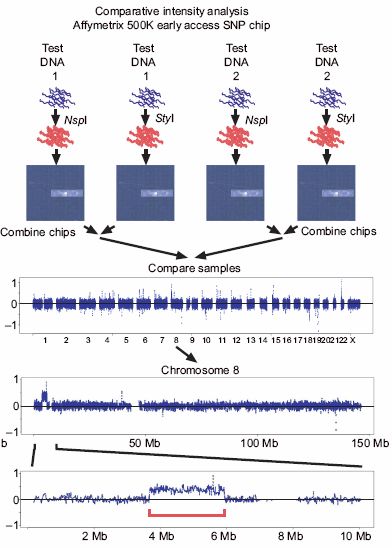
08.02.2007Affymetrix 500K EA
SNP Chip
• 474.642 SNPs
• je 2 Chips pro Probe mit
unterschiedlichen
Restriktions-Endonukle-
asen (NspI und StyI)
• Experimente für 15
Individuen
• log2 (Test1/Test2) gegen
Genom (Chromosomen)
auftragen
• Analyse mit SW-ARRAY
08.02.2007Strategien zum Finden von CNVs
Vergleich mit Test Referenz
Referenz
CNV
Vergleich mit
Test Ref1 Ref2 Ref3
gemittelten
Referenzen
CNV
Multiple paarweise Test1 Test2 Test3
Vergleiche (ohne Test2 CNV
Referenz) ⇒ Zusammen-
Test3 CNV CNV fassung
Test4 CNV CNV CNV
08.02.2007Wunsch und Wirklichkeit
• Rauschen in realen
Daten sehr groß
• selbst nach Entfernen
von Artefakten,
Normalisierung und
Mittelwertbildung
• Beispiel rechts:
Vergleich von
Chromosom 21 zweier
Individuen aus der
HapMap-Datenbank
• CGH-Daten frei
verfügbar am Sanger
Center
08.02.2007Problem mit Array CGH
• Sensitivität und Spezifität niedrig
⇒ für Einsatz in klinischer Diagnostik noch ungeeignet!?
CNV vorhergesagt Keine CNV vorhergesagt
CNV True positive False negative
Keine CNV False positive True negative
• Sensitivität: Fähigkeit, korrekterweise ein positives Ergebnis
vorherzusagen (hit rate, Trefferrate)
= True positive / (True positive + False negative)
• Spezifität: Fähigkeit, korrekterweise ein negatives Ergebnis
abzulehnen (false alarm rate, Selektivität)
= True positive / (True positive + False positive)
08.02.2007SW-ARRAY
T.Price et al., SW-ARRAY: a dynamic programming solution for the identification of copy-
number changes in genomic DNA using array comparative genome hybridization data.
Nucleic Acids Research, 33(11):3455-64, 2005
• Entwicklung einer Variante des Smith-Waterman Algorithmus
(paarweises lokales Sequenzalignment mit Dynamischer
Programmierung)
• zur Detektion von Deletionen und Duplikationen (bzw.
Vervielfältigungen), die als „Inseln“ bezeichnet werden
• Außerdem Angabe einer statistischen Signifikanz für die beste
„Insel“
• öffentlich verfügbares Programmpaket, in R implementiert
08.02.2007SW-ARRAY
• Subtraktion eines Schwellwertes von allen Intensitäts-Log-
Ratios
• Sei X(p) dann der justierte Score von Probe p.
• Der Score von Probe p bis Probe q ist dann:
• S(p) sei der Score einer „Insel“, die in Probe p endet und
B(p) die Probe, an der diese „Insel“ beginnt.
• Sei S(0) = 0
>0
• Rekursion:
08.02.2007SW-ARRAY
Beispiel
• Berechnungsformel des
Schwellwertes beruht
auf empirischen Daten
• Mittelwert der neuen
Werte soll < 0 sein
• neue Werte =
log ratios - Schwellwert
08.02.2007SW-ARRAY
Beispiel
• Berechnung der
statistischen
Signifikanz p einer
Insel
• zufälliges Permu-
tieren der Daten,
1000 mal
• bei jedem Durchlauf
die Anzahl t der
Inseln mit einem
höheren Score
zählen
• p = t / 1000
08.02.2007SW-ARRAY
• “Robustness” oder “Reliability”
• Schwellwert 100-mal kontinuierlich verändert
• zwischen median(X) und median(X)+0,4*mad(X)
• dabei für jede Position berechnen, wie oft sie sich in einer Insel
befindet
• Wert nahe 0: keine Änderung der Copy Number an dieser
Position, unabhängig vom Schwellwert
• Wert nahe 1: Änderung der Copy Number an dieser Position,
unabhängig vom Schwellwert
08.02.2007SW-ARRAY
Beispiel zum Mitrechnen
2
1
Score
0
-1
-2
Probe / Position
08.02.2007SW-ARRAY
Beispiel zum Mitrechnen
Insel
2
1
Score
0
-1
-2
Probe / Position
08.02.2007SW-ARRAY
• Wie muß man vorgehen, um mit diesem Verfahren Deletionen zu
finden?
• Die Ursprungsdaten mit -1 multiplizieren!
• Wie muß man vorgehen, um nicht nur eine Insel, sondern
weitere kleinere Inseln zu finden?
• Die Scores der bisher gefundenen Insel(n) auf 0 setzen und dann
das Verfahren wiederholen!
08.02.2007Weitere Methoden zum Finden von “Inseln”
• CBS (Circular Binary Segmentation):
– A.Olshen et al., Circular binary segmentation for the analysis of array-based
DNA copy number data. Biostatistics, 5(4):557-72, 2004
– Suche nach Positionen, an denen sich die Copy Number ändert;
Verwendung von t-Test und permutierten Referenzdaten
• CLAC (Cluster along chromosomes):
– P.Wang et al., A method for calling gains and losses in array CGH data.
Biostatistics, 6(1):45-58.2005
– Erzeugen eines Baums (hierarchisches Clustering) entlang jedes
Chromosoms; Auswahl “interessanter” Cluster
• HMM (Hidden Markov Model):
– S.P.Shah, Integrating copy number polymorphisms into array CGH analysis
using a robust HMM. Bioinformatics, 22(14):e431-9, 2006
•...
08.02.2007Experimentelle Untersuchung des Humangenoms
L.Feuk et al., Structural variation in the human genome.
Nature Reviews Genetics, 7(2):85-97, 2006
• Probleme:
– was soll als Standard- bzw. Referenz-Genom verwendet
werden?
– gerade die noch vorhandenen Lücken im menschlichen
Referenz-Genom befinden sich in der Nähe von strukturellen
Varianten
– selbst zwischen dem Referenz-Genom am NCBI und dem bei
Celera Genomics bestehen große Unterschiede
⇒ Anteil struktureller Varianten zum jetzigen Zeitpunkt schwer
einzuschätzen
08.02.2007Database of Genomic Variants on Human Genome
08.02.2007• projects.tcag.ca/variation/
• am "The Center for Applied Genomics", Kanada
• Sammlung struktureller Varianten im menschlichen Genom
• momentan auf phänotypisch gesunde Personen beschränkt
• Datenbestand am 1.Februar 2007:
– CNVs: 5150
– Inversionen: 77
– Daten aus 39 Publikationen
08.02.2007HapMap
08.02.2007HapMap
• www.hapmap.org
• Haplotyp-Mapping
• Arbeitsgruppen aus Kanada, China, Japan, Großbritannien,
Nigeria und den USA
• Auffinden von Genen, die mit Krankheiten des Menschen
assoziiert sind und der Wirkung von Medikamenten
• 270 Individuen aus 4 Populationen aus Europa, Afrika und Asien
• 30 Eltern-Kind-Trios der Yoruba aus Nigeria
• 30 Eltern-Kind-Trios europäischer Herkunft aus Utah, USA
• 45 nicht-verwandte Japaner aus Tokio
• 45 nicht-verwandte Han-Chinesen aus Peking
08.02.2007ENSEMBL
08.02.2007ENSEMBL
• www.ensembl.org
• Kooperation aus EMBL European Bioinformatics Institute (EBI)
und Wellcome Trust Sanger Center (WTSC)
• Datenbank und Softwaresystem zur Verwaltung und
(automatischen) Annotation kompletter (eukaryotischer)
Genome
• inzwischen 27 Genome - von der Mücke bis zum Elefanten
08.02.2007http://www.sanger.ac.uk/humgen/cnv/data/
(Länge)
(Häufigkeit)
08.02.2007Weitere Quellen
• Vortragsfolien von Tom Price, 2003
“Locating deletions and polysomy in genomic DNA microarray
data using the Smith-Waterman algorithm.”
http://itmat.upenn.edu/~tsprice/talks/Heidelberg.pdf
• Vortragsfolien von Chris Barnes, 2006
“Techniques for the detection of copy number variation using
SNP genotyping arrays”
http://www.newton.cam.ac.uk/webseminars/pg+ws/2006/scb/scbw02/1212/barnes/all.pdf
08.02.2007Sie können auch lesen

















































