LIQUORPROTEOMANALYSE BEI DER VILIUISK-ENZEPHALOMYELITIS - OPARU
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Universitätsklinikum Ulm
Klinik für Neurologie
Ärztlicher Direktor: Prof. Dr. med. A. C. Ludolph
LIQUORPROTEOMANALYSE
BEI DER
VILIUISK-ENZEPHALOMYELITIS
Dissertation
zur Erlangung des Doktorgrades der Medizin
der Medizinischen Fakultät der Universität Ulm
vorgelegt von
Daniel Grözinger
aus Heidenheim
2012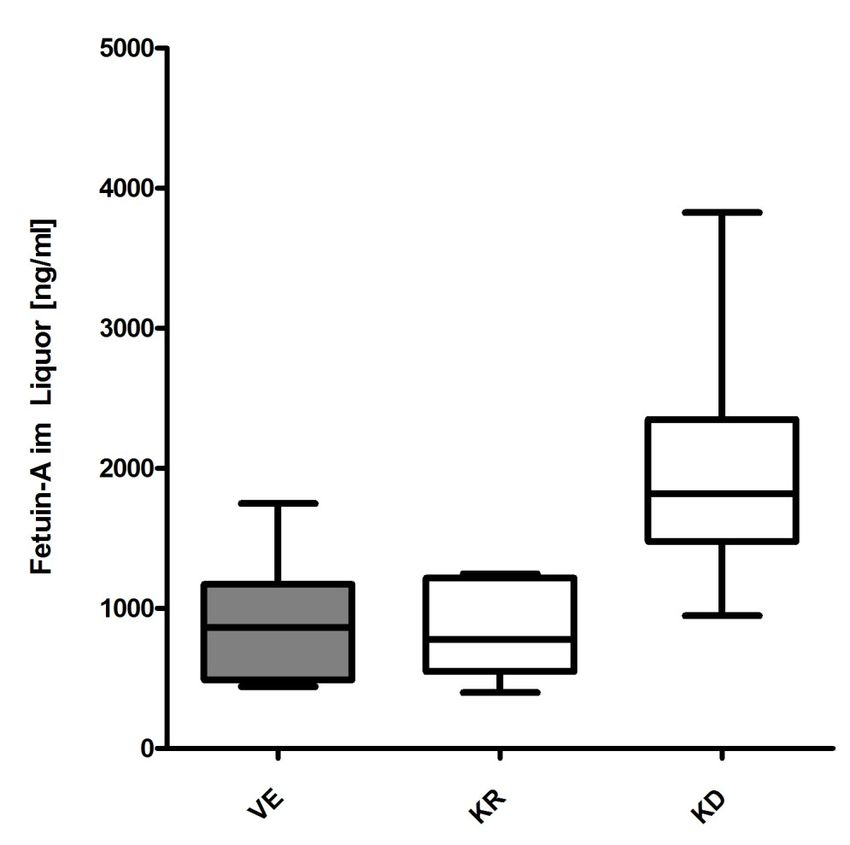
Amtierender Dekan: Prof. Dr. Thomas Wirth 1. Berichterstatter: Prof. Dr. Hayrettin Tumani 2. Berichterstatter: Prof. Dr. Thomas Mertens Tag der Promotion: 18. Oktober 2012
Seite I
INHALTSVERZEICHNIS
Inhaltsverzeichnis .................................................................................................... I
Abkürzungsverzeichnis ......................................................................................... III
1. Einleitung............................................................................................................ 1
1.1 Viliuisk-Enzephalomyelitis ............................................................................. 1
1.1.1 Geschichte und Epidemiologie ............................................................... 1
1.1.2 Klinik und Neuropathologie ..................................................................... 3
1.1.3 Pathogenese ........................................................................................... 5
1.2 Liquorproteomanalyse mit 2-D DIGE ............................................................ 6
1.2.1 Liquor cerebrospinalis ............................................................................. 6
1.2.2 Proteomanalyse ...................................................................................... 7
1.2.3 2-D DIGE ................................................................................................ 8
1.3 Aufgabenstellung und Zielsetzung ................................................................ 9
2. Material und Methoden ..................................................................................... 10
2.1 Material ....................................................................................................... 10
2.1.1 Chemikalien .......................................................................................... 10
2.1.2 Kits ........................................................................................................ 10
2.1.3 Geräte ................................................................................................... 11
2.1.4 Software ................................................................................................ 11
2.1.5 Verbrauchsmaterial ............................................................................... 11
2.2 Patientenproben .......................................................................................... 12
2.2.1 Patientenproben mit chronischer Viliuisk-Enzephalomyelitis ................ 12
2.2.2 Kontrollen aus Jakutien......................................................................... 12
2.2.3 Kontrollen aus Deutschland zur Validierung mittels ELISA ................... 13
2.3 Methoden .................................................................................................... 15
2.3.1 Vorbereitungen ..................................................................................... 15
2.3.2 2-D differenzielle fluoreszierende Gelelektrophorese (2-D DIGE) ........ 16
2.3.3 Validierung von Fetuin-A mit ELISA ...................................................... 24
3. Ergebnisse ....................................................................................................... 27
3.1 2-D DIGE Proteomanalyse .......................................................................... 27
3.1.1 Ausgewertete Gele ............................................................................... 27
3.1.2 Identifizierte Proteine ............................................................................ 28
Seite II
3.1.3 Dreidimensionale Darstellung eines Spots am Beispiel von Fetuin-A ... 30
3.1.4 Identifizierte Proteine ............................................................................ 31
3.2 Validierung von Fetuin-A mittels ELISA ...................................................... 36
3.2.1 Vergleich der Fetuin-A-Liquorkonzentration zwischen allen Gruppen... 36
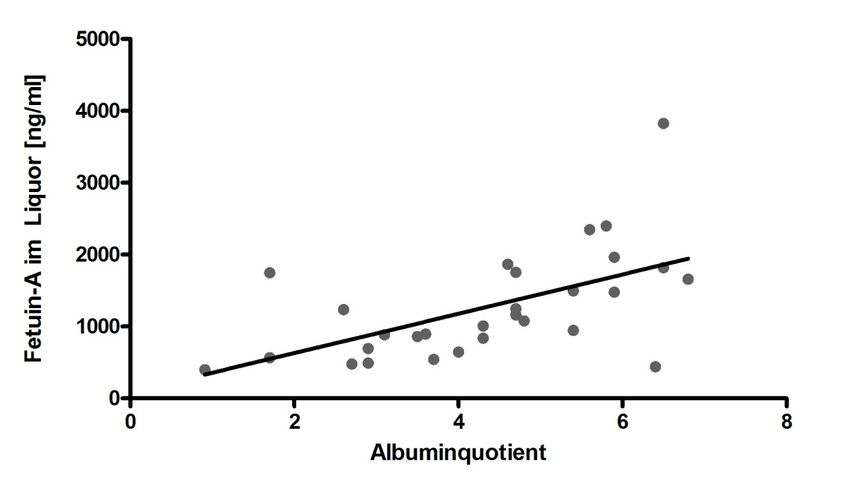
3.3.2 Korrelation der Fetuin-A-Liquorkonzentration mit Qalb........................... 37
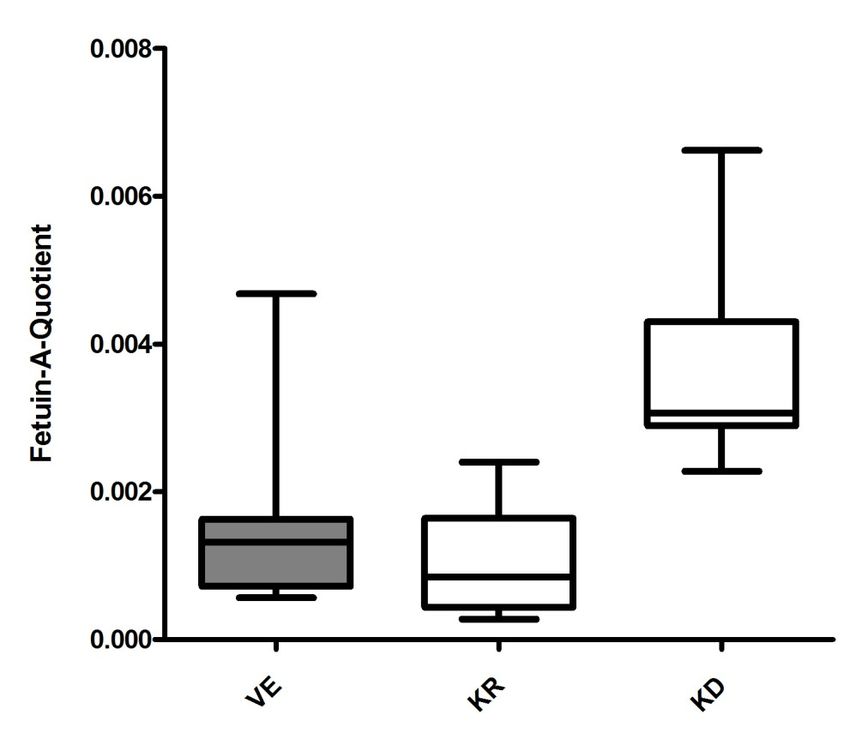
3.3.2 Vergleich des Fetuin-A-Quotienten zwischen allen Gruppen ................ 39
3.3.2 Vergleich des Fetuin-A-Index zwischen allen Gruppen......................... 40
4. Diskussion ........................................................................................................ 41
4.1 Liquoranalyse bei der Viliuisk-Enzephalomyelitis ........................................ 41
4.2 Methodik der 2-D DIGE ............................................................................... 42
4.3 Methodik des Fetuin-A ELISA ..................................................................... 46
4.4 Qualität der Proben und Auswahl der klinischen Gruppen .......................... 46
4.5 Bewertung der identifizierten Proteine ........................................................ 48
4.5.1 Transthyretin ......................................................................................... 48
4.5.2 Transferrin ............................................................................................ 49
4.5.3 Ceruloplasmin ....................................................................................... 51
4.5.4 Ectropic viral integration site 2B (EVI-2B) ............................................. 53
4.5.5 Factor VII active site mutant immunoconjugate .................................... 54
4.5.6 Apolipoprotein E.................................................................................... 55
4.5.7 α2-HS-Glycoprotein (Fetuin-A) ............................................................. 56
4.5.8 α-1-Antichymotrypsin Isoform 1 und α-1-Antitrypsin Isoform 1 ............. 58
4.5.9 Thyroid hormone receptor α.................................................................. 58
4.5.10 ADIR1, Torsin family 3 member A und ADIR2 .................................... 60
4.5.11 Keratin, Microfibril-associated Glycoprotein 4, Actinin α 1 Isoform 3,
Immunglobulinderivate und Serum-Albumin .................................................. 61
4.6 Schlussfolgerung und Ausblick ................................................................... 62
5. Zusammenfassung ........................................................................................... 64
6. Literaturverzeichnis .......................................................................................... 66
Danksagung ......................................................................................................... 86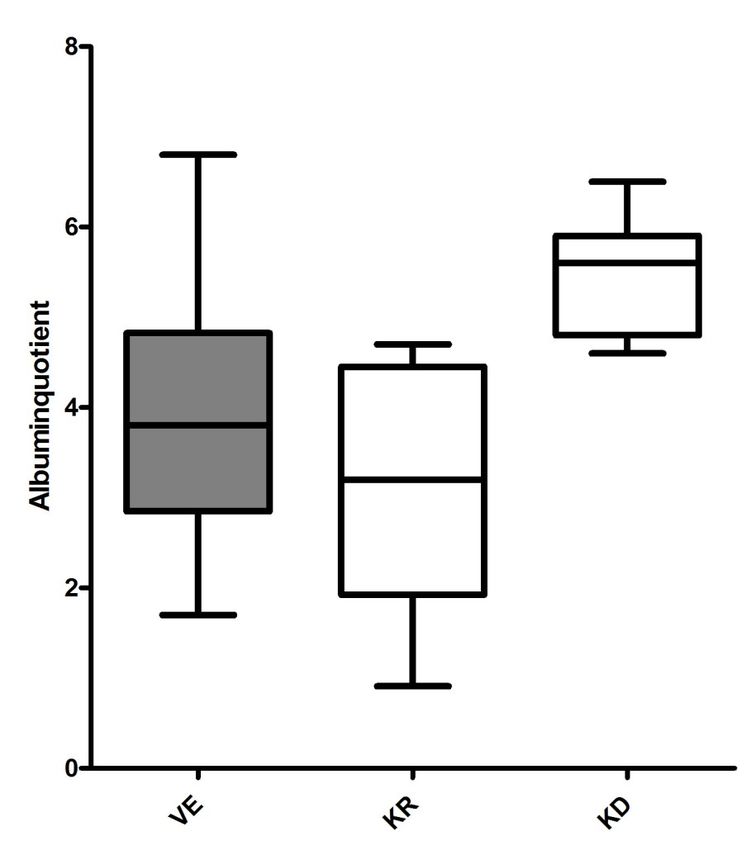
Seite III
ABKÜRZUNGSVERZEICHNIS
µg Mikrogramm
µl Mikroliter
°C Grad Celcius
2-D zweidimensional
2-D DIGE zweidimensionale differenzielle fluoreszierende
Gelelektrophorese
Adir ATP-dependent interferon responsive gene
APS Ammoniumperoxidsulfat
ATP Adenosintriphosphat
cAMP zyklisches Adenosinmonophosphat
cDNA komplementäre Desoxyribonukleinsäure
CHAPS 3-[(3-Cholamidopropyl)-dimethyl-ammonio]-1-propane
sulfonate
cm Zentimeter
COPD chronisch obstruktive Lungenerkrankung
dH2O destilliertes Wasser
DHB 2,5-Dihydroxybenzoesäure
DNA Desoxyribonukleinsäure
DTT Dithiothreitol
ELISA Enzyme-Linked Immunosorbent Assay
EVI-2B Ectropic viral integration site 2B
g Gramm
HCl Salzsäure
HDL High Density Lipoprotein
HIV-1 Humanes Immundefizienz-Virus 1
IL InterleukinSeite IV
IPG immobilisierter pH-Gradient
kDa Kilodalton
L Liter
LDL Low Density Lipoprotein
M Mol
m/z-Wert mass-to-charge-Wert
MALDI-TOF-MS matrix-assisted laser desorption/ionisation time-of-flight
Massenspektrometrie
mg Milligramm
ml Milliliter
mM Millimol
mm Millimeter
mm³ Kubikmillimeter
ng Nanogramm
nm Nanometer
NMWL nominal molecular weight limit
pH potentia Hydrogenii
ppm parts per million
Qalb Albuminquotient
RNA Ribonukleinsäure
rpm Umdrehungen pro Minute
SDS Sodium Dodecyl Sulfate
Temed N,N,N’,N’-Tetramethylethylendiamin
TNF-α Tumornekrosefaktor α
Tris Tris[hydroxymethyl]aminomethan NH2C(CH2OH)3
V Volt
VLDL Very Low Density LipoproteinSeite 1 1. EINLEITUNG 1.1 Viliuisk-Enzephalomyelitis 1.1.1 Geschichte und Epidemiologie Es war Richard Maak, ein russischer Ethnologe und Geograph, der vor über hundert Jahren erstmals über die Viliuisk-Enzephalomyelitis berichtete. Er entdeckte die Erkrankung in der ländlichen Region Sacha in Ost-Sibirien unter dem Volk der Jakuten [49]. Zunächst beschränkte sich die Verbreitung der Viliuisk-Enzephalomyelitis ausschließlich auf die Gegend um Wiljuisk am Fluss Wiljui, doch im Zuge von Bevölkerungsbewegungen nach dem Zweiten Weltkrieg breitete sich die Erkrankung entlang des Wiljui weiter aus und wurde später auch in den dichter besiedelten Gebieten um Jakutsk im Flussgebiet der Lena beschrieben [162]. Die Prävalenz der Viliuisk-Enzephalomyelitis betrug Mitte des vergangenen Jahrhunderts in einigen Dörfern in der Gegend um Wiljuisk über 1% und die jährliche Mortalität über 1 Promille [49]. Die jährliche Inzidenz belief sich zeitweise auf bis zu 8,8 pro 100.000 Einwohner, heute nur noch auf etwa 0,5 pro 100.000 Einwohner [94]. Insgesamt wird in der Literatur von etwa 400 nachgewiesenen und registrierten Fällen seit dem Beginn offizieller Aufzeichnungen 1950 berichtet [51]. Das Erkrankungsalter der dokumentierten Patienten schwankt zwischen 11 und 68 Jahren. Zu Beginn der Aufzeichnungen 1950 begann eine Erkrankung an der Viliuisk-Enzephalomyelitis durchschnittlich im Alter von 30,2 Jahren. Heute ist das Erkrankungsalter im Durchschnitt etwas höher, etwa bei 35 Jahren. Die Erkrankungshäufigkeit von Männern und Frauen ist heute ungefähr gleich hoch, früher trat die Viliuisk-Enzephalomyelitis bei Frauen häufiger auf [94].
Seite 2 Abbildung 1. Karte von Russland und Nachbarländern. Die Region im Rahmen ist in Abbildung 2 in Vergrößerung dargestellt. Abbildung 2. Karte der Region um Wiljuisk. Die Viliuisk-Enzephalomyelitis tritt vor allem in den ländlichen Gebieten um Wiljuisk entlang des Flusses Wiljui auf. Beschrieben wurden auch Fälle in der Nähe von Jakutsk, der dichter besiedelten Hauptstadt der Region Sacha.
Seite 3 1.1.2 Klinik und Neuropathologie Die Viliuisk-Enzephalomyelitis zeigt sehr vielfältige Symptome, deshalb ist die Abgrenzung zu anderen neuroentzündlichen und neurodegenerativen Erkrankung nicht immer leicht, gelingt jedoch dennoch meist im Verlauf der Krankheit oder nach dem Tod durch neuropathologische Untersuchungen. Um die Beschreibung der Symptomatik zu erleichtern, ist eine klinische Klassifikation in einen subakuten, einen langsam fortschreitenden und einen chronischen Verlauf hilfreich [49]. Goldfarb et al. beschreiben, dass die subakute Verlaufsform in der Regel mit hohem und lange anhaltendem Fieber beginnt. Neben Muskel- und Gelenkschmerzen treten Meningismus-Zeichen wie starke Kopfschmerzen, Nackenmuskelsteife und Hirnnervenzeichen auf. Nach knapp einem Monat findet sich eine zunehmende Bradykinesie, Dysarthrie und schließlich eine spastische Tetraparese. Die Patienten werden komatös und sterben innerhalb von zwei bis 24 Monaten [49, 51]. Neuropathologischer Hauptbefund sind zahlreiche kleine nekrotische Herde mit bis zu 0,4 mm Durchmesser, die vor allem in der grauen Substanz des gesamten Zentralnervensystems auftreten. In den Meningen und perivaskulär finden sich inflammatorische Infiltrate, vor allem durch T- Lymphozyten [114]. McLean et al. konnten weder mit Spezialfärbungen noch durch Elektronenmikroskopie Erreger wie zum Beispiel Protozoen, Bakterien oder Viren nachweisen [114]. Die meisten Neudiagnosen der Viliuisk-Enzephalomyelitis zeigen einen langsam fortschreitenden Krankheitsverlauf. Auch diese Form der Erkrankung beginnt laut Goldfarb et al. mit einer akuten Phase mit Fieber und Enzephalitis ähnlich der subakuten Verlaufsform. Nach einigen Wochen bilden sich die Symptome beinahe vollständig zurück, um dann nach einiger Zeit wieder aufzutreten und zu persistieren [51]. Es finden sich Zeichen einer Beteiligung des gesamten zentralen Nervensystems, wie zum Beispiel häufig eine spastische Tetraparese, Gang- und Gangartstörungen und eine Sphinkterinkontinenz. Es treten Veränderungen der Persönlichkeit und eine zunehmende Apathie auf. Im weiteren Verlauf entwickelt sich eine schwere Demenz. Die Krankheit führt innerhalb weniger Jahre zum Tod,
Seite 4
meist durch Niereninsuffizienz oder Pneumonien [49]. Neuropathologisch tauchen
nekrotische Herde etwas weniger häufig auf als bei der subakuten Verlaufsform,
sind jedoch trotzdem meist von ähnlichem Charakter. Einige Herde sind zentral
eingeschmolzen und zeigen eine Reaktion der Gliazellen in der Umgebung. Zum
Teil beginnen die Herde zu konfluieren. In der Nähe der Entzündungsherde findet
sich teilweise eine sekundäre Demyelinisation [114].
In einigen Fällen verläuft die Viliuisk-Enzephalomyelitis chronisch. Dann ist die
akute Phase nicht oder nur sehr milde ausgeprägt. Klinisch findet sich ein dem
langsam fortschreitenden Typ sehr ähnlicher Verlauf. Die Progression der
Krankheit stoppt jedoch in einem meist späten Stadium. Die Patienten leiden in
der Regel unter schwerster Demenz. Sie leben teilweise noch viele Jahre bis
Jahrzehnte [49, 51]. Im Spätstadium zeigen sich neuropathologisch kleine,
multifokale Zysten mit gliotischem Saum. Es gibt keine aktiven nekrotisierenden
Herde mehr. Der zerebrale Kortex ist atroph [114].
Tabelle 1: Übersicht der verschiedenen Verlaufsformen und der wichtigsten Symptome der
Viliuisk-Enzephalomyelitis. ++++ = Symptom immer vorhanden. +++ = Symptom meistens
vorhanden. ++ = Symptom gelegentlich vorhanden. + = Symptom selten vorhanden. - = Symptom
nie vorhanden.
subakut langsam chronisch
fortschreitend
Fieber ++++ +++ ++
Meningismus +++ ++ -
Koma +++ ++ +
Euphorie - ++ +++
Demenz - +++ +++
Dysarthrie - +++ +++
Pyramidenbahnzeichen ++++ ++++ ++++Seite 5 1.1.3 Pathogenese Obwohl die Viliuisk-Enzephalomyelitis in den letzten Jahrzehnten von vielen verschiedenen Forschungsgruppen bearbeitet wurde, konnte die Pathogenese bis heute nicht eindeutig geklärt werden. Vladimirtsev et al. haben eine Häufung der Erkrankung in Familien nachgewiesen, fanden aber keinen Hinweis auf einen bestimmten Vererbungsmodus. Außerdem gibt es Berichte über eine Übertragung auch auf Nicht-Verwandte [94]. Risikofaktor scheint deshalb weniger die Vererbung, sondern eher der prolongierte Kontakt mit einem Erkrankten zu sein [162]. Auch eine Untersuchung von 17 häufigen Polymorphismen in acht mit Entzündungen assoziierten Genen zeigte keine Assoziation zwischen Genpolymorphismen und dem Risiko, an der Viliuisk- Enzephalomyelitis zu erkranken [127]. Obwohl epidemiologische Daten auf eine infektiöse Genese der Viliuisk- Enzephalomyelitis hindeuten [162], ließ diese sich bisher nicht zweifelsfrei bestätigen. Eine Infektion oder der Kontakt mit Borrelia burgdorferi korreliert nicht mit der Erkrankung an der Viliuisk-Enzephalomyelitis [150]. Ein Kandidat für eine virale Ursache der Viliuisk-Enzephalomyelitis ist das Viliuisk-Virus, ein Cardiovirus, das im Liquor eines Erkrankten nachgewiesen wurde [28, 98, 99]. Wahrscheinlich ist der Nachweis von diesem Virus jedoch auf eine Kontamination zurückzuführen, denn es wurden ausgedehnte in-vivo-Maus- und Zellkultur-Passagen zum Nachweis benötigt und es konnte eine phylogenetisch enge Verwandtschaft zwischen dem Viliuisk-Virus und dem Theilovirus der Nagetiere nachgewiesen werden [135]. Ein weiterer Kandidat für eine virale Genese der Viliusik- Enzephalomyelitits war das KPN-Virus, das sich jedoch als 5S-RNA von Acanthamoeba castellanii heraustellte [35] und wahrscheinlich auch eine Kontamination ist [49]. Obwohl die multiplen nekrotischen Herde in der Neurohistologie von Patienten, die an der Viliuisk-Enzephalomyelitis verstorbenen sind, denen einer viralen Enzephalitis ähneln, passt das histologische Gesamtbild dennoch zu keiner bekannten viralen neurologischen Erkrankung [114].
Seite 6 Lee et al. legen dar, dass die Viliuisk-Enzephalomyelitis zeitweise in Bevölkerungsgruppen auftrat, die sich sowohl genetisch als auch geographisch von der ursprünglich betroffenen Population um Wiljuisk unterschieden [94]. Damit scheinen die in der russischen Literatur diskutierte geobiochemische Hypothese, die eine Assoziation der Erkrankung mit der Zusammensetzung des Erdbodens im Gebiet des Sees Mastakhs beim Fluss Wiljui und die ökologische Hypothese, die einen Zusammenhang der Viliuisk-Enzephalomyelitis mit dem lokalen Ökosystem und dem dadurch erleichterten zirkulieren eines auslösenden Agens postuliert, unwahrscheinlich [94]. Ein weiterer Vorschlag zur Pathogenese ist in der russischen Literatur die große Verbreitung von Immundefizienzerkrankungen in der Bevölkerung, die anfälliger für die Viliuisk-Enzephalomyelitis machen solle. Auch diese Hypothese lehnen Lee et al. aus den oben genannten Gründen ab [94]. Die neuropathologischen Veränderungen bei der Viliuisk-Enzephalomyelitis passen nicht zu bekannten entzündlichen Mikroinfarkt-Syndromen wie dem Systemischen Lupus erythematodes, dem Antiphospholipidsyndrom und anderen autoimmunen systemischen Bindegewebserkrankungen und Vaskulitiden [114]. 1.2 Liquorproteomanalyse mit 2-D DIGE 1.2.1 Liquor cerebrospinalis Liquor cerebrospinalis wird größtenteils im Plexus choroideus als Ultrafiltrat des Blutes gebildet [113]. Er zirkuliert in den inneren und äußeren Liquorräumen des Gehirns und des Rückenmarks und wird überwiegend durch die Arachnoidalzotten rückresorbiert. Etwa 80 % der Proteine im Liquor stammen aus dem Blut [157]. Sie werden durch zahlreiche Transportsysteme über die Blut-Hirn-Schranke transportiert [42]. Der dadurch entstehende Blut-Liquor-Gradient einzelner Proteine ist von verschiedenen Faktoren wie zum Beispiel Molekülgröße, Molekülladung und Blutkonzentration der Proteine abhängig [157]. Ein weiterer wichtiger Einflussfaktor auf die Proteinkonzentration im Liquor ist die Funktion der Blut-Liquor-Schranke [42]. Weil Albumin beinahe vollständig in der Leber und kaum im Gehirn gebildet wird, kann der Liquor-Serum-Quotient von Albumin Qalb die Schrankenfunktion gut beschreiben [16, 157].
Seite 7 Etwa 20 % der Liquorproteine werden intrathekal oder im Hirngewebe gebildet. Liquor hat engen anatomischen Kontakt mit der interstitiellen Flüssigkeit des Gehirns. Biochemische Veränderungen der Proteinzusammensetzung im Gehirn bilden sich daher auch im Liquor ab [11, 157]. Viele neurologische Erkrankungen zeigen sowohl eine Veränderung der Proteinexpression und des Proteinumsatzes, als auch eine post-translationale Modifikation einzelner Proteine im Gehirngewebe und damit auch im Liquor [138]. Weil manche dieser Veränderungen relativ typisch und spezifisch für einzelne Erkrankungen sind [96, 97], ist die Liquoruntersuchung ein wichtiges Instrument in der neurologischen Diagnostik und Forschung. Liquor wird meistens durch Lumbalpunktion gewonnen, ein zwar invasives, aber bei korrekter Durchführung mit wenig Komplikationen behaftetes Verfahren [12]. 1.2.2 Proteomanalyse Proteine haben in lebenden Organismen zahlreiche essentielle Funktionen, unter anderem als Enzyme, zur Kommunikation und als Strukturelemente. Das Proteom, ein von Marc L. Wilkins geprägter Begriff [169], besteht aus der Gesamtheit aller Proteine eines Lebewesens, einer Zelle oder eines anderen lebendigen Kompartiments zu einem bestimmten Zeitpunkt. Im Gegensatz zum Genom, das relativ statisch ist, ändert sich das Proteom ständig in seiner Zusammensetzung. Es ist vielen komplexen Regulationsmechanismen unterworfen und wird von externen Faktoren wie zum Beispiel Krankheit, Belastung und Medikamenten beeinflusst. In den Veränderungen der Zusammensetzung des Proteoms bildet sich darum die Interaktion zwischen dem lebenden Organismus und seiner Umwelt dynamisch ab [104]. Die Proteomanalyse ist ein vielversprechender Forschungszweig, um sowohl die Pathogenese von Krankheiten als auch Möglichkeiten der Diagnostik und der Therapie zu erforschen [72, 73, 177].
Seite 8 1.2.3 2-D DIGE Mit der zweidimensionalen Gelelektrophorese lassen sich viele Proteine gleichzeitig nach ihrem isoelektrischen Punkt, ihrer Löslichkeit und ihren Massenunterschieden auftrennen. Durch eine anschließende Massen- spektrometrie können sie identifiziert werden. Damit ist die zweidimensionale Gelelektrophorese eine der effektivsten Techniken der Proteomic-Forschung [53]. Eine weitere Verbesserung stellt die 2-D DIGE dar. Bei ihr werden die Proteine vor der Gelelektrophorese mit fluoreszierenden Farbstoffen markiert. Dadurch können bis zu drei Proben gleichzeitig in einem einzigen Gel elektrophoretisch aufgetrennt werden. Dies erhöht die Leistungsfähigkeit, die Reabilität und die Sensitivität der Methode [111]. Abbildung 3: Ablaufschema einer Proteomanalyse mittels zwei dimensionaler differenzieller fluoreszierender Gelelektrophorese.
Seite 9 1.3 Aufgabenstellung und Zielsetzung Viele wissenschaftliche Fragestellungen in Bezug auf die Viliuisk- Enzephalomyelitis sind noch nicht beantwortet. Obwohl die Erkrankung immer tödlich ist und lokal eine sehr hohe Prävalenz erreicht, ist die Pathogenese nicht geklärt und es gibt weder ursächliche Behandlungsmethoden noch Möglichkeiten der Prävention. In der Region, wo die Viliuisk-Enzephalomyelitis gehäuft auftritt, finden sich viele andere neurodegenerative Erkrankungen. Eine Abgrenzung zu klinisch ähnlichen Krankheiten und eine sichere Frühdiagnose gestalten sich deshalb oft schwierig [50]. Ziel der vorliegenden Arbeit ist es, das Liquorproteom von Erkrankten mittels der 2-D DIGE Methode mit dem Proteom einer Kontrollgruppe zu vergleichen und dadurch in ihrer Konzentration signifikant veränderte Proteine zu entdecken. Denkbar wäre es, diese Proteine dann einzeln oder in Kombination als Biomarker zur frühen, sensitiven und spezifischen Diagnostik und Verlaufsprognose der Viliuisk-Enzephalomyelitis zu verwenden. Außerdem könnten Konzentrationsunterschiede einzelner Proteine Ansatzpunkte zur weiteren Erforschung der Pathogenese liefern und vielleicht dadurch langfristig zur Entwicklung therapeutischer und präventiver Optionen beitragen.
Seite 10
2. MATERIAL UND METHODEN
2.1 Material
2.1.1 Chemikalien
1-Butanol Merck
2-Iodacetamid Merck
Aceton AppliChem
Acrylamid-Bis, Fertiglösung 30% Merck
Agarose Amersham
0,8% Ampholyt (pH 3-10) Merck
APS (Ammoniumperoxidsulfat) Merck
Bromphenolblau Merck
CHAPS (3-[(3-Cholamidopropyl)-dimethyl-
ammonio]-1-propane sulfonate) 4% Amersham
DTT (Dithiothreitol) Sigma-Aldrich
Ethanol Merck
Glycin AppliChem
Glycerin Roth
Harnstoff Riedel-de Haën
L-Lysin Sigma
Methanol Merck
RotiBlue colloidal Roth
Salzsäure (HCl) Merck
SDS (Sodium Dodecyl Sulfate) Serva
Temed (N,N,N’,N’-Tetramethylethylendiamin) Merck
Thioharnstoff Merck
Tris (Tris[hydroxymethyl]aminomethan NH2C(CH2OH)3) USB Corporation
Ecotainer ® Aqua B. Braun (destilliertes Wasser) B. Braun
2.1.2 Kits
Aurum Serum Protein Mini Kit Bio-Rad
CyDye DIGE Fluor minimal dyes (Cy2, Cy3, Cy5) AmershamSeite 11 Human Fetuin-A ELISA Bio-Vendor Ultrafree®-0.5, Centrifugal Filter Devices (5K NMWL) Millipore ImmobilineTM DryStrip (IPG-Streifen, pH = 3-10, 18 cm) Amersham 2.1.3 Geräte Biofuge fresco (Kühlzentrifuge) Heraeus BN ProSpec® Nephelometer Siemens Electrophorese Power Supply EPS 601 Amersham Ettan DALTsix (Elektrophoresekammer) Amersham Ettan DIGE Imager GE Healthcare Feinwaage Sartorius Gelgießkammer Amersham Labofuge 400R Heraeus MultiTemp III (Kühlanlage für Elektrophorese) Pharmacia Biotech Magnetrührer Janke & Kunkel Mikrowelle Philips pH-Indikatorstäbchen, pH = 6,5-10 Merck Protean IEF Cell Bio-Rad Vortex MS2 Minishaker IKA Voyager DE-STR MALDI/TOF Massenspektrometer Biosystems 2.1.4 Software DeCyder™ Differential Analysis Software (Version 5.0) Amersham 2.1.5 Verbrauchsmaterial Pipetten Eppendorf Pipettenspitzen Eppendorf Safe Lock Tubes (Eppendorfreaktionsgefäß) Eppendorf
Seite 12 2.2 Patientenproben 2.2.1 Patientenproben mit chronischer Viliuisk-Enzephalomyelitis Es standen Liquores und Seren von zehn Patienten aus Jakutien mit der Diagnose einer chronischen Viliuisk-Enzephalomyelitis zur Verfügung. Sowohl die Probengewinnung als auch die neurologische Evaluation der Patienten erfolgte im Rahmen einer Expedition nach Jakutien vom 23. August bis zum 9. September 2006 durch Prof. A. C. Ludolph, Prof. J. Kassubek, Prof. H. Tumani von der Universität Ulm und Prof. A. Storch und Dr. A. Hermann von der Technischen Universität Dresden. Die Diagnose wurde nach sorgfältiger neurologischer Untersuchung und Ausschluss ähnlicher neurologischer Krankheiten gestellt. Alle Patienten litten unter einer chronisch progressiven sensorischen Ataxie, jeweils neun von zehn unter Gangapraxie, Harninkontinenz und spastischen Paresen. Von zwei Erkrankten war das Alter nicht eindeutig eruierbar, die restlichen waren im Mittel 53,5 Jahre, der jüngste 44 Jahre und der älteste 69 Jahre alt. Neun der zehn Proben stammten von männlichen Patienten. Der Mittelwert des Albuminqotienten der Proben betrug 3,98, der Median 3,8 mit einer Spannbreite von 1,7 bis 6,8. 2.2.2 Kontrollen aus Jakutien Als Kontrollen dienten insgesamt acht nach Alter und Albuminquotient gematchte Serum- und Liquorproben aus Jakutien, die ebenfalls von dem Expeditionsteam im Sommer 2006 gewonnen wurden. Drei der Probanden waren neurologisch unauffällig, zwei litten unter einer subkortikalen vaskulären Enzephalopathie. Jeweils einer der Probanden hatte Migräne, Multiple Sklerose und eine autosomal- dominante hereditäre spastische Paraplegie. Die zwei Frauen und sechs Männer in der Kontrollgruppe aus Jakutien waren zwischen 17 und 67 Jahre alt, das Durchschnittsalter betrug 46 Jahre. Der Albuminquotient bewegte sich im Bereich zwischen 0,91 und 4,7. Im Mittel betrug er 3,1, der Median belief sich auf 3,2.
Seite 13 Abbildung 4: Das Expeditionsteam in Jakutien. Die Gewinnung der Proben und die neurologische Evaluation erfolgten im Sommer 2006 durch Prof. A. C. Ludolph, Prof. A. Storch, Paul Ludolph, Prof. J. Kassubek, Dr. A. Hermann und Prof. H. Tumani. 2.2.3 Kontrollen aus Deutschland zur Validierung mittels ELISA Zur Validierung von Fetuin-A mittels ELISA wurde eine zusätzliche Kontrollgruppe gesunder Patienten aus Deutschland herangezogen. Von den insgesamt 11 zwischen 36 und 63 Jahren alten Probanden war einer weiblich. Das Durchschnittsalter betrug 50,6 Jahre. Die Albuminquotienten mussten mit den Proben aus Jakutien nicht gematcht werden, da sie später in der Ergebnisdiskussion rechnerisch berücksichtigt werden können. Der Albuminquotient der deutschen Kontrollgruppe bewegte sich zwischen 4,6 und 6,5 und betrug im Schnitt 5,6 und im Median ebenfalls 5,6.
Seite 14
Tabelle 2. Demographische Daten und Liquorparameter der Probandengr uppen. VE =
Patienten mit Erkrankung an der Viliuisk -Enzephalomyelitis. KR = Kontrollprobanden aus
Russland. KD = Kontrollprobanden aus Deutschland. Q alb = Albuminquotient.
VE KR KD
Anzahl der
Probanden 10 8 11
(weiblich/männlich) (1 / 9) (2 / 6) (1 / 10)
Alter
Median 52 45,5 53
(Spannbreite) (44 - 69) (17 - 67) (36 - 63)
Qalb (x 0,001)
Median 3,8 3,2 5,6
(Spannbreite) (1,7 - 6,8) (0,91 - 4,7) (4,6 - 6,5)
Oligoklonale 7 von 10 3 von 8 0 von 11
Banden im LiquorSeite 15 2.3 Methoden 2.3.1 Vorbereitungen Herstellung des Labellingpuffers Um 5 ml Labellingpuffer herzustellen wurden 2,4 g Harnstoff, 0,2 g CHAPS und 18,2 g Tris zusammengegeben und mit dH2O auf 5 ml aufgefüllt. Anschließend wurde der Puffer mit HCl auf pH 8,5 titriert. Herstellung des Fokussierpuffers für die isoelektrische Fokussierung Es wurden durch sorgfältiges Mischen von 4,2 g Harnstoff mit 1,5 g Thioharnstoff, 0,2 g CHAPS und 80 µl Ampholyt insgesamt 10 ml Fokussierpuffer hergestellt. Um den Puffer etwas anzufärben wurden etwa 20 µl 1%-ige Bromphenolblaulösung (1% Bromphenolblau in 50% Ethanol und 50% dH2O gelöst) dazugegeben. Der Fokussierpuffer wurde bis zur weiteren Verwendung bei -25 °C eingefroren. Herstellung des Laufpuffers für die Gelelektrophorese Um 5 L 10-fachen Laufpuffer herzustellen wurden 151 g Tris und 721 g Glycin in etwa 4 L destilliertem Wasser aufgelöst und dann unter dem Abzug 50 g SDS zugegeben. Anschließend wurde die Lösung mit destilliertem Wasser auf 5 L aufgefüllt und für einige Stunden gerührt. Da für die Gelelektrophorese 2-facher und 1-facher Laufpuffer benötigt werden, wurde der 10-fache Laufpuffer entsprechend mit destilliertem Wasser verdünnt. Gießen der Acrylamid-Gele Für die Gele wurde zunächst ein 1,5 M Tris-HCl-Puffer hergestellt. Dazu wurden 182 g Tris in 700 ml destilliertem Wasser vollständig gelöst und dann unter pH- Kontrolle mit konzentrierter Salzsäure auf einen pH-Wert von 8,8 titriert. Anschließend wurde der Puffer mit destilliertem Wasser auf ein Gesamtvolumen von 1 L aufgefüllt. Bis zur weiteren Verwendung konnte der 1,5 M Tris-HCl-Puffer im Kühlschrank gelagert werden.
Seite 16 Außerdem wurde noch eine 10%-ige SDS-Lösung aus SDS und destilliertem Wasser hergestellt. Dann wurde die Gelgießkammer mit gründlich gereinigten Glasplatten bestückt. Anschließend wurden 150 ml des 1,5 M Tris-HCl-Puffers mit 237 ml destilliertem Wasser, 6 ml 10%-iger SDS-Lösung und 200 ml Bis-Acrylamid-Stammlösung gut gemischt. Zum Schluss wurden noch 10%-ige Ammoniumperoxidisulfat-Lösung (1 g Ammoniumperoxidisulfat in 9 ml dH2O gelöst) und frisch angesetzte 10%-ige Temed-Lösung (100 µl Temed mit 900 µl dH2O gemischt) dazugegeben und das Ganze kurz gerührt. Sofort danach wurden mit Hilfe eines Trichters die Kammern zwischen den Glasplatten zügig mit der noch flüssigen Mischung befüllt und dünn mit n-Butanol überschichtet. Nach knapp einer Stunde waren die Gele ausgehärtet und konnten nach vorsichtigem Abgießen des n-Butanols mit destilliertem Wasser sorgfältig gereinigt werden. Die Gele wurden bis zur weiteren Verwendung in feuchte Tücher eingewickelt und in einem Kühlschrank bei 4 °C gelagert. 2.3.2 2-D differenzielle fluoreszierende Gelelektrophorese (2-D DIGE) Unter gleichem Vorgehen wurden drei Gele mit jeweils Patienten- und Kontrollproben angefertigt. Dadurch ließ sich eine hohe Reabilität der Ergebnisse erreichen. Patienten- und Kontrollliquores wurden jeweils gepoolt, aufbereitet, mit Farbstoff markiert und dann durch 2-D Gelelektrophorese aufgetrennt. Anschließend wurden die Gele gescannt, die Daten am Computer ausgewertet und interessante Spots gepickt. Durch Massenspektrometrie und Vergleich der Werte mit Referenzdatenbanken konnten die Proteine identifiziert werden. Konzentrierung und Entsalzung des Liquors Die Liquorproben wurden aufgetaut und die Kühlzentrifuge auf 4 °C gekühlt. Zur Aufkonzentrierung der Liquores wurden Ultrafree Centrifugal Filter Devices verwendet. Sie bestehen aus einer Filtereinheit mit einer senkrechten 5K-NMWL- Membran, die Proteine unter 5 kDa passieren lässt. Damit der Filter nicht austrocknet, wurden 400 µl destilliertes Wasser in das Sammelgefäß vorgelegt. Anschließend konnten in die Filtereinheit 500 µl Liquor
Seite 17 eingefüllt werden. Das Gefäß wurde gut verschlossen und in der Kühlzentrifuge bei 10.000 rpm für etwa zehn Minuten zentrifugiert. Das Filtrat im Sammelgefäß wurde verworfen und erneut durch 400 µl dH2O ersetzt. Dann wurde die Filtereinheit wieder mit Liquor derselben Probe befüllt und anschließend zentrifugiert. Diese Arbeitsschritte wurden sooft wiederholt, bis jede Probe um etwa das 20- fache konzentriert war. Danach wurden die Proteine durch vorsichtiges Auf- und Abpipettieren von der Filtermembran gelöst und jede Probe gesondert bei -80 °C eingefroren. Aufreinigung des Liquors Im Liquor finden sich relativ große Mengen an Albumin und Immunglobulinen, welche nach der 2-D Gelelektrophorese möglicherweise andere interessante Proteine mit ähnlichem isoelektrischen Punkt und ähnlicher Masse überdecken. Deshalb wurden Albumin und Immunglobuline aus den Proben weitestgehend entfernt. Dazu wurde das Aurum Serum Protein Mini Kit verwendet. Zuerst mussten die Säulen vorbereitet werden. Sie wurden für fünf Minuten senkrecht stehen gelassen, bis sich durch die Schwerkraft die blaue Lösung gesetzt hatte. Dann wurde der untere Deckel entfernt, damit die Flüssigkeit ablaufen konnte. Anschließend musste die Säule zweimal mit jeweils 1 ml Bindungspuffer gewaschen und dann in einem leeren Eppendorfreaktionsgefäß 20 Sekunden lang bei 10.000 rpm trockenzentrifugiert werden. Zum Schluss wurde die Säule mit einem Deckel unten wieder verschlossen. Nun wurden 100 µl der Probe mit 140 µl Bindungspuffer gut vermischt. 200 µl davon wurden auf die Säule gegeben. Nach vorsichtigem Schütteln wurde die Säule senkrecht stehen gelassen. Nach fünf und nach zehn Minuten wurde sie erneut leicht geschüttelt. Als insgesamt 15 Minuten verstrichen waren, wurde der untere Deckel entfernt und die Säule in einem Eppendorfreaktionsgefäß 20 Sekunden lang bei 10.000 rpm in der Kühlzentrifuge zentrifugiert. Die 40 µl Restmischung wurden nochmals mit 160 µl Bindungspuffer gemischt und mit der gleichen Säule und dem gleichen Vorgehen wie die ersten 200 µl aufgereinigt. Am
Seite 18 Ende wurden die beiden Fraktionen jeder Probe jeweils zusammengegeben. Aus allen Proben wurden auf diese Art und Weise Albumin und Immunglobuline entfernt. Proteinbestimmung Nun konnten sowohl die Proben der Patientengruppe als auch die der Kontrollgruppe jeweils in ein Gefäß gepoolt werden. Um die genaue Proteinmenge bestimmen zu können, wurden aus jedem Pool 120 µl für das Nephelometer entnommen. Die gemessene Proteinkonzentration wurde in das Gesamtgewicht der Proteine in jedem Pool umgerechnet. Proteinpräzipitierung mit Aceton Um störende Komponenten aus den Probenpools zu entfernen, wurden die Proteine mit Aceton gefällt. Dazu wurde das dreifache Volumen -30 °C kalten Acetons zu den Pools gegeben und das Ganze gut gemischt. Über Nacht und bei -30 °C präzipitierten die Proteine. Anschließend wurden die Probenpools in der Kühlzentrifuge bei 4 °C und 4500 rpm für 45 Minuten zentrifugiert. Der Überstand wurde vorsichtig abpipettiert und die entstandenen Proteinpellets einige Minuten offen stehen gelassen, bis sie etwas getrocknet waren. Schließlich wurden zu den Proteinpellets jeweils Labellingpuffer gegeben, bis sich die Pellets vollständig gelöst hatten und die Endkonzentration der in Puffer gelösten Proteine 4 µg/µl betrug. Fluoreszenzlabelling der Probenpools Insgesamt sollten drei 2-D DIGE-Gele hergestellt werden, zwei identische und eines, bei dem die Fluoreszenzmarkierungen der Patienten und Kontrollen vertauscht waren. Außerdem wurde ein interner Standard aus den beiden Pools gebildet, der je zur Hälfte aus dem Patienten- und dem Kontrollpool bestand. Ein Pipettierschema wurde errechnet und für jedes Gel die zwei Pools und der interne Standard nach diesem Schema pipettiert und anschließend zusammengemischt.
Seite 19
Insgesamt ergaben sich so dreimal 35,19 µl Probe, die jeweils mit 305 µl
Fokussierpuffer zu einem Gesamtvolumen von 340 µl aufgefüllt und sofort
weiterverwendet wurden.
Pipettierschemata für drei DIGE-Gele
Tabelle 3: Pipettierschema für Gel 1. In jedem Pool befindet sich die gleiche Menge an Protein
(30 µg). Jeder Probenpool wurde unterschiedlich gefärbt. Gemeinsam mit dem Fa rbstoff (CyDye)
und Lysin ergibt sich ein Endvolumen von 11,73 µl pro Probenpool. Gel 1 wurde gepickt. µg =
Mikrogramm. µl = Mikroliter. NK = Normalkontrollen. VE = Viliuisk-Enzephalomyelitis-Patienten. IS
= interner Standard aus je gleichen Teilen Protein en von Kontrollen und Viliuisk-
Enzephylomyelitis-Patienten. Cy3 = rot. Cy5 = grün. Cy2 = gelb.
Protein Probenpool 250 pmol/µl Lysin Endvolumen
[µl] CyDye [µl] [µl]
[µg] [µl]
30 NK 8,33 1 Cy3 rot 2,4 11,73
30 VE 8,33 1 Cy5 grün 2,4 11,73
30 IS (15 NK 4,17 NK 1 Cy2 gelb 2,4 11,73
und 15 VE) 4,17 VE
Tabelle 4: Pipettierschema für Gel 2. In jedem Pool befindet sich die gleiche Menge an Protein
(30 µg). Jeder Probenpool wurde unterschiedlich gefärbt. Gemeinsam mit dem Fa rbstoff (CyDye)
und Lysin ergibt sich ein Endvolumen von 11,73 µl pro Probenpool. µg = Mikrogramm. µl =
Mikroliter. NK = Normalkontrollen. VE = Viliuisk -Enzephalomyelitis-Patienten. IS = interner
Standard aus je gleichen Teilen Proteinen von Kontrollen und Viliuisk-Enzephylomyelitis-
Patienten. Cy3 = rot. Cy5 = grün. Cy2 = gelb.
Protein Probenpool 250 pmol/µl Lysin Endvolumen
[µl] CyDye [µl] [µl]
[µg] [µl]
30 NK 8,33 1 Cy3 rot 2,4 11,73
30 VE 8,33 1 Cy5 grün 2,4 11,73
30 IS (15 NK 4,17 NK 1 Cy2 gelb 2,4 11,73
und 15 VE) 4,17 VESeite 20
Tabelle 5: Pipettierschema für Gel 3. In jedem Pool befindet sich die gleiche Menge an Protein
(30 µg). Jeder Probenpool wurde unterschiedlich gefärbt. Gemeinsam mit dem Farbstoff (CyDye)
und Lysin ergibt sich ein Endvolumen von 11,73 µl pro Probenpool. µg = Mikrogramm. µl =
Mikroliter. NK = Normalkontrollen. VE = Viliuisk -Enzephalomyelitis-Patienten. IS = interner
Standard aus je gleichen Teilen Proteinen von Kontrollen und Viliuisk -Enzephylomyelitis-
Patienten. Cy3 = rot. Cy5 = grün. Cy2 = gelb.
Protein Probenpool 250 pmol/µl Lysin Endvolumen
[µl] CyDye [µl] [µl]
[µg] [µl]
30 NK 8,33 1 Cy5 grün 2,4 11,73
30 VE 8,33 1 Cy3 rot 2,4 11,73
30 IS (15 NK 4,17 NK 1 Cy2 gelb 2,4 11,73
und 15 VE) 4,17 VE
Rehydratisierung
Der Fokussierpuffer wurde aufgetaut und mit 4 mg DTT pro 1 ml versetzt. Jede
der drei Proben wurde anschließend vorsichtig in jeweils ein Fokussiertray des
Protean IEF Cell pipettiert. Dann konnten die IPG-Streifen mit der Gelseite nach
unten aufgelegt werden. Es wurde darauf geachtet, dass die Streifen korrekt
ausgerichtet und gleichmäßig luftblasenfrei mit der Probe benetzt waren.
Nachdem sie mit etwa 1,5 ml Mineralöl überschichtet worden waren, wurden sie
über Nacht bei 50 V und 20 °C Raumtemperatur rehydratisiert.
Isoelektrische Fokussierung
Nach der Rehydratisierung über Nacht konnte am nächsten Tag die isoelektrische
Fokussierung gestartet werden. Um störende Salze der Probe zu entziehen
wurden kleine, mit destilliertem Wasser befeuchtete Filterpapierstreifen in jeden
Tray an beiden Polen zwischen IPG-Streifen und Elektrode eingelegt.
Anschließend konnte das Fokussierprotokoll gestartet werden, zunächst für
anderthalb Stunden mit 500 V, dann für weitere anderthalb Stunden mit 1000 V.
Danach stieg die Spannung innerhalb von zwei Stunden auf 8000 V, um dann
nochmals für vier Stunden bei 8000 V zu bleiben. Am Ende schaltete das Gerät in
den Schonmodus mit 500 V. Nach der Fokussierung wurden die Streifen mit 1-
fachem Laufpuffer kurz gespült und eingefroren.Seite 21 Äquilibrierung der IPG-Streifen Um nun die Proteine in der Gelelektrophorese nach dem Molekulargewicht bzw. der molekularen Größe auftrennen zu können, mussten sie zunächst äquilibriert werden. Eine Äquilibrierung erfolgt in zwei Schritten: die Proteine werden zuerst mit einem SDS-DTT-Puffer reduziert und dadurch entfaltet und dann mit einem SDS-Iodacetamid-Puffer alkyliert, damit sie stabil bleiben. Der SDS-Äquilibrierungspuffer wurde aus 6,7 ml des für die Acrylamid-Gele hergestellten 1,5 M Tris-HCl-Puffers, 73 g Harnstoff und 69 ml Glycerin gemischt. Anschließend wurden vorsichtig 4 g SDS dazugegeben, dann einige Tropfen Bromphenolblaulösung und schließlich das Ganze mit dH2O auf 200 ml aufgefüllt. Aus jeweils 10 ml dieses SDS-Äquilibrierungspuffers wurden durch Zugabe von 100 mg DTT ein DTT-Puffer und durch Zugabe von 250 mg Iodacetamid ein Iodacetamid-Puffer hergestellt. Die aufgetauten IPG-Streifen wurden in einem Röhrchen zuerst mit dem DTT- Puffer, dann mit dem Iodacetamid-Puffer für jeweils 15 Minuten unter vorsichtigem Schwenken äquilibriert. Danach wurden die Streifen kurz in 1-fachen Laufpuffer getaucht. Polyacrylamid-Gelelektrophorese Die fertig äquilibrierten IPG-Streifen wurden vorsichtig in die vorher gut gereinigten und getrockneten Geltaschen der Gelplatten bis direkt an das Gel geschoben. 50 mg Agarose wurden mit 50 ml 1-fachen Laufpuffer gemischt, einige Tropfen Bromphenolblaulösung dazugegeben und das Gemisch in der Mikrowelle vorsichtig geschmolzen. Gleichzeitig wurden die Gelplatten in das Gelelektrophoresesystem eingebaut. Als die Agarose-Lösung auf etwa 50 °C abgekühlt war, wurde sie über die IPG- Streifen in den Geltaschen pipettiert und gewartet, bis sie fest war. Dann wurde die untere Kammer des Gelelektrophoresesystems mit 1-fachem Laufpuffer, die obere Kammer mit 2-fachem Laufpuffer befüllt und das Gerät gestartet.
Seite 22 Kurz bevor die Lauffront das untere Ende der Gele erreicht hatte, wurde die Gelelektrophorese beendet. Die Gelplatten wurden ausgebaut und gut gereinigt. Scannen der Gele Sofort nach der Gelelektrophorese wurden die Gele noch in den Glasplatten mit dem Ettan DIGE Imager gescannt. Der Scanner nutzt drei verschiedene Anregungswellenlängen und Emissionsfilter und nimmt deshalb von jedem Fluoreszenzfarbstoff ein gesondertes Bild auf. Statistische Auswertung der Gele Zur statistischen Auswertung der gescannten Gele wurde die DyCyder Differential Analysis Software 5.0 verwendet. Die notwendigen Berechnungen erfolgten für jedes Gel gesondert und durch die Software weitgehend automatisiert. Die Software kombinierte zunächst alle drei (Cy2, Cy3 und Cy5) vom Scanner erzeugte Bilder, um alle Proteinspots zu detektieren. Dann wurden von jeder der drei Aufnahmen gesondert die Volumina der einzelnen detektierten Spots berechnet. Hintergrundrauschen und Artefakte konnten mit Hilfe der Software zum größten Teil automatisch entfernt werden. Nun wurden die Spotvolumen der reinen Patienten- und der reinen Kontrollprobe (Cy3 und Cy5) jeweils ins Verhältnis mit den entsprechenden Spots des internen Standards (Cy2) gesetzt. Dieser Schritt führte zu einer deutlich verbesserten Vergleichbarkeit zwischen verschiedenen Gelen, weil dadurch versuchsbedingte unterschiedliche Gesamtproteinkonzentrationen in den einzelnen Gelen keine Rolle mehr spielten. Bevor die Volumenverhältnisse endgültig verwendbar waren, wurden sie normalisiert. Dazu erstellt das Programm ein Häufigkeitshistogramm der logarithmierten Volumenverhältnisse zwischen dem internen Standard und der Probe. Dieses Histogramm nähert sich bei ausreichend vielen Proteinspots ungefähr einer normalverteilten Kurve. Unter der Annahme, dass die Volumina der meisten Spots sowohl im internen Standard als auch in der Probe gleich sind, entspricht der Hochpunkt dieser Kurve, das häufigste Volumenverhältnis, eigentlich dem Volumenverhältnis 1:1. Deshalb wird von der Software das
Seite 23 Häufigkeitshistogramm so verschoben, das der Hochpunkt der Kurve auf der y- Achse liegt (log (1/1)=0). Schließlich wurden alle Werte, die nach Normalisierung eine signifikante Volumenabweichung zum internen Standard hatten, als Spots markiert. Coomassie-Färbung der Gele Um die Proteinspots sichtbar zu färben, wurde jedes Gel nach dem Scan aus den Glasplatten gelöst und über Nacht in 500 ml Coomassie-Färbelösung unter langsamem Schwenken inkubiert. 500 ml Coomasie-Färbelösung wurden aus 300 ml destilliertem Wasser, 100 ml Methanol und 100 ml RotiBlue colloidal hergestellt. Am nächsten Morgen wurden die Gele für etwa 10 Minuten in 25%-iger Methanollösung gewaschen und sofort danach mit etwas destilliertem Wasser luftdicht in eine Folie eingeschweißt und zur massenspektrometrischen Analyse verschickt. Massenspektrometrische Auswertung der Gele Die Firma TOPLAB GmbH in Martinsried leistete die massenspektrometrische Auswertung der 2-D DIGE-Gele. Dort wurden die ausgewählten Proteinspots manuell aus dem Gel gepickt und anschließend dreimal in 100 µl einer 50 mM (NH4)2HCO3-Lösung gewaschen. Dann mussten die einzelnen Proben fünf Minuten lang mit 50 µl Acetonitril dehydratisiert werden. Danach wurde der Überstand vorsichtig entfernt und die Probe einige Minute offen stehen gelassen. Mit 1 µl einer Proteaselösung (0,05 µl/µg Trypsin in 10 mM (NH 4)2HCO3) wurden die Proteine in den Gelstücken über Nacht bei 37 °C verdaut. Die MALDI-TOF-MS (matrix-assisted laser desorption/ionisation time-of-flight Massenspektrometrie) wurde mit 0,1 - 0,5 ml der verdauten Peptidlösung vorgenommen. Die Lösung wurde mit DHB-Matrix (2,5-Dihydroxybenzoesäure (DHB) in einer Lösung aus Acetonitril und Trifluoroacetic Säure) gemischt und auf eine MALDI-Platte gegeben, auf der die Mischung auskristallisierte.
Seite 24 Das Massenspektrometer, ein Voyager DE-STR MALDI-TOF- Massenspektrometer, ionisierte nun durch Laserstrahlen zunächst die Matrix, die wiederum ein Teil ihrer Ladung an die zu untersuchende Peptidmischung weitergab. Dann wurden die durch den Laser ionisierten und vaporisierten Peptide durch ein elektrisches Feld beschleunigt. Leichtere Moleküle waren schneller und erreichten den Detektor deshalb früher, der dann die Ladungen maß. Nun ließen sich über die einzelnen Peaks sowohl qualitative (m/z-Wert, also die Masse pro Ladung) und quantitative Aussagen (die Gesamtladung) über die Proteinzusammensetzung machen. Proteinidentifizierung Um die Proteine zu identifizieren wurden alle mass-to-charge-Werte (m/z-Werte) zwischen 700 und 4200 bei ProFound und Mascot zur Spektrenanalyse eingegeben. Gesucht wurden alle Proteine im Massenbereich von 5 bis 200 kDa mit einem isoelektrischen Punkt zwischen pH 2 und 14 und der Gattung Homo sapiens. Als Massentoleranz wurden 100 ppm eingegeben. Datenquelle war die unabhängige Proteindatenbank des National Centre for Biotechnology Information (NCBI) und der Internationale Protein Index. 2.3.3 Validierung von Fetuin-A mit ELISA Da die 2-D DIGE nur ein semiquantitatives Verfahren ist, muss zur Validierung der gefundenen Proteine eine andere Methode angewandt werden. Ein ELISA wurde durchgeführt, um das in Patientenproben signifikant erniedrigte Protein Fetuin-A zu validieren. Dazu wurden die Proteinkonzentrationen in den Patientenproben, in Kontrollen aus Russland und in Normalkontrollen aus Deutschland gemessen und analysiert. Durchführung Zunächst wurden sowohl das Wash Buffer Konzentrat als auch das Dilution Buffer Konzentrat zehnfach mit destilliertem Wasser verdünnt. Die Proben wurden ausgefroren und die Seren im Verhältnis 1:10.000 und die Liquores im Verhältnis 1:50 mit dem Dilution Buffer vermischt. Eine Standardverdünnungsreihe wurde aus dem Master Standard nach folgendem Pipettierschema hergestellt:
Seite 25 Tabelle 6: Pipettierschema für die Standardverdünnungsreihe des Fetuin -A-ELISA. Durch wiederholte Verdünnung mit Dilution Buffer wurden Fetuin -A-Proben mit definierter Endkonzentration hergestellt. µl = Mikroliter. ELISA = Enzyme -Linked Immunosorbent Assay. ml = Milliliter. ng = Nanogramm. Fetuin-A-Probe Dilution Buffer [µl] Endkonzentration [ng/ml] Master Standard 800 100 200 µl von 100 ng/ml 300 40 250 µl von 40 ng/ml 250 20 250 µl von 20 ng/ml 250 10 250 µl von 10 ng/ml 250 5 200 µl von 5 ng/ml 300 2 Desweiteren wurden zwei Qualitätskontrollproben mit hoher und mit niedriger Fetuin-Konzentration vorbereitet. Auf die ELISA-Platte wurden jeweils zweifach 100 µl der Standardverdünnungsreihe, der Qualitätskontrollproben und der vorbereiteten Patientenproben pipettiert. Nach einer Stunde Inkubationszeit wurde die Platte sorgfältig mit dem Wash Buffer gewaschen und anschließend in jedes Well 100 µl Conjugate Solution gegeben. Nach einer weiteren Stunde und sorgfältigem Spülen wurden jeweils 100 µl der Substrate Solution in die Wells pipettiert. Die Platte musste nun nochmals zehn Minuten bei Raumtemperatur in Dunkelheit inkubiert werden. Anschließend beendeten 100 µl Stop Solution in jedem Well die Reaktion. Mit dem ELISA-Leser wurden bei 450 nm die Extinktion jedes Wells bestimmt und anschließend anhand der Referenzkurve der Standardverdünnungsreihe und der Verdünnungsverhältnissen der Proben zu Beginn auf die ursprünglichen Fetuin- Konzentrationen umgerechnet.
Seite 26 Abbildung 5: Referenzkurve der Standardverdünnungsreihe des Fetuin -A-ELISAs. Als Referenzkurve zur Berechnung der Fetuin -A-Konzentrationen in den Proben aus den gemessenen Extinktionswerten wurde im gleichen Versuch eine definierte Standardverdünnungsreihe gemessen. Auf der x-Achse ist die Konzentration des Fetuin -A in ng/ml aufgetragen. Die y-Achse zeigt die Extinktion bei 450 nm. Die gemessenen Werte sind mit S1-S6 markiert. ELISA = Enzyme-Linked Immunosorbent Assay. ng= Nanogramm. nm = Nanometer. ml = Milliliter. Auswertung Die Auswertung der Messdaten des ELISAs wurde mit GraphPad Prism 5 for Windows (Version 5.04) von GraphPad Software Inc. vorgenommen. Die drei Gruppen (Patientenproben, Kontrollen aus Russland, Kontrollen aus Deutschland) wurden mit dem Kruskal-Wallis-Test auf signifikante Unterschiede in der Fetuin-A- Konzentration getestet.
Seite 27 3. ERGEBNISSE 3.1 2-D DIGE Proteomanalyse 3.1.1 Ausgewertete Gele Es wurden drei unabhängige 2-D DIGE-Läufe mit jeweils gepoolten Liquores von Patienten und Kontrollen durchgeführt. In diesen drei Gelen konnten von der DeCyder Differential Analysis Software insgesamt über 2000 Proteinspots detektiert werden. Abbildung 6. Vergleichende Darstellung des Kontrollpools (links) und des Viliuisk - Enzephalomyelitis-Pools (rechts) nach Durchführung einer zweidimensionalen differenziellen fluoreszierenden Gelelektrophorese (2 -D DIGE) und Auswertung durch die DeCyder Differenz Analysis Software. Die weiß markierten Spots zeigen in allen drei unabhängigen 2-D DIGE-Gelen Unterschiede um mindestens das 1,5 -fache zwischen dem Kontrollpool und dem Patientenpool und wurden zur weiteren Analyse gepickt. Die grau umrandeten Spots wurden nicht gepickt. kDa = Kilodalton. Mw = Molekülmasse. pH = pH -Wert. VE = Viliuisk-Enzephalomyelitis.
Seite 28 3.1.2 Identifizierte Proteine Insgesamt 24 Proteinspots wiesen einen Volumenunterschied zwischen Patientenpool und Kontrollen um mindestens das 1,5-fache auf. Diese Spots wurden markiert, gepickt und durch MALDI-TOF-Massenspektrometrie und Datenbankrecherche identifiziert. Im Patientenpool um mindestens das 1,5-fache hochreguliert zeigten sich Microfibril-associated Glycoprotein 4, Transthyretin, Apolipoprotein E, Immunglobulin κ light chain VLJ region und Immunglobulin κ chain constant region. Um mindestens das 1,5-fache erniedrigt waren Ceruloplasmin, Actinin α 1 Isoform 3, Transferrin, Serum Albumin, α-1-antichymotrypsin, α-1-antitrypsin, Immunglobulin heavy variable 4-31, Immunglobulin heavy constant µ, Immunglobulin heavy constant γ 1, Immunglobulin γ-4 chain C region, Factor VII active site mutant immunoconjugate, Ecotropic viral integration site 2B, cDNA FLJ55606 (sehr ähnlich zu α-2-HS-glycoprotein), Thyroid hormone receptor α, Torsin family 3 member A (ADIR1 und ADIR2), λ-Immunglobulin und Immunglobulin λ variable 2-14. Zwei Proteinspots (Spotnummer 1864 und 1937) konnten nicht identifiziert werden. Spotnummer 1874 und 1904 bestanden beide aus Keratin und waren einmal erhöht und einmal erniedrigt. Das spricht dafür, dass es sich hier um Verunreinigungen handelt, die wahrscheinlich beim Picken der Spots entstanden sind.
Seite 29 Abbildung 7. Für die massenspektrographische Analyse gepickte Proteinspots. Alle weiß markierten und mit Nummern versehenen Spots zeigen einen mindestens 1,5 -fachen Volumenunterschied zwischen dem Kontrollpool und dem Patientenpool und wurden für die weitere Analyse mit dem Massenspektrometer gepickt.
Seite 30 3.1.3 Dreidimensionale Darstellung eines Spots am Beispiel von Fetuin -A Abbildung 8. Dreidimensionale Darstellung des Volumens des Fetuin -A-Spots bei der Viliuisk-Enzephalomyelitis (VE) im Vergleich zum Kontrollpool. Dreidimensionale graphische Darstellung durch die DeCyder Differential Analysis Software. Der Proteinspot ist weiß umrandet.
Seite 31
3.1.4 Identifizierte Proteine
Tabelle 7. Tabellarische Übersicht der identifizierten Liquorproteine mit mindestens 1,5-
fachem Spotvolumenunterschied zwischen Patientenpool und Kontrollpool. Die
Liquorproteine wurden mit zweidimensionaler differenzieller fluoreszierender Gelelektrophorese
(2-D DIGE) aufgetrennt und mittels Massenspektrographie und Abgleich mit dem Internationale
Protein Index (die Identifikationsnummer beginnt mit IPI) und der unabhängigen Proteindatenbank
des National Centre für Biotechnology Information (die Identifikationsnummer beginnt mi t Gi)
identifiziert. Der Regulationsfaktor beschreibt das Spotvolumenverhältnis Patientenpool versus
Kontrollpool. Da = Dalton. ID = Identifikationsnummer. Mw = Molekülmasse. pH = pH -Wert. Pi =
isoelektrischer Punkt. ÜE = Übereinstimmung der Sequenz in Pro zent.
Proteinname Spot Faktor ÜE ID Mw Pi
[Da] [pH]
Ceruloplasmin 823 -10,1 38 IPI00017601 122983 5,4
Ceruloplasmin 824 -1,51 58 IPI00017601 122983 5,4
Ceruloplasmin 826-1 -1,73 46 IPI00017601 122983 5,4
Actinin α 1 Isoform 3 826-2 -1,73 29 IPI00921118 107644 5,4
Transferrin 1095 -4,74 47 IPI00022463 79280 6,8
Serum Albumin 1242 -7,1 70 IPI00745872 71317 5,9
Isoform 1
α-1-antichymotrypsin 1274 -1,65 61 IPI00847635 47792 5,3
Isoform 1
α-1-antitrypsin 1385 -9,76 34 IPI00553177 46878 5,4
Isoform 1
Ecotropic viral 1436-1 -1,76 25 Gi462028 49000 4,7
integration site 2B
precursor
Ecotropic viral 1436-2 -1,76 24 Gi13543536 48900 4,7
integration site 2BSie können auch lesen

















































