Workshop Medizinprodukterecht Peter Pieper Vistec AG - Workshop Medizinprodukterecht
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

Workshop
Medizinprodukterecht
Workshop Medizinprodukterecht
Peter Pieper
Vistec AG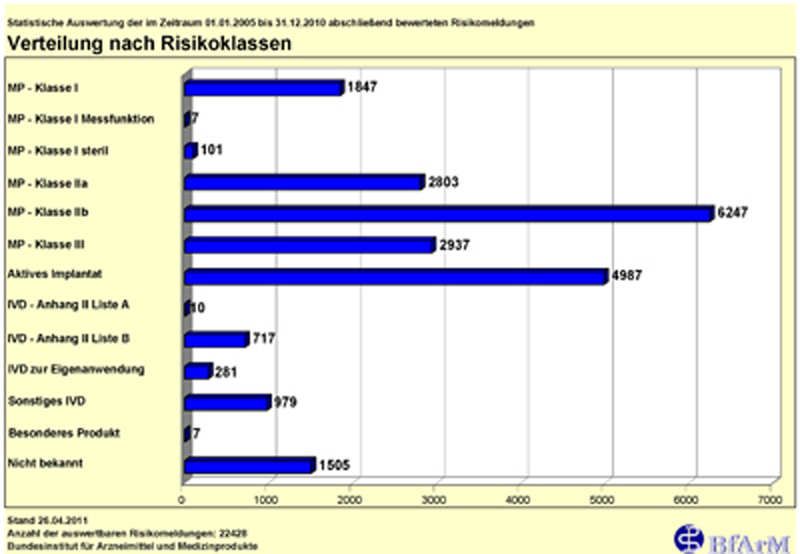
Workshop
Medizinprodukterecht
Was ist neu?
Bisher: Richtlinie 93/42 EWG (Inverkehrbringen und Inbetriebnahme v. Med.-Produkten)
Neu: MDR 2017 (Medical Device Regulation) (Europ. Vorschrift)
Medizinproduktegesetz (MpG) weiterhin gültig
MDR:
Inkrafttreten geplant für Mitte 2017
Anwendung 3 Jahre später verbindlich
Neuerungen betreffen in erster Linie die Hersteller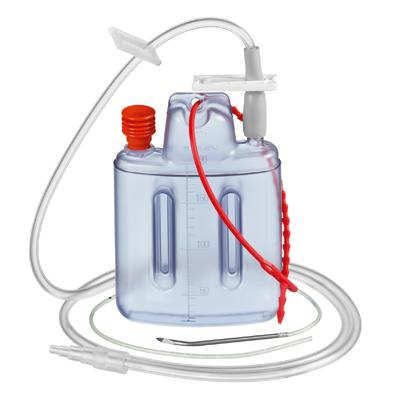
Workshop
Medizinprodukterecht
Um was geht es?
Alle einzelnen oder miteinander verbunden, verwendeten Instrumente, Apparate, Vorrichtungen,
Stoffe oder andere Gegenstände, die vom Hersteller zur Anwendung für Menschen für folgende
Zwecke bestimmt sind:
Erkennen, Überwachung, Behandlung, Linderung oder Kompensierung
von Verletzungen oder Behinderungen
Untersuchung, Ersatz oder Veränderung des anatomischen Aufbaus oder
eines physiologischen Vorgangs
Empfängnisverhütung (sofern kein Arzneimittel)
Workshop
Medizinprodukterecht
Welche Anforderungen enthält die Verordnung?
Grundsätzlich dürfen Medizinprodukte nur in den Verkehr gebracht und in Betrieb genommen
werden, wenn sie die Sicherheit und die Gesundheit der Patienten, der Anwender und
gegebenenfalls Dritter bei sachgemäßer Installation, Instandhaltung und ihrer Zweck-
bestimmung entsprechenden Verwendung nicht gefährden.
Verschärfung der Anforderungen!
Änderungen in der Klassifizierung
Neuregelung der Marktüberwachung mit kürzeren Meldefristen
Höhere Anforderungen an klinische Bewertung
Workshop
Medizinprodukterecht
Klassifizierung der Produkte (Anhang VIII Kapitel 3 MDR)
Medizinprodukte werden entsprechend ihres Gefährdungspotentials nach den Regeln
gemäß Anhang VIII MDR in die Klassen I, II a, II b und III eingestuft
Die Klassifizierungsregeln basieren auf der Verletzbarkeit des menschlichen Körpers
und berücksichtigen die potentiellen Risiken im Zusammenhang mit der technischen
Auslegung der Produkte und ihrer Herstellung
Workshop
Medizinprodukterecht
Klasse I
Keine methodischen Risiken
Geringer Invasivitätsgrad
Kein oder unkritischer Hautkontakt
Vorübergehender Hautkontakt max. 60 min
Workshop
Medizinprodukterecht
Klasse IIa
Geringes Anwendungsrisiko
Mässiger Invasivitätsgrad
Kurzzeitige Anwendung im Körper oder chirurgisch geschaffenen Körperöffnungen
Kurzzeitiger bis max. 30 Tage ununterbrochener oder wiederholter Einsatz des
gleichen Produkts
Redon Vacuum Drainage
Workshop
Medizinprodukterecht
Klasse IIb
Mässiges methodisches Risiko
Mässiger Invasivitätsgrad
Kurzzeitige Anwendung im Körper oder chirurgisch geschaffenen Körperöffnungen
Kurzzeitiger bis max. 30 Tage ununterbrochener oder wiederholter Einsatz des
gleichen Produkts
Workshop
Medizinprodukterecht
Klasse III (entspricht hohem Gefährdungspotential)
Erhöhtes methodisches Risiko
Zur langfristigen Medikamentenabgabe
Inhaltsstoffe tierischer Herkunft in Wunden oder im Körper
Unmittelbare Anwendung am Herzen, zentralem Kreislaufsystem,
zentralem Nervensystem
Künstliche, biologische Herzklappe (Schwein)
Workshop
Medizinprodukterecht
CE-Kennzeichnung / Konformitätserklärung
Kennzeichnung mit nur dem CE-Kennzeichen: Nur Produkte der Klasse I
Nummer der
Bei Produkten der Klasse Im, Is IIa, IIb, III: CE1275
benannten Stelle
Voraussetzung zur Kennzeichnung ist die Konformitätserklärung bzw.
die Zertifizierung durch eine benannte Stelle
Ziel: Sicherstellung der Gleichartigkeit von Produkten in EuropaWorkshop
Medizinprodukterecht
Eine rechtmäßig nach Bestimmungen des MPG auf Med.Produkten angebrachte
CE-Kennzeichnung ist grundsätzlich die Garantie für die Beachtung folgender
Sicherheitsaspekte:
Durchführung einer Risikoanalyse für jedes Produkt,
Etablierung eines Risikomanagements
Erstellung einer aktuellen technischen Dokumentation über den Herstellungsablauf
und die technische Spezifikation des Produktes
Errichtung und Erhaltung eines Qualitätsmanagementsystems –
Wiederholung des kompletten Zertifizierungsverfahrens durch regelmäßige
Überwachungsaudits
Beachtung europäisch harmonisierter Normen –MEDDEV Guidelines,EK-MED,
Beschlüsse der ZLG usw.Workshop
Medizinprodukterecht
Medizinprodukteberater (§ 31 MPG) :
Medizinprodukteberater (§ 31 MPG) Ist wer berufsmäßig Fachkreise fachlich informiert
oder
in die sachgerechte Handhabung der Medizinprodukte einweist
Nach MPBetreibV ist dies beim Betreiber bisher der „Geräteverantwortliche“
Neu:
Beauftragter für MedizinproduktesicherheitWorkshop
Medizinprodukterecht
Qualifikation des Medizinprodukteberaters (MPB) :
Der Nachweis der erforderlichen Sachkenntnis als MPB für Med.Produkte
wird erbracht durch:
eine erfolgreich abgeschlossene Ausbildung in einem
naturwissenschaftlichen, medizinischen oder technischen Beruf
und mindestens einjähriger Tätigkeit mit ausreichender Produktkenntnis
die zur Einweisung befähigt.
Die Sachkenntnis ist auf Verlangen der zuständigen Behörde nachzuweisen
Der MPB hält sich auf dem neuesten Erkenntnisstand über die jeweiligen Medizinprodukte,
um sachkundig beraten zu können.
Der Auftraggeber hat für eine regelmäßige Schulung des MPB zu sorgen.Workshop
Medizinprodukterecht
Beauftragter für Medizinproduktesicherheit (§6 MPBetreibV )
Gesundheitseinrichtungen mit regelmäßig mehr als 20 Beschäftigten haben sicherzustellen,
dass ein Beauftragter für Medizinproduktesicherheit bestimmt ist
Beauftragter für Medizinproduktesicherheit ist eine sachkundige und zuverlässige
Person mit medizinischer, naturwissenschaftlicher, pflegerischer, pharmazeutischer
oder technischer Ausbildung.
Aufgabenbereiche:
Der Beauftragte für Medizinproduktesicherheit:
Ist Kontaktperson bei Meldungen über Risiken oder Vorkommnisse
gegenüber Hersteller, Vertreiber und Behörden
Koordiniert Maßnahmen bei Rückrufmaßnahmen durch den Verantwortlichen nach
§5 MPG (Hersteller – Bevollmächtigter)Workshop Medizinprodukterecht Klasse Im z.B. Sehtestgeräte
Workshop
Medizinprodukterecht
Sicherheitsbeauftragter für Medizinprodukte §30 MPG
(Medical Device Safety Officer- MDSO)
vom Hersteller bestimmte Person mit erforderlicher Sachkenntnis und Ausbildung
regelt Meldungen über bekanntgewordene Risiken
Der Hersteller meldet den Sicherheitsbeauftragen für
Medizinprodukte der DIMDIWorkshop
Medizinprodukterecht
Behörden, Meldestellen, Informationsquellen
DIMDI – Deutsches Institut für Med.Dokumentation
BfArM – Bundesinstitut für Arzneimittel und Medizinprodukte
ZLG – Zentralstelle der Länder für Gesundheitsschutz bei
Arzneimitteln und Medizinprodukten
ZLS Bayern – Zentralstelle der Länder für SicherheitstechnikWorkshop
Medizinprodukterecht
MPBetreibV Verordnung über das Errichten, Betreiben und
Anwenden von Medizinprodukten:
Regelt, wer Medizinprodukte betreiben darf und wofür der Betreiber verantwortlich ist
Dokumentationspflicht:
Führen von Medizinproduktebüchern (§12)
Protokolle über Geräteeinweisungen und MPG-Einweisungen
Protokolle über Instandhaltungen (§7)
Protokolle über sicherheitstechnische Kontrollen (§11)
Protokolle über messtechnische Kontrollen (§14)Workshop
Medizinprodukterecht
Meldepflicht § 6 MPBetreibV :
(2) Der Beauftragte für Medizinproduktesicherheit nimmt als zentrale Stelle
in der Gesundheitseinrichtung folgende Aufgaben für den Betreiber wahr:
Die Aufgaben einer Kontaktperson für Behörden, Hersteller und Vertreiber
im Zusammenhang mit Meldungen über Risiken von Medizinprodukten sowie bei der
Umsetzung von notwendigen korrektiven Maßnahmen,
die Koordinierung interner Prozesse der Gesundheitseinrichtung zur Erfüllung der Melde-
und Mitwirkungspflichten der Anwender und Betreiber und
die Koordinierung der Umsetzung korrektiver Maßnahmen und der Rückrufmaßnahmen
durch den Verantwortlichen nach § 5 des Medizinproduktegesetzes in den
Gesundheitseinrichtungen.Workshop
Medizinprodukterecht
Dabei gilt:
(3) Der Beauftragte für Medizinproduktesicherheit darf bei der Erfüllung der nach
Absatz 2 übertragenen Aufgaben nicht behindert und wegen der Erfüllung der Aufgaben
nicht benachteiligt werden.Workshop
Medizinprodukterecht
Anforderungen lt. MPG :
Meldepflicht von Vorfällen (§25)
Medizinprodukte-Beobachtungs- und Meldesystem (§29)
Medizinprodukteberater (§31)
Sicherheitsbeauftragter für Medizinprodukte (§30)Workshop
Medizinprodukterecht
Verordnung über die Erfassung, Bewertung und Abwehr von Risiken bei
Medizinprodukten MPSV (BGBl. IS. 2131 u. 2203):
Regelt Verfahren zur Erfassung, Bewertung und Abwehr von Risiken
Meldepflichten in §3 durch Verantwortliche gem. §5 MPGWorkshop
Medizinprodukterecht
Wartung und Instandhaltung von Medizinprodukten:
Nach STK – MTK – DGUV V3
Verantwortlich ist immer der Betreiber (§2 MPBetreibV und §5 MPG)
§2 MPBetreibV (Allgemeine Anforderungen)
Vorschriften gem. DGUV bleiben unberührt es sei denn der Prüfumfang ist in den
sicherheitstechnischen Kontrollen STK (§11 MPBetreibV) enthaltenWorkshop
Medizinprodukterecht
DGUV V3 (nicht MPG - EN 61140 (Deutschland: VDE 0140-1)
Darf von autorisierten Personen mit zugelassenen Prüfgeräten durchgeführt werden
Ortsveränderliche Geräte alle 6 bzw. 12 Monate
Ortsfeste Geräte spätestens nach 24 Monaten
DGUV V3 regelt Prüfungen aller technischen Geräte unabhängig vom
Medizinproduktegesetz
Entspricht nicht einer Wartung / Instandhaltung nach §7 MPBetreibV!Workshop
Medizinprodukterecht
§ 7 Absatz 1 MPBetreibV
Hersteller empfiehlt Wartungs- / Instandhaltungsintervalle
Verantwortlich für die Instandhaltung ist der Betreiber!
§14 MPBetreibV: Messtechnische Kontrolle MTK
für alle Geräte in Anlage 2 und Geräte die vom Hersteller
dafür vorgesehen sindWorkshop
Medizinprodukterecht
Fachkraft für Arbeitssicherheit – FASI (alt: Sicherheitsfachkraft):
nicht Sicherheitsfachkraft für Medizinprodukte!
Unterstützt zusammen mit dem Arbeitsmediziner Unternehmen oder Behörden
ab einem Mitarbeiter
ASiG (Arbeits-Sicherheits-Gesetz):
Umsetzungen der EG-Rahmenrichtlinie 89/391Workshop
Medizinprodukterecht
Vielen Dank für Ihre
AufmerksamkeitSie können auch lesen

















































