2019 ZAR-SeminAR RINDERZUCHT AUSTRIA
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
RINDERZUCHT AUSTRIA
ZAR – Zentrale Arbeitsgemeinschaft österreichischer Rinderzüchter
Zuchtdata edv-dienstleistungen gmbh
ZAR-Seminar
2019
10 Jahre genomische Selektion
Rückblick und Ausblick
Zentrale Arbeitsgemeinschaft österreichischer Rinderzüchter - ZAR
ZuchtData EDV-Dienstleistungen GmbH
Dresdner Straße 89/19, 1200 Wien
+43 1 334 17 21 11
www.rinderzucht-austria.at I www.zuchtdata.at www.rinderzucht-austria.at I www.zuchtdata.at
Inhaltsverzeichnis
Verzeichnis der ReferentInnen 2
Univ.-Prof. Dr. Johann Sölkner
Genomische Selektion – Hintergrund und Geschichte 3
Dr. Hermann Schwarzenbacher
Genomische Zuchtwertschätzung und Herdentypisierung 7
Dr. Christa Egger-Danner
Aspekte zur Optimierung von genomischen Zuchtprogrammen 16
Dr. Christian Fürst
Status quo und Analyse der genomischen Zuchtprogramme in
Österreich und Deutschland 25
DI Peter Stückler
Genomisches Zuchtprogramm – Herausforderungen der Umsetzung in der
Praxis 39
Dr. Stefan Rensing
Blick über die Grenzen – Genomische Selektion bei Holstein international 45
Dr. Jørn Rind Thomasen
Genomic Selection in the Nordic Countries 46
Dr. Gábor Mészáros
Genomische Selektion in kleinen Rassen: Erfahrungen aus drei Ländern 48
Dr. Kay-Uwe Götz
Zukunftsperspektiven der Rinderzucht vor dem Hintergrund neuer
züchterischer Entwicklungen 53
ZAR-Seminar 2019 – Inhaltsverzeichnis
1
Verzeichnis der ReferentInnen
Dr. Christa Egger-Danner ZuchtData EDV-Dienstleistungen GmbH
Dresdner Straße 89/19, 1200 Wien
egger-danner@zuchtdata.at
www.zuchtdata.at
Dr. Christian Fürst ZuchtData EDV-Dienstleistungen GmbH
Dresdner Straße 89/19, 1200 Wien
fuerst@zuchtdata.at
www.zuchtdata.at
Prof. Dr. Kay-Uwe Götz Bayerische Landesanstalt für Landwirtschaft
Institut für Tierzucht
Prof.-Dürrwaechter-Platz 1, 85586 Poing
Kay-Uwe.Goetz@lfl.bayern.de
www.lfl.bayern.de
Assoc.Prof. Dr. Gábor Mészáros Universität für Bodenkultur Wien (BOKU)
Institut für Nutztierwissenschaften (NUWI)
Gregor-Mendel-Straße 33, 1180 Wien
gabor.meszaros@boku.ac.at
www.boku.ac.at
Dr. Stefan Rensing Vereinigte Informationssysteme Tierhaltung w.V. (VIT)
Heinrich-Schröder-Weg 1, 27283 Verden/Aller
stefan.rensing@vit.de
www.vit.de
Dr. Hermann Schwarzenbacher ZuchtData EDV-Dienstleistungen GmbH
Dresdner Straße 89/19, 1200 Wien
schwarzenbacher@zuchtdata.at
www.zuchtdata.at
Univ.-Prof. Dr. Johann Sölkner Universität für Bodenkultur Wien (BOKU)
Institut für Nutztierwissenschaften (NUWI)
Gregor-Mendel-Straße 33, 1180 Wien
johann.soelkner@boku.ac.at
www.boku.ac.at
DI Peter Stückler GENOSTAR Rinderbesamung GmbH
Am Tieberhof 6, A-8200 Gleisdorf
peter.stueckler@lk-stmk.at
www.genostar.at
Dr. Jørn Rind Thomasen VikingGenetics
Ebeltoftvej 16, Assentoft,
DK-8960 Randers SØ
jotho@vikinggenetics.com
www.vikinggenetics.com
ZAR-Seminar 2019 – Verzeichnis der ReferentInnen
2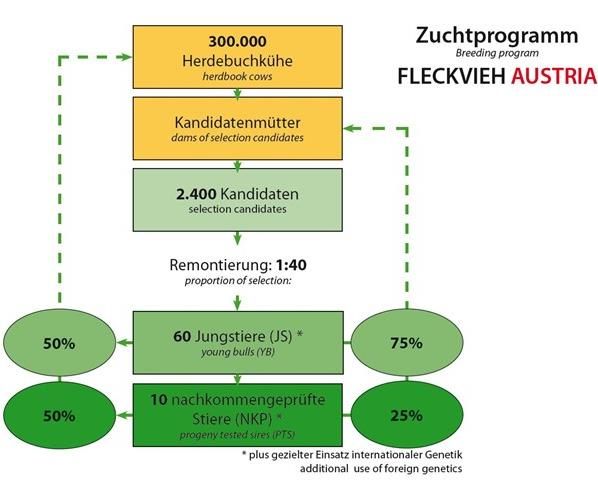
Genomische Selektion – Hintergrund und Geschichte
Johann Sölkner
Universität für Bodenkultur Wien (BOKU), Department für Nachhaltige Agrarsysteme,
Institut für Nutztierwissenschaften (NUWI)
und Wechselwirkung akkumuliert werden
Einleitung könnte, um den Zuchtwert von Tieren ausrei-
Die Rinderzucht hat eine lange Tradition von chend gut zu beschreiben.
sehr systematischer Selektion und damit ver-
bundenen Leistungssteigerungen. Nach dem
Das Genom und molekulare
Aufkommen der künstlichen Besamung in den
1950er Jahren, die eigentlich ein Mittel zur Marker
Deckseuchenbekämpfung war, haben Rinder- Die genetische Information ist beim Rind in
züchter rasch erkannt, dass die systematische den 30 Chromosomenpaaren jeder Zelle ge-
Nutzung der Informationen vieler Töchtern speichert, der genetische Code ist simpel. Vier
einzelner Stiere extrem nützlich für die Zucht Basen-Buchstaben (A, C, G, T) reichen aus, um
ist. Die Nachkommenprüfung war geboren in Dreierkombinationen „Worte“ oder „An-
und dominierte für die nächsten 60 Jahre das weisungen“ zur Bildung von Aminosäuren und
Zuchtgeschehen. Leistungsprüfung und Pe- Start- und Stop-Codons zu bilden, die für die
digree waren die zentralen Informationen im Bildung von Enzymen, Proteinen und letztlich
System der Zuchtwertschätzung. In dieser Zeit allen anderen Bausteinen des Lebens verant-
erkannte man, dass das Zuchtziel komplex wortlich sind. Das Genom besteht aus rund 3
sein muss und dass Informationen über sehr Milliarden solcher Buchstaben. Das Genom
viele Merkmale erhoben und genutzt werden wurde im Jahr 2001 beim Menschen vollstän-
müssen. Der ökonomische Gesamtzuchtwert dig gelesen (sequenziert), seit 2005 liegt die
wurde Mitte der 1990er Jahre in Österreich Sequenz auch beim Rind vor. Die Molekular-
entwickelt (Miesenberger, 1997) und ist mit genetik ist dabei zu lernen, die Sprache des
Modifikationen nach wie vor die zentrale Zahl Genoms besser zu verstehen. Aktuell weiß
bei der Zuchtwahl von Rindern. man, dass es rund 50.000 Gene gibt, die abge-
lesen und in Aminosäureketten übersetzt
Molekulargenetiker haben früh auf den po- werden. Proteine sind aneinander gereihte
tentiellen Nutzen molekularer Marker hinge- Aminosäuren. Die regulatorischen Mechanis-
wiesen. Prof. Martin Förster von der LMU men der DNA sind Gegenstand vieler Unter-
München postulierte im Jahr 1995, dass In- suchungen.
formationen zur Wirkung von Genen auf Leis-
tungsmerkmale recht bald die Leistungsprü- Molekulare Marker sind einzelne Basen oder
fung ersetzen würden. Die quantitativen Ge- kurze Basen-Ketten, die mit molekularen Me-
netiker, zu denen auch ich mich zähle, hielten thoden eindeutig wiederzufinden und örtlich
das für übertrieben. Mein Vorgänger Prof. zuzuordnen sind. Sie können als eine Art Mar-
Alois Essl replizierte, dass, selbst wenn Gene kierung für das jeweils rundum liegende Stück
mit großer Wirkung gefunden würden, nie- des Genoms dienen. Ein wesentlicher Faktor
mals genug Information über deren Aktion für die genomische Revolution in der Rinder-
zucht war die Entwicklung von Hochdurch-
ZAR-Seminar 2019 – Sölkner – Genomische Selektion – Hintergrund und Geschichte
3
satz-Technologien für die Genotypisierung gemessen, weil es eben zu teuer war, ein
von SNPs (gesprochen: „Snips“, single nucleo- dichtes Marker-Netz zu knüpfen.
tide polymorphisms) bezeichnet. Ein SNP be-
zeichnet die Position einer einzigen Base (von Meine Zeit an der University of
rund 3 Milliarden!), die in einer Population in
Sydney, 2006-2007
mehr als einer Variante vorliegt. Fast 99 % des
Im Jahr 2006 war eine einjährige Gastprofes-
Genoms sind für alle Individuen einer Art
sur „Molecular Animal Breeding“ an der Uni-
identisch, also nicht SNP. Die Technologie zur
versity of Sydney in Australien ausgeschrie-
Auffindung von SNP hat sich rasant entwi-
ben. Weil ich zwar einen guten Ruf als quanti-
ckelt, heute kann man beim Rind und anderen
tativer Genetiker hatte, aber noch wenig er-
Nutztieren rund 50.000 SNP um € 20-30 ge-
fahren mit der Analyse molekularen Daten
notypisieren lassen.
war, bewarb ich mich und bekam diese Stelle.
Im September 2006 übersiedelte ich mit mei-
Genomische Selektion ner Familie nach Sydney und fand Daten von
Der erste Ansatz, genomische Marker in der 15.000 SNP Markern von 1.500 sicher geprüf-
Zucht zu verwenden, wurde „Marker- ten Holstein Friesian Stieren vor, die von ei-
gestützte Selektion“ benannt. Man suchte nem Wissenschafter-Team unter Leitung von
Marker in der Nähe von Genen, für welche Prof. Herman Raadsma bearbeitet wurden.
eine große Wirkung auf ein Merkmal postu- Ich fügte mich in dieses Team ein und war
liert wurde, schätzte den Effekt verschiedener dort so etwas wie die kreative Qualitäts-
Allele und fügte diese Marker in die konventi- Kontrolle des Teams. Die Überprüfung der
onelle BLUP-Schätzgleichung ein. Dies wurden von Meuwissen, Hayes und Goddard (2001)
von manchen Zuchtorganisationen in den vorgeschlagenen Methoden und auch die
1990er und frühen 2000er Jahren implemen- Entwicklung alternativer statistischer Ansätze
tiert, mit recht wenig Erfolg. Das lag wohl der genomischen Selektion waren zentrales
auch daran, dass Marker teuer waren und das Thema. Die Ergebnisse waren hervorragend.
Genom mit vergleichsweise wenigen Markern Im März 2007 kam ich für zwei Wochen nach
sehr schlecht abgedeckt werden konnte. Österreich und präsentierte erste Ergebnisse
im Rahmen des ZAR-Seminars. Die zentrale
In einer Schlüssel-Publikation schlugen Meu-
Folie dieser Präsentation ist in Tabelle 1 wie-
wissen, Hayes und Goddard (2001) einen al-
dergegeben.
ternativen Weg zur Nutzung genetischer Mar-
ker in der Selektion vor, und benannten diese Tabelle 1: Sicherheiten genomischer Zuchtwerte
Gruppe von Methoden „Genomische Selekti- aus Tests beim australischen Holstein Friesian Rind
on“. In einer Simulation zeigten sie, dass ein (Sölkner, 2007)
dichtes Netz von Markern verteilt über das
Genom effektiv für die Vorhersage von
Zuchtwerten genutzt werden kann, ohne vor-
her die Effekte der einzelnen Marker zu prü-
fen und einen großen Teil wegen nicht gesi-
cherter Effekte auszuschließen. Dieser Publi-
kation wurde anfangs wenig Bedeutung zu-
ZAR-Seminar 2019 – Sölkner – Genomische Selektion – Hintergrund und Geschichte
4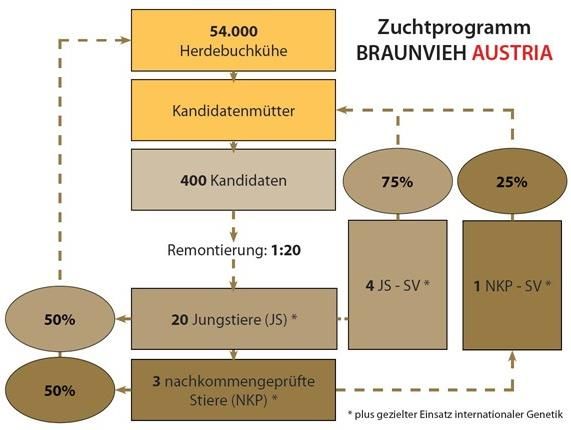
Die Vorstellung, Zuchtwerte mit solchen Si- nicht nur eine große Zahl genetischer Marker,
cherheiten aus dem Blut (also der DNA) jun- sondern auch eine große Referenzpopulation
ger männlicher Kälber lesen zu können, war für die Erstellung von Schätzformeln für die
für die Verantwortlichen der österreichischen genomische Selektion erforderlich ist (z.B. Liu
Rinderzucht, ganz besonders der Fleckvieh- et al., 2011). Bei Holstein Friesian bildeten
zucht, faszinierend. Noch vor meiner Rückrei- sich zwei große Konsortien, die Länder über-
se nach Australien setzten wir uns zusammen greifend genomische Selektion betrieben. Für
und beschlossen gemeinsam, ein Projekt zur die Rasse Braunvieh bildete sich ein einziges
genomischen Selektion für Fleckvieh in Öster- internationales Konsortium mit dem Namen
reich zu entwickeln. Ich nahm Kontakt mit der „Intergenomics“, an dem alle führenden
ZuchtData GmbH auf und Christa Egger- Braunvieh-Länder teilnehmen, darunter na-
Danner war meine wichtigste Partnerin bei türlich auch Österreich. Für die lokalen Rassen
der Projektentwicklung. Zurück in Sydney ar- Tiroler Grauvieh und Pinzgauer führten wir
beitete ich wieder an verschiedenen Metho- gemeinsam mit den Zuchtverbänden ein vom
den der genomischen Selektion, bis hin zu BMLFUW (jetzt BMNT) finanziertes Projekt
Machine Learning, die Ergebnisse waren je- zur potentiellen Einführung der genomischen
doch für viele verschiedene Ansätze ähnlich Selektion bei diesen Rassen durch. Die Ergeb-
gut. Es gab keine Methode, die besser als alle nisse mit jeweils rund 220 sicher geprüften
anderen war. Stieren zeigten keine sehr starke Verbesse-
rung, weshalb zu dieser Zeit (2014) auf eine
Anfänge der genomischen Implementierung verzichtet wurde.
Selektion in Österreich
Anfang 2008 konnten wir bereits mit einem Aktueller Stand zur genomischen
von der Österreichischen Forschungsförde- Selektion
rungsgemeinschaft (FFG) sowie der AGÖF und Zuchtwertschätzung ist traditionell ein übli-
deren Mitgliedsverbänden finanzierten Pro- cherweise höchst komplexes statistisches Ver-
jekt „Entwicklung einer genomischen Zucht- fahren, mit dem man die Effekte des Geno-
wertschätzung für Fleckvieh“ beginnen. Birgt typs und verschiedener Umweltfaktoren mög-
Gredler und Hermann Schwarzenbacher wa- lichst gut trennen will. Die Hinzufügung von
ren durch dieses Projekt finanzierte hervorra- rund 50.000 SNP Markern pro genotypisier-
gende Wissenschafter und wir konnten zei- tem Tier hat das Verfahren weiter kompli-
gen, dass genomische Selektion auch bei ziert. Bis vor recht kurzer Zeit war es nicht
Fleckvieh funktioniert und deshalb implemen- möglich, genotypisierte und nicht genotypi-
tiert werden sollte. Zeitgleich gab es auch ein sierte Tiere in einem Rechengang gemeinsam
Projekt zur genomischen Selektion bei Fleck- zu berücksichtigen. Mit genotypisierten Tiere
vieh in Deutschland und die Vorbereitungen wurde eine genomische Zuchtwertschätzung
zur Implementierung der genomischen Selek- durchgeführt und diese Zuchtwerte wurden
tion liefen gemeinsam bei allen drei Zucht- mit konventionellen Zuchtwerten aus der tra-
wertschätz-Partnern. Im Dezember 2010 ditionellen Pedigree-basierten Zuchtwert-
wurde die erste genomische Zuchtwertschätz- schätzung zu so genannten genomisch opti-
liste bei Fleckvieh veröffentlicht. Internationa- mierten Zuchtwerten verknüpft. Ein in den
le Untersuchungen in dieser Zeit zeigten, dass USA lebender polnischer Wissenschafter, Ig-
ZAR-Seminar 2019 – Sölkner – Genomische Selektion – Hintergrund und Geschichte
5
nacy Misztal, hat gemeinsam mit Kollegen ei- Ich denke auch, dass wir uns für die kleinen
ne Methode entwickelt, eben diese Trennung Rassen überlegen müssen, ob der Zuchtfort-
aufzuheben und die Zuchtwerte genotypisier- schritt genomisch zu verbessern ist. Meine
ter und nicht genotypisierter Tiere in einem Meinung is „ja“ und ich verweise auf den Bei-
Rechengang zu ermitteln, die Single-Step Me- trag von Gabor Meszaros.
thode. Hermann Schwarzenbacher wird diese
und auch andere technische Aspekte der ge- Literatur
nomischen Zuchtwertschätzung in seinem Liu, Z., Seefried, F.R., Reinhardt, F., Rensing, S., Thaller,
Beitrag erörtern. G., Reents, R. (2011). Impacts of both reference
population size and inclusion of a residual polygenic
effect on the accuracy of genomic predic-
Schlussfolgerung tion. Genetics Selection Evolution, 43, 19.
Die Technologie der genomischen Selektion Meuwissen, T.H.E., Hayes, B.J., Goddard, M. E. (2001).
Prediction of total genetic value using genome-wide
hat die bereits zuvor extrem effiziente Rin- dense marker maps. Genetics, 157, 1819-1829.
derzucht noch einmal deutlich verbessert. Na-
Miesenberger, J. (1997). Zuchtzieldefinition und In-
türlich stellt sich wie immer im Zusammen- dexselektion für die österreichische Rinderzucht.
hang mit verbesserten Züchtungstechniken Dissertation, Universität für Bodenkultur Wien.
auch die Frage, ob denn die Richtung der Sölkner, J. (2007). Möglichkeiten der molekularen Rin-
derzucht. ZAR-Seminar 2007, Heffterhof, Salzburg.
Zucht stimmt. Das möchte ich aus meiner
Sicht für die österreichische Rinderzucht be-
jahen.
ZAR-Seminar 2019 – Sölkner – Genomische Selektion – Hintergrund und Geschichte
6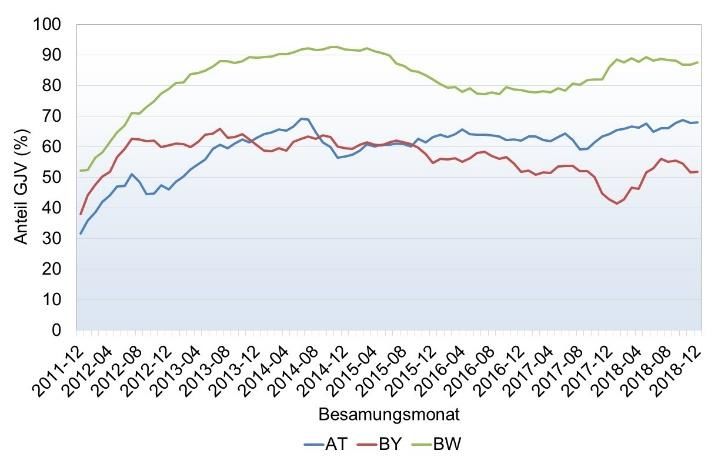
Genomische Zuchtwertschätzung und
Herdentypisierung
Hermann Schwarzenbacher1, Christian Edel2
1ZuchtData EDV-Dienstleistungen GmbH, Wien
2Bayerische Landesanstalt für Landwirtschaft, Institut für Tierzucht
lange Zeit völlig unklar wie verbreitet solche
Einleitung Hauptgene bei züchterisch relevanten Merk-
Wissenschaftler in der Nutztierzüchtung ar- malen sind.
beiten schon viele Jahrzehnte an Methoden
zur Einbeziehung von Information aus geneti- Zuchtprogramme mit markergestützter Selek-
schen Markern in den Selektionsprozess (Sol- tion wurden in einigen Ländern eingeführt,
ler at al. 1976). Dies ist vergleichsweise ein- konnten sich jedoch nicht durchsetzen, da die
fach bei sogenannten „Mendelschen Merk- Kosten-Nutzen Relation meist ungünstig war
malen“, da hier häufig nur ein Genort an der (Bennewitz et al., 2004; Boichard et al. 2006).
Ausprägung des Erscheinungsbildes beteiligt
Die Jahre 2001 mit den Schlüsselpublikatio-
ist. Beispiele hierfür sind rezessive Erbfehler
nen von Meuwissen et al. (2001) zur Metho-
oder genetische Besonderheiten wie die
dik der genomischen Selektion und 2008 mit
Hornlosigkeit.
der Einführung von SNP Chips (Van Tassel et.,
Viel schwieriger gestaltet sich dieses Vorha- 2008) haben dann aber der Molekulargenetik
ben bei polygenen Merkmalen. Solche Merk- in der Tierzüchtung endgültig zum Durch-
male werden durch eine Vielzahl von Genva- bruch verholfen. Was waren die entscheiden-
rianten beeinflusst. Die weitaus größte Grup- den Faktoren, die dies möglich gemacht ha-
pe der Phänotypen, die Tierzüchter interes- ben?
sieren, fällt in diese Kategorie. Erschwerend
kommt hinzu, dass hier Umwelteinflüsse Genomische Zuchtwertschätzung
meist mehr zu den beobachtbaren Unter- Die Zuchtwertschätzung (ZWS) über das Tier-
schieden am Tier beitragen als die Genetik. modell ermöglicht die Abschätzung der gene-
tischen Veranlagung eines Tieres anhand ei-
Da die Genotypisierung vor der Einführung
gener Leistungen bzw. der Leistungen von
der sog. SNP Chips ein kostspieliges und zeit-
verwandten Tieren. Um einschätzen zu kön-
aufwändiges Unterfangen war, war es damals
nen welche Aussagekraft die Eigenleistung
nicht leistbar Nutztiere mit 1000en von Mar-
oder auch Verwandteninformation auf den
kern zu untersuchen. Ausgangspunkt der
Zuchtwert eines Tieres hat, braucht man die
Hoffnungen lag deshalb in der Hauptgenhypo-
Erblichkeit, auch Heritabilität genannt. Un-
these, welche unterstellt, dass auch bei poly-
glücklicher Weise können wir diese nicht mes-
genen Merkmalen einzelne Genvarianten
sen, sondern sind auf statistische Verfahren
existieren, die einen relevanten Anteil der
angewiesen, die uns einen Schätzwert liefern.
Tierunterschiede erklären. Obwohl man ein-
Dabei werden die Leistungen verwandter Tie-
zelne solcher Varianten kannte (z.B. DGAT1,
re miteinander verglichen und darüber ermit-
Grisart et al. 2002, Winter et al. 2002) war
ZAR-Seminar 2019 – Schwarzenbacher – Genomische Zuchtwertschätzung und Herdentypisierung
7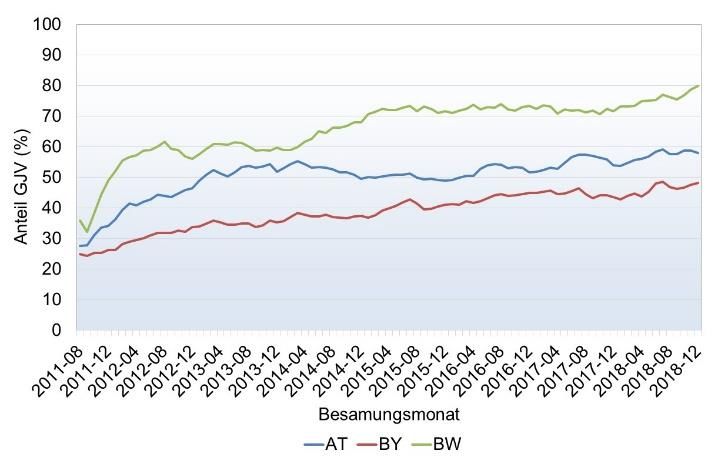
telt, welcher Anteil der beobachtbaren Unter- nomischen Selektion, da darüber der jährliche
schiede auf genetische Einflüsse zurückzufüh- Zuchtfortschritt vor allem über die Verkür-
ren ist. zung des Generationsintervalls erhöht wer-
den kann.
In der eigentlichen ZWS werden nun nicht nur
die Leistungen des einzelnen Tieres betrach- Der Vollständigkeit halber darf nicht uner-
tet sondern es werden Leistungsinformatio- wähnt bleiben, dass es mit dem SNP-BLUP
nen von allen verwandten Tieren im Pedigree Modell („Markermodell“) eine alternative
gleichzeitig herangezogen, um den Zuchtwert Formulierung zum G-BLUP gibt. Im Gegensatz
eines Tieres zu schätzen. Je höher die Erblich- zum G-BLUP werden in diesem Modell Zucht-
keit eines Merkmals dabei ist und umso enger werte für Markereffekte geschätzt. Der
die Verwandtschaft, umso höher ist die Ge- Zuchtwert eines genotypisierten Tieres ergibt
wichtung einer verwandten Leistungsinforma- sich durch simples Multiplizieren der Marker-
tion. Die Verwandtschaft zwischen Tieren effekte mit dem jeweiligen Markergenotyp,
entspricht jedoch nicht dem tatsächlichen An- und anschließendes Aufsummieren über alle
teil herkunftsgleicher Gene, sondern dem Marker in der Schätzung. Obwohl intuitiv
durchschnittlichen Wert aufgrund der Ab- überraschend, führt das SNP-BLUP Modell un-
stammung. So teilen sich bei diesem Verfah- ter bestimmten Umständen zu identischen
ren Enkel beispielsweise mit Großeltern im Genomzuchtwerten wie das G-BLUP Modell.
Durchschnitt 25% gleiche Genvarianten (ohne Diese Modellformulierung hat in Schätzsys-
Berücksichtigung von Inzucht), während der temen mit einer großen Anzahl von genotypi-
tatsächliche Schwankungsbereich aber zwi- sierten Stieren rechentechnische Vorteile.
schen 10 und 40% liegen kann.
Die genomische ZWS als genomisches BLUP In zwei Schritten zum
Modell (G-BLUP) kann nun als Erweiterung Genomzuchtwert
dieser herkömmlichen ZWS verstanden wer- Obwohl durch die Einführung der SNP Chips
den. Da über SNP Chips für jedes genotypi- die Preise für die Erzeugung eines Genotyps
sierte Tier tausende Markergenotypen zur drastisch sanken und der Labordurchsatz ver-
Verfügung stehen, ist es nun möglich die tat- vielfacht wurde, war die Genotypisierung in
sächlichen Verwandtschaften zwischen Tieren den Anfangszeiten der genomischen Selektion
mit extrem hoher Genauigkeit zu bestimmen, immer noch ein teures Unterfangen. Es war
anstatt sich auf die aus der bekannten Ab- daher effizient nur Tiere mit genau geschätz-
stammung ermittelten Durchschnittswerte zu ten Phänotypen zu genotypisieren – beim
verlassen. Dadurch können Leistungsinforma- Rind sind das geprüfte Stiere mit hunderten
tionen von verwandten genotypisierten Tie- bis tausenden von Nachkommenleistungen.
ren nun wesentlich genauer gewichtet wer-
Diese Gruppe bildet die sogenannte Lern-
den. Erstmals war hierduch möglich bei Jung-
stichprobe (LSP), die oft auch als Kalibrierung
tieren ohne Eigen- und Nachkommenleistung
bezeichnet wird. Die Übertragung der Leis-
Zuchtwerte zu schätzen, die vom Mittelwert
tungsinformation von den Nachkommen auf
der Eltern abweichen (=Zufallshälfte). Die
die Stiere der Lernstichprobe geschieht über
frühzeitige Abschätzung der Zufallshälfte ist
eine vorgeschaltete konventionelle Zucht-
beim Rind der entscheidende Vorteil der ge-
wertschätzung. Aus dieser Schätzung werden
ZAR-Seminar 2019 – Schwarzenbacher – Genomische Zuchtwertschätzung und Herdentypisierung
8
dann sogenannte „Pseudo-Phänotypen“ für Phänotypen der LSP nicht repräsentiert ist.
die Tiere der LSP abgeleitet. Diese Informati- Das betrifft beispielsweise Eigenleistungen
onen sind die zusammengefassten und um von Kühen, deren Kälber als Kandidaten ge-
systematische Umwelteinflüsse bereinigten schätzt werden. Der gdZW wird dadurch zum
durchschnittlichen Leistungen der Töchter- offiziell anerkannten genomisch optimierten
gruppen der Stiere. ZW (goZW).
In der G-BLUP ZWS nach dem oben beschrie-
benen Prinzip gehen daher zunächst nur Stie-
re der LSP ein. Die Zuchtwerte aus diesem
Schätzlauf werden als genomisch direkte
Zuchtwerte (gdZW) bezeichnet. Der Vollstän-
digkeit halber darf nicht unerwähnt bleiben,
dass grundsätzlich auch genotypisierte Kühe
mit Eigenleistung in die LSP aufgenommen
werden könnten. Dass genotypisierte Kühe in
der Anfangszeit kaum in nennenswertem Um-
fang anfielen ist wohl der Grund dafür, dass
sie in der Regel in der LSP bis heute nicht be-
rücksichtigt werden. Wollte man dies ändern,
müsste man im Hinblick auf eine zuverlässige
Schätzung jedoch zusätzlich darauf zu achten, Abbildung 1: Schematischer Ablauf der zweistufi-
ob die wenigen bisher sehr selektiv ausge- gen genomischen Zuchtwertschätzung (G-BLUP)
wählten genotypisierten Kühe tatsächlich re- Aus dieser Ablaufbeschreibung wird klar, dass
präsentativ für die zu schätzende Population das aktuelle zweistufige Schätzsystem aus vie-
sind. len Arbeitsschritten besteht. Daraus ergeben
Als Kandidaten werden alle genotypisierten sich neben organisatorischen Erfordernissen
Tiere bezeichnet, die nicht in der Lernstich- auch methodische Schwächen:
probe sind. Das können Kälber sein aber auch Keine Berücksichtigung der genomi-
Kühe mit mehreren abgeschlossenen Laktati- schen Vorselektion:
onen. Da wir keinen Informationsübertrag
Die Genomzuchtwerte eines Kandidaten er- aus der Genomik haben, geht die kon-
rechnen sich aus den genomischen Zuchtwer- ventionelle ZWS wie bisher davon aus,
ten aller Stiere in der LSP sowie deren geno- dass Genomische Jungvererber (GJV)
mischer Verwandtschaften zum jeweiligen zufällige Stichproben ihrer Eltern sind.
Tier. Ein wichtiger Aspekt dabei ist, dass Leis- Der Durchschnitt der Elternzuchtwerte
tungsinformationen aus Kandidaten nicht ist daher der vermeintlich beste
zum Schätzwert aus der Lernstichprobe bei- Startwert und sobald verfügbar wird
tragen. Ebenso beeinflussen sich Genom- Nachkommeninformation herangezo-
zuchtwerte von Kandidaten nicht gegenseitig. gen um die tatsächliche Elternabwei-
Im letzten Schritt wird schließlich die Leis- chung abzuschätzen. Tatsächlich wer-
tungsinformation ergänzt, die in den Pseudo- den aber Kandidaten an den Genom-
ZAR-Seminar 2019 – Schwarzenbacher – Genomische Zuchtwertschätzung und Herdentypisierung
9zuchtwerten streng vorselektiert, was
dazu führt, dass die wahren Zuchtwer- Lektionen aus 10 Jahren Genomik
te von selektierten GJV im Durch- Hunderttausende, beim Holstein gar Millio-
schnitt deutlich über dem wahren El- nen von Genotypen erlauben völlig neue Ein-
ternmittel liegen. Es ist zu erwarten, sichten in die genetische Fundierung von po-
dass konventionelle Zuchtwerte bei lygen bedingten Merkmalen. Diese Befunde
Merkmalen, die unter strenger Selek- sind wichtig um Wege zu identifizieren, wie
tion stehen, im weiteren Verlauf zu- die Leistungsfähigkeit der genomischen ZWS
nehmend unterschätzt werden, vor al- zukünftig gesteigert werden kann.
lem solange noch wenig Information Es gibt (fast) keine wichtigen Genvari-
aus Nachkommen vorliegt. Es gibt in- anten!
zwischen Hinweise aus vielen Ländern, Die Genomforschung hat gezeigt, dass
dass dies zunehmend zum Problem in fast allen züchterisch interessanten
werden dürfte (z.B. Masuda et al., Merkmalen tausende genetische Vari-
2018). Da konventionelle Zuchtwerte anten an der Merkmalsausprägung be-
wiederum Grundlage der G-BLUP teiligt sein dürften. Einzelne Hauptge-
Schätzung sind, ist mit einer Fehler- ne sind in den vielen Merkmalen von
fortpflanzung in die Genomik zu rech- Bedeutung, erklären aber zusammen-
nen. genommen selten mehr als 5 oder
Kein Rückfluss von Information auf 10% der genetischen Variation (God-
nicht genotypisierte Tiere: dard et al. 2016, Weller et al., 2017).
Der SNP-Genotyp eines Kalbes bein- Sehr viele SNPs bringen keine Verbes-
haltet Information zum Genotyp der serung!
Mutter, auch wenn diese selbst nicht Dieser Befund ist auch in Zusammen-
genotypisiert ist. Weist ein Kalb an ei- hang mit den weitgehend fehlenden
nem Marker den Genotyp AC auf und Hauptgenen zu sehen. Vereinfacht ge-
dessen Vater ist reinerbig AA, dann ist sagt ist es nicht entscheidend eine
beispielsweise klar, dass die Mutter kausale Genvariante über dichte Ge-
ein C Allel vererbt hat. Diese indirekte notypisierung zielgenau festzunageln,
Herleitung von Genotypen wird als wenn diese nur 0,01% der genetischen
Genotypen-Imputation bezeichnet Unterschiede erklärt. Zudem ist die
und ist relevant, da hergeleitete Geno- genetische Variabilität unserer Nutz-
typeninformation natürlich auch rinder aufgrund der strengen Selektion
zuchtwertrelevante Informationen im Vergleich zur Tierzahl sehr klein
darstellen. (die effektive Populationsgröße liegt
Im aktuellen zweistufigen Schätzssys- meist zwischen 50 und 200). Das be-
tem bleibt diese jedoch unberücksich- deutet, dass gleiche und vorteilhafte
tigt. Chromosomenabschnitte bei sehr vie-
len Zuchttieren zu finden sind. Es ist
nachvollziehbar, dass es wenig bringt
die Vererbung eines solchen Ab-
schnitts mit hunderten Markern zu
ZAR-Seminar 2019 – Schwarzenbacher – Genomische Zuchtwertschätzung und Herdentypisierung
10verfolgen, da in der genomischen ZWS Die Wiedervereinigung:
jeweils nur ein Effekt für den gesam- Single-Step ZWS
ten Chromsomenabschnitt schätzbar Im Jahr 2009 haben Legarra und Misztal einen
ist. Für das Fleckvieh wurden sowohl Vorschlag zur Zusammenführung von genomi-
mit 780.000 HD Genotypen als auch scher und konventioneller ZWS in ein Verfah-
mit ~12 Mio imputierten Sequenzvari- ren gemacht (Legarra at al. 2009, Misztal et
anten nur marginale Anstiege der Si- al. 2009). Herzstück dieses Ansatzes ist die
cherheiten von Genomzuchtwerten Berücksichtigung der Verwandtschaft aus Ge-
gefunden (Ertl et. al. 2014, Erbe et al. nominformation nicht nur für Genotypisierte
2016). sondern für alle Tiere in der Zuchtwertschät-
zung. Dies wird durch einen Informations-
Teuer aber effektiv: Genotypen und
übertrag erreicht, bei dem beobachtete Ge-
Phänotypen in großer Zahl
notypen und Abstammungsinformationen
Plieschke et al. (2016) zeigen in einer
miteinander kombiniert werden. Dabei wer-
aufwändigen Simulationsstudie, dass
den mit einer bestimmten Genauigkeit Geno-
bei Merkmalen mit 40% Erblichkeit
typen, ausgehend von genotypisierten Tieren,
durch das gezielte Typisieren von bis
ins Pedigree ‚hineingerechnet‘. Dieser Prozess
zu 100 Töchtern pro GJV über mehrere
wird als Genotypenimputation bezeichnet.
Stierjahrgänge die Sicherheiten der
Dadurch entfällt vom Prinzip her die Notwen-
goZW auf 80% gesteigert werden kön-
digkeit, genomische Zuchtwerte und nicht be-
nen. Die große Zahl der Töchtergeno-
rücksichtigte konventionelle Zuchtwerte
typen mit den zugehörigen Leistungs-
nachträglich miteinander zu kombinieren.
informationen erlauben eine wesent-
lich genauere Abschätzung der Unter- Analog zum zweistufigen SNP-BLUP Verfahren
schiede zwischen den einzelnen alter- gibt es auch im Single-Step eine äquivalente
nativen Chromosomenabschnitten die Modellformulierung über die Zuchtwerte zu-
dieser Vater an seine Töchter weiter- nächst für SNP Marker geschätzt werden.
gegeben hat. Dies gilt aber auch für Tierzuchtwerte können dann wie oben be-
die Zuchtwerte einzelner Chromoso- schrieben aus Markereffekten errechnet wer-
menabschnitte dieses Stieres. Da diese den.
Chromosomenabschnitte über zwei bis
drei Generationen relativ stabil wei- Der Rechenaufwand für die Lösung einer Sin-
tergegeben werden ist es dadurch gle-Step ZWS ist stark abhängig von der An-
besser möglich einzuschätzen, wie sich zahl der Genotypen. Problematisch ist vor al-
die Weitergabe von Chromosomen an lem, dass sich der Rechenaufwand sowie der
die Nachkommen auf deren Zuchtwert Speicherbedarf nicht linear zur Anzahl der
auswirkt. Die Autoren zeigen in ihrer Genotypen verhält. Anwendungen in großen
Studie aber auch auf, dass es wichtig Populationen sind daher nur über leistungs-
ist repräsentative Töchterinformatio- fähige Rechner und vor allem hochentwickel-
nen zu bekommen. Bei vorselektierten te Software wie MiX99 (Lidauer et al. 2017)
Töchtergruppen geht der Sicherheits- möglich.
zugewinn fast vollständig verloren.
ZAR-Seminar 2019 – Schwarzenbacher – Genomische Zuchtwertschätzung und Herdentypisierung
11figen Verfahren bei Merkmalen unter
starker genomischer Vorselektion eine
Verzerrung konventioneller und in
weiterer Folge auch genomischer
Zuchtwerte zu erwarten. Im Single-
Step Verfahren wird jedoch der geno-
mischen Vorselektion Rechnung getra-
gen, da in diesem Verfahren nicht der
konventionelle sondern der genomi-
sche Ahnenindex der beste Startwert
eines vorselektierten Besamungsstie-
res ist, bevor Töchterleistungen auf-
laufen. Zu berücksichtigen ist jedoch,
dass es bisher noch keine langfristigen
Abbildung 2: Schematischer Ablauf der Single- Erfahrungen aus Routineanwendun-
Step Zuchtwertschätzung gen mit diesem Zuchtwertschätzver-
fahren gibt.
Vorteile des neuen Verfahrens Bereits in diesem Jahr wird es voraussichtlich
Optimale Nutzung der verfügbaren In- zur Routineeinführung des neuen Verfahrens
formation für die genomische Vorher- bei allen Exterieurmerkmalen in der Rasse
sage Fleckvieh kommen. Auch bei den anderen
Jedes Tier mit Leistungsinformation Merkmalen und beim Braunvieh ist in den
trägt in diesem Verfahren zur Schät- nächsten Jahren mit einer stufenweise Um-
zung bei. Dies betrifft vor allem geno- stellung auf das neue Verfahren zu rechnen.
typisierte Stiere und Kühe, aber, auf-
grund der Genotypenimputation, auch
Herdentypisierung
untypisierte Tiere unter Leistungsprü-
Seit Einführung der Genomischen Selektion
fung. Indirekt können sogar genotypi-
(GS) vor 10 Jahren hat sich der Schwerpunkt
sierte Tiere ohne Leistung zur Schät-
der Genotypisierung zunehmend auf weibli-
zung beitragen, indem sie helfen den
che Tiere verlagert. Die Nutzung von GS zur
Genotyp von verwandten Tieren mit
genomischen Vorselektion von Stieren wurde
Leistung zu imputieren. Ein Beispiel
daher zusehends erweitert auf die Selektion
wäre eine untypisierte Kuh mit mehre-
interessanter Kalbinnen für die Hochzucht
ren genotypisierten Nachkommen.
(z.B. Embryotransfer) und in weiterer Folge
Daher ist dieses Verfahren auch opti-
auf die innerbetriebliche Selektion der Nach-
mal um Informationen aus Herdenty-
zucht. Die Frage ob und wie umfangreich
pisierungsprojekten wie FoKUHs oder
weibliche Tiere mit Phänotypendaten geno-
Braunvieh Vision in die genomische
typisiert werden, ist heute zu einem ent-
ZWS zu integrieren.
scheidenden Faktor im Wettbewerb der Rin-
Berücksichtigung der genomischen derpopulationen geworden. Vorreiter dieser
Vorselektion Entwicklung sind wie so oft die USA mit aktu-
Wie oben beschrieben ist im zweistu- ell 2,43 Mio Genotypen, 91% davon weiblich
ZAR-Seminar 2019 – Schwarzenbacher – Genomische Zuchtwertschätzung und Herdentypisierung
12(https://queries.uscdcb.com/Genotype/cur_ctry.h
tml). Auch in unserer genomischen ZWS ist
diese Entwicklung in abgeschwächter Form zu
beobachten (Abbildung 3).
Abbildung 3: Entwicklung der Genotypisierungszahlen beim Fleckvieh (links) und Braunvieh
(rechts) nach Geschlecht und Geburtsjahrgang in der gemeinsamen genomischen ZWS (Stand
03/19).
Besonders wichtig wird die Genotypisierung verwundert daher nicht, dass in vielen Län-
von weiblichen Tieren bei Merkmalen die erst dern Projekte zur systematischen Genotypi-
seit kurzer Zeit erhoben werden. Ein Beispiel sierung von weiblichen Tieren initiiert wur-
hierfür sind direkte Gesundheitsmerkmale auf den. Das Ziel dieser Programme ist der Auf-
der Basis von tierärztlichen Diagnosen bau einer Kuh-Lernstichprobe, meist mit dem
und/oder Beobachtungen durch den Tierhal- Schwerpunkt auf Gesundheitsmerkmalen. Die
ter. Hier ist der Aufbau einer Kalibrierung Tabelle 1 gibt einen Überblick über laufende
über geprüfte Stiere ein langwieriger Prozess Herdentypisierungsprojekte in Österreich und
der mindestens 10 bis 15 Jahre in Anspruch Deutschland. Im Rahmen des Projektes
nimmt. Über die direkte Einbeziehung von FoKUHs wurden bisher 13.500 FV, 1.400 BV
genotypisierten Kühen, die für das Merkmal und 1.700 HF Typisierungen durchgeführt
unter Leistungskontrolle stehen, kann hinge- (Stand März 2019). Die Vollständigkeit der
gen innerhalb weniger Jahre eine genomische Phänotypenerfassung wird im Projekt laufend
Zuchtwertschätzung etabliert werden. Es überprüft.
ZAR-Seminar 2019 – Schwarzenbacher – Genomische Zuchtwertschätzung und Herdentypisierung
13Tabelle 1: Übersicht über derzeit laufende Herdentypisierungsprojekte in Österreich und Deutschland
(Stand 03/2019).
Projekt FoKUHs Braunvieh Vision Fleckfficient KuhVision
Land Österreich Bayern, Baden- Baden- Deutschland (incl.
Württemberg Württemberg AT)
Laufzeit 01.18-12.22 07.17-06.20 01.19-12.21 06.16-09.20
Anzahl 463 182 200 1.250
Betriebe 346-FV (Stand 02.19) (Stand 01.19,
57-BV inkl.
60-HF (Teiln. an Herdentypisierung
Kuhvision)
Rassen FV, BV, HF BV FV HF
Anzahl Geno- 35.000 (FV) 38.000 20.000 Ziel 250.000
typisierungen 5.500 (BV) aktuell>300.000
5.500 (HF) (Stand 01.19)
Erfasste Ein Programm für Ein Programm für Mehrere Mehrere
Phänotypen alle alle Programme: Programme:
Basis, Premium, Basis, Basis+,
Premium+ Basis++
Standardmerkmale Standardmerkmale Standardmerkmale Standardmerkmale
Gesundheitsdaten Gesundheitsdaten Gesundheitsdaten Gesundheitsdaten
über GMON als über ProGesund über GMON; Kälbe- über GMON;
tierärztl. Diagnosen und GMON als Di- rerkrankungen
agnosen und Be- incl.Trinkschwäche
obachtungen; Käl-
bererkrankungen
incl. Trinkschwäche
Stoffwechsel Verhalten: (Saug-
(Ketotests) verhalten, Melkver-
halten, Kuhcharak-
ter)
Lebendgewichte
und Körpermaße
Klauenpflegedaten Klauenpflegedaten Klauenpflegedaten
lineare lineare lineare lineare
Beschreibung Beschreibung Beschreibung Beschreibung
In Bayern ist der Start des Fleckviehprojekts „FleQS“ für den 1. Mai 2019 geplant.
Die Herausforderungen die sich aus der Viel- Zum einen müssen weibliche Tiere mit Leis-
zahl Projekte für die genomische Zuchtwert- tungsinformationen für bestehende Merkma-
schätzung ergeben sind beträchtlich. le sukzessive in die Lernstichprobe integriert
werden. Dies wird aus heutiger Sicht über die
ZAR-Seminar 2019 – Schwarzenbacher – Genomische Zuchtwertschätzung und Herdentypisierung
14Umstellung auf die Single-Step ZWS gesche- Grisart et al., (2002): Positional candidate cloning of a
QTL in diry cattle. Identification of a missense muta-
hen. Es gibt aber auch eine Reihe von Merk- tion in the bovine DGAT1 gene with major effect on
malen aus dem Gesundheitsbereich, für die es milk yield and composition. Genome Ges. 12:222-
entweder noch keine genomischen Zuchtwer- 231.
te gibt (Mastitis, frühe Fruchtbarkeitsstörun- Legarra, A. et al. (2014): Single step, a general approach
for genomic selection. Livest. Sci. 166:54-65.
gen, Zysten, Milchfieber), oder für die völlig
Legarra, A., et al. (2009): A relationship matrix including
neue ZWS entwickelt werden müssen
full pedigree and genomic information. J. Dairy Sci.
(Klauenbereich, Kälbererkrankungen, Verhal- 92:4656-4663.
tensmerkmale). Lidauer, M., K. Matilainen ,E. Mäntysaari, T. Pitkänen,
M. Taskinen und I. Strandén. 2017 MiX99. General
Entscheidend für den langfristigen Erfolg der program for solving large mixed model equations
Projekte wird sein, ob es gelingt eine Verste- with preconditioned conjugate gradient method.
Relase XI/2017, Jokioinen, Finland.
tigung der Herdentypisierung in Österreich
Masuda et al. (2017): Differing genetic trend estimates
und Deutschland nach dem Auslaufen der from traditional and genomic evaluations of geno-
Fördermittel zu erreichen. Betriebe werden typed animals as evidence of preselection bias in US
nach nüchternen Kosten-Nutzen Überlegun- Holsteins. J. Dairy Sci. 101:5194-5206.
gen entscheiden. Daher ist es wichtig einen Meuwissen THE. et al. (2001): Prediction of total genet-
ic value using genome-wide dense marker maps.
unmittelbaren Nutzen für den Betrieb zu ge- Genetics 157. 1819-1829.
nerieren. Dafür sind neben aussagekräftigen Misztal, I., et al. (2009): Computing procedures for ge-
Genomzuchtwerten, dem einfachen Zugang netic evaluation including phenotypic, full pedigree,
zur genomischen Untersuchung und günsti- and genomic information. J. Dairy Sci. 92.4648-
4655.
gen Typisierungspreisen auch Software Lö-
Plieschke L. et al. (2016): Systematic genotyping of
sungen im RDV notwendig, die die Nutzung groups of cows to improve genomic estimated
von Genomik für die Zuchtarbeit am Betrieb breeding values of selection candidates. Genet. Sel.
unterstützen. Evol. 48:73.
Soller et al. (1976): On the power of experimental de-
signs for the detection of linkage between marker
Literatur: loci and quantitative loci in crosses between inbred
Bennewitz J. et al. (2004): Top down preselection using lines. Theor. Appl. Genet. 47(1):35.9.
marker assisted estimates of breeding values in VanTassel, C.P., et al. (2008): SNP discovery and allele
dairy cattle. J. Anim. Breed. Genet. 121:307-318. frequendy estimation by deep sequencing of re-
Boichard D. et al. (2006): Implementation of marker- duced representation libraries. Nat. Methods 5:
assisted selection: Practical lesion from dairy cattle. 247.252.
Proc. 8th World Cong. Genet. Appl. Livest. Prod., Be- Weller et al. (2017): Invited review: A perspective on
lo Horizonte, MG Brazil. the future of genomic selection in dairy cattle. J.
Erbe et al. (2016): Genomic prediction using imputed Dairy Sci. 100: 8633-8644.
sequence data in dairy and dual purpose breeds. Winter et al. (2002): Association of a lysine-232/alanine
Journal of Animal Science 94(supplement5):198. polymorphism in a bovine gene encoding acyl-
Ertl et al. (2014): On the limited increase in validation CoA:diacylglycerol acyltransferase (DGAT1) with var-
reliability using high-density genotypes in genomic iation at a quantitative trait locus for milk fat con-
best linear unbiased prediction: observations from tent Proc. Natl. Acad. Sci. USA99: 9300-9305.
Fleckvieh cattle. J Dairy Sci. 2014;97(1):487-96. doi:
10.3168/jds.2013-6855.
Goddard et al. (2016): Genetics of complex traits: pre-
diction of phenotype, identification of causal poly-
morphisms and genetic architecture. Proc. Biol. Sci.
283:20160569.
ZAR-Seminar 2019 – Schwarzenbacher – Genomische Zuchtwertschätzung und Herdentypisierung
15Aspekte zur Optimierung von genomischen Zucht-
programmen
Egger-Danner1, Alfons Willam2
1ZuchtData EDV-Dienstleistungen GmbH, Wien
2Universität für Bodenkultur Wien (BOKU), Institut für Nutztierwissenschaften (NUWI)
teils auf Analysen im Rahmen des For-
1. Einleitung schungsprojektes OptiGene (Egger-Danner et
Preisstürze bei Genomanalysen ermöglichten al., 2015a). Im Beitrag von (Fürst et al., 2019)
die Entwicklung und Einführung der genomi- wird der aktuelle Stand der Umsetzung der
schen Selektion in den Routinebetrieb inner- Zuchtprogramme evaluiert.
halb weniger Jahre. Weltweit wurden die
Zuchtprogramme aufbauend auf diesen Mög-
2. Evaluierung eines
lichkeiten umstrukturiert. Durch die Möglich-
keiten der genomischen Selektion liegen von Zuchtprogrammes
Jungtieren ohne Eigenleistung oder Nach- 2.1 Methode
kommenprüfung bereits Zuchtwerte mit Si- Die Analyse von Optimierungsschritten bzw.
cherheiten von 50-70% vor. Große Steigerun- Auswirkungen von verschiedenen Strategien
gen bei den Zuchtfortschritten von bis zu bei der Gestaltung von Zuchtprogrammen
100% wurden oftmals prognostiziert (u.a. wurde im Rahmen des Projekte OptiGene
Schaffer, 2006). Zuchtziele, Möglichkeiten der (Egger-Danner et al. 2015a) mit dem Compu-
Leistungsprüfung, Zuchtwertschätzung und terprogramm ZPLAN (Willam et al., 2008)
Zuchtprogramme wurden überarbeitet und durchgeführt. ZPLAN optimiert Selektionsstra-
werden laufend an die neuen Möglichkeiten tegien in der Tierzucht bei Verwendung eines
angepasst. Um die laut Zuchtziel angestrebten deterministischen Ansatzes aufbauend auf
Zuchtfortschritte zu realisieren, ist es wichtig, der Genflussmethode und einem Selektions-
dass aussagekräftige Merkmale aus der Leis- index. Die genetische und ökonomische Effizi-
tungsprüfung für die Zuchtwertschätzung zur enz von Zuchtprogrammen kann evaluiert
Verfügung stehen. Im Gesamtzuchtwert sind werden. Selektionsgruppen mit unterschiedli-
die Merkmale nach ihren Erheblichkeiten, ge- chen Selektionsintensitäten und individuellen
netischen Beziehungen untereinander und Informationsquellen können im Index defi-
den wirtschaftlichen Gewichten kombiniert niert werden. Zusätzlich müssen für alle Se-
um die gewünschten Zuchtfortschritte zu er- lektionsgruppen verschiedene biologische
zielen. Die Informationen aus der Zuchtwert- Kennzahlen, Populations- und Kostenparame-
schätzung sind die Grundlage für die Auswahl ter definiert werden. Informationen zu den
der interessantesten Zuchttiere gemäß verwendeten biologischen, genetischen und
Zuchtprogramm. Im vorliegenden Beitrag ökonomischen Parametern sind in Egger-
werden verschiedene Möglichkeiten zur Op- Danner et al. (2012b) dargestellt. Als Kriteri-
timierung eines genomischen Zuchtprogram- um für die Evaluierung wird in diesem Beitrag
mes am Beispiel Fleckvieh AUSTRIA aufge- der monetäre Zuchtfortschritt pro Jahr
zeigt. Die Ausführungen beziehen sich groß-
ZAR-Seminar 2019 – Egger-Danner – Aspekte zur Optimierung von genomischen Zuchtprogrammen
16(monZF/J herangezogen. Dieser ist folgen- 2.3 Genomisches Zuchtprogramm
dermaßen definiert: Fleckvieh AUSTRIA
Monetärer Zuchtfortschritt (ZF): Der Zucht- Abbildung 2: Zuchtprogramm Fleckvieh AUSTRIA
fortschritt ist die durchschnittliche monetäre 2012
bzw. naturale Überlegenheit der Nachkom-
men der selektierten Tiere einer Selektions-
runde gegenüber der Elterngeneration in der
Zuchtstufe (pro Generation oder pro Zeitein-
heit).
2.2 Faktoren (Hebel) im Zuchtprogramm
für Zuchtfortschritt
Die Formel zur Berechnung des Zuchtfort-
schrittes ist in Abbildung 1 dargestellt.
Abbildung 1: Mathematische Formulierung des
Zuchtfortschrittes (ZF) pro Zeiteinheit (T) und Fak-
toren für Zuchtfortschritt
Das genomische Zucht-
programm Fleckvieh
AUSTRIA wurde 2012
von der Arbeitsgemein-
schaft österreichischer
Fleckviehzüchter mit ei-
nem angestrebten Anteil
an Jungstierbesamungen
bei den Herdebuchkü-
hen von 50% und von
75% in der gezielten
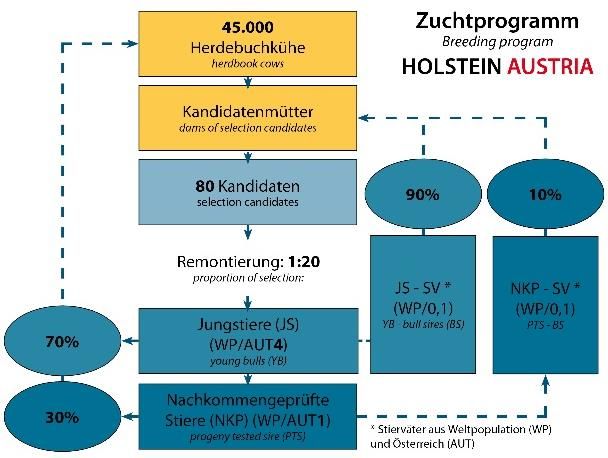
Der Zuchtfortschritt pro Jahr wird durch die Paarung beschlossen. Als weiteres Ziel wurde
Remontierung (Selektionsintensität (i)), die festgelegt, dass aus 20 Kandidaten jeweils ein
genetische Streuung des Merkmals (sa), die Jungstier selektiert wird (1:20). Die 50% der
Genauigkeit der Zuchtwertschätzung (rAgA) Jungstierbesamungen an den Herdebuchkü-
und das Generationsintervall (T) bestimmt. hen sollen mit 60 jedes Jahr selektierten
Der Zuchtfortschritt wird wiederum für die Jungstieren durchgeführt werden, die besten
verschiedenen Selektionsgruppen berechnet. 10 Nachkommen geprüften Stiere sollen jähr-
lich selektiert werden. Die Remontierungsrate
wurde inzwischen angepasst und liegt aktuell
bei 1:40.
ZAR-Seminar 2019 – Egger-Danner – Aspekte zur Optimierung von genomischen Zuchtprogrammen
173. Optimierungsschritte im dargestellt. Bei GS50 werden 50 % aller Be-
Zuchtprogramm samungen mit Jungstieren mit genomisch op-
timierten Zuchtwerten durchgeführt, bei
Im vorliegenden Kapitel wird auf die Möglich- GS100 werden nur mehr Jungstiere einge-
keiten von Optimierungsschritten eingegan- setzt.
gen. Die Diskussion orientiert sich an den all-
gemeinen Stellschrauben in einem Zuchtpro- Tabelle 1: Monetärer Zuchtfortschritt pro Jahr in
Abhängigkeit von der Sicherheit der genomisch
gramm.
optimierten Zuchtwerte für drei Varianten eines
Begriffe bei den Darstellungen: genomischen Zuchtprogrammes (GS50, GS75,
GS50: Genomisches Zuchtprogramm, bei dem GS100)
50% der Besamungen der Herdebuchkühe und GS50 GS75 GS100
Stiermütter mit Jungstieren durchgeführt wer-
GZW-Si-49% 97,0 (-3,0) 101,8 (-4,3) 107,5 (-6,1)
den.
GZW-Si-58% 100,0 106,1 113,6
GS75: Genomisches Zuchtprogramm, bei dem
75% der Besamungen der Herdebuchkühe und GZW-Si-69% 103,0 (+3,0) 110,6 (+4,5) 120,0 (+6,4)
Stiermütter mit Jungstieren durchgeführt wer-
GZW-Si-80% 105,5 (+5,5) 114,3 (+8,2) 125,5 (+11,9)
den.
GI (Jahre) 4,69 4,13 3,57
GS100: Genomisches Zuchtprogramm, bei
dem 100% der Besamungen der Herdebuch-
kühe und Stiermütter mit Jungstieren durchge- Wie Tabelle 1 zeigt, erhöht sich der monetäre
führt werden. Zuchtfortschritt pro Jahr bei einer Steigerung
der Sicherheit der goGZW von 49% auf 58%
3.1 Genauigkeit und Zuverlässigkeit der um 3%, wenn 50% aller Kühe mit Jungstieren
Zuchtwerte mit genomisch optimierten Zuchtwerten be-
Durch die genomische Zuchtwertschätzung samt werden. Je höher der Anteil der Jung-
liegen bereits von Jungstieren Zuchtwerte mit stierbesamungen ist, desto positiver wirkt
Sicherheiten für den genomisch optimierten sich eine höhere Sicherheit aus. Bei GS100,
Gesamtzuchtwert (goGZW) von 60-65% vor. einem rein genomischen Zuchtprogramm,
Im Vergleich zum vorgeschätzten Zuchtwert bringt eine Steigerung der Sicherheit von ca.
aus den Elternzuchtwerten (Pedigree-Index) 10- Prozentpunkten einen jeweils um ca. 6%
wird hier ein Anstieg der Sicherheit um 20-30 höheren Zuchtfortschritt pro Jahr. Die Sicher-
Prozentpunkte je nach Merkmal erzielt. Ist heit der genomisch optimierten Zuchtwerte
der Vater von diesem Jungstier wiederum ein spielt allerdings auf Ebene der Gesamtpopula-
Jungstier, so liegen die Sicherheiten ca. 7 Pro- tion eine geringere Rolle als z.B. die Effekte
zentpunkte niedriger. der Reduktion des Generationsintervalls (Ab-
In Tabelle 1 sind die Auswirkungen von höhe- bildung 3).
ren Sicherheiten der genomisch optimierten Durch die Möglichkeiten der genomischen Se-
Zuchtwerte auf den monetären Zuchtfort- lektion gewinnt die weibliche Seite im Zucht-
schritt pro Jahr bei unterschiedlichen Anteilen programm stärker an Bedeutung. Durch die
Jungstierbesamungen im Zuchtprogramm
ZAR-Seminar 2019 – Egger-Danner – Aspekte zur Optimierung von genomischen Zuchtprogrammen
18Genotypisierung kann auch auf der weibli-
3.2 Erblichkeit/Variabilität der
chen Seite strenger selektiert werden. Die
Merkmale
Genotypisierung von Stiermüttern gewinnt
Wesentlich für die Erzielung von Zuchtfort-
deshalb an Interesse. Basierend auf den An-
schritt für die gewünschten Merkmalsberei-
nahmen im Zuchtprogramm Fleckvieh AUS-
che sind aussagekräftige Parameter aus der
TRIA wurden Berechnungen zur Genotypisie-
Leistungsprüfung mit hohen Erblichkeiten. Die
rung von Stiermüttern durchgeführt. Wenn
Erblichkeiten für die Milch-, Fleisch- und Exte-
Kalbinnen-Stiermütter genotypisiert werden,
rieurmerkmale liegen üblicherweise im mitt-
kann dadurch die Sicherheit deutlich gestei-
leren bis hohem Bereich (20-50%), Merkmale
gert werden (ohne Genotypisierung ca. 35%
wie Zellzahl oder Nutzungsdauer liegen zwi-
Si, mit Genotypisierung ca. 61% Si). Tabelle 2
schen 10 und 15% und Gesundheitsmerkmale
zeigt, dass der Nutzen der alleinigen Genoty-
weisen meist nur eine Erblichkeit von unter
pisierung der Stiermütter sehr gering ist. Je
5% auf. Verschiedene Studien zeigen jedoch,
nach Anteil Jungstierbesamungen ist mit einer
dass es sehr wohl Unterschiede in den Er-
Erhöhung von 1,8 bis 2,4% monetärer Zucht-
blichkeiten bei einzelnen Merkmalen in Ab-
fortschritt zu rechnen. Wenn die Genotypisie-
hängigkeit von der Datenquelle und Datener-
rung mit einer Verkürzung des Generationsin-
fassung gibt, z.B. umfassend und standardi-
tervalles verbunden ist, weil mehr Kalbinnen
siert dokumentierte Klauenpflegedaten von
bereits gezielt angepaart werden, steigt der
ausgebildeten Klauenpflegern lassen höhere
zu erzielende relative Zuchtfortschritt auf 4,5
Erblichkeiten erwarten. Die neuen Möglich-
bzw. 6,5%. Ein weiterer positiver Effekt ist die
keiten aus den Automatisierungen lassen
Nutzung von Reproduktionstechnologien bei
auch Fortschritte u.a. im Bereich der Gesund-
den genetisch interessantesten weiblichen
heit erwarten. Gelingt es nicht von Zielmerk-
Tieren.
malen eine umfangreiche Leistungsprüfung
Tabelle 2: Auswirkungen der Genotypisierung von aufzubauen, sind Hilfsmerkmale das Mittel
Stiermüttern auf den monetären Zuchtfortschritt der Wahl.
pro Jahr
GS50 GS75 GS100 3.3 Generationsintervall
Ø GI Jahre 4,69 4,13 3,57
Die Verkürzung des Generationsintervalls ist
die wirkungsvollste Maßnahme in einem ge-
Basis 100,0 106,1 113,6 nomischen Zuchtprogramm (Schaeffer, 2006).
Basis+STM 101,8 108,2 (+2,1) 116,0
In Abbildung 3 ist die Auswirkung der Reduk-
(+1,8) (+2,4) tion des Generationsintervalls durch Erhö-
Basis+STM-GI 104,5 111,5 (+5,4) 120,1 hung des Anteils an Jungstierbesamungen auf
reduz. Ø GI (+4,5) 4,01 (+6,5) den monetären Zuchtfortschritt pro Jahr dar-
Jahre 4,57 3,45 gestellt. Bei GS20 werden 20% der Besamun-
gen mit Jungstieren durchgeführt, bei GS50
50% und bei GS100 werden nur mehr Jung-
stiere eingesetzt. Dieser Prozentanteil bezieht
sich auf die Herdebuchkühe als auch auf die
Stiermütter. Durch die Steigerung des Anteils
ZAR-Seminar 2019 – Egger-Danner – Aspekte zur Optimierung von genomischen Zuchtprogrammen
19Jungstierbesamungen von 20 auf 100% wird Zuchtfortschritt haben die Pfade der Mütter
der monetäre Zuchtfortschritt pro Jahr um und Väter der Stiere. Je strenger hier selek-
20,8% gesteigert werden. Das Generationsin- tiert wird, desto höher ist der zu erwartende
tervall wird durch diese Maßnahme insgesamt Zuchtfortschritt. Wenn jedoch Tiergruppen
um 1,8 Jahre reduziert. aufgrund von bestimmten Eigenschaften aus
der Zucht ausgeschlossen werden, so redu-
Abbildung 3: Monetärer Zuchtfortschritt pro Jahr
ziert das den Zuchtfortschritt. Eine sehr
und durchschnittliches Generationsintervall nach
strenge Selektion auf bestimmte Linien er-
Anteil Jungstier-Besamungen (Egger-Danner et al.
2015) höht die Inzucht. Die breite Genotypisierung
von Kühen bietet
auch die Chance,
dass interessante
Kühe gefunden
werden, die bisher
nicht aufgefallen
sind. Die Selekti-
onsintensität kann
auch über die
Vermehrungsrate
gesteigert werden.
Hier bieten Embry-
otransfer und wei-
Bei Fleckvieh AUSTRIA lag der Anteil an Jung- tere Reproduktionstechniken Möglichkeiten
stierbesamungen im Kontrolljahr 2018 bei den Zuchtfortschritt über die Selektionsinten-
50%. Dies ist der Anteil, wie er im Jahr 2012 sität aber auch über das Generationsintervall
beschlossenen Zuchtprogramm Fleckvieh zu steigern.
AUSTRIA festgelegt wurde. Das Alter der Vä-
Vorselektion der Jungstiere
ter der genomischen Jungstiere hat sich bei Durch die genomische Selektion können die
Fleckvieh in Österreich von 2010 von 7 Jahren zukünftigen Vererber bereits als Kalb selek-
auf 3,4 Jahre 2018 reduziert; auf der weibli- tiert werden. Wurden früher Teststiere auf-
chen Seite von 4,8 auf 3,2 Jahre. Um den grund des durchschnittlichen Elternzuchtwer-
Zuchtfortschritt zu steigern, ist es empfeh- tes (Pedigree-Index) und Eigenleistungsinfor-
lenswert den Anteil an Jungstierbesamungen mationen selektiert, so werden jetzt gene-
zu erhöhen. tisch interessante Kälber genotypisiert und in
erster Linie aufgrund des genomisch optimier-
3.4 Selektionsintensität ten Zuchtwertes für den Besamungseinsatz
ausgewählt. Diese Möglichkeit der Vorselekti-
3.4.1 Allgemein
on bietet die Chance den Zuchtfortschritt pro
Um die Selektionsintensität zu steigern bzw.
Jahr zu steigern. Tabelle 3 zeigt die Auswir-
hoch zu halten, ist es wichtig, dass in allen Se-
kungen auf den monetären Zuchtfortschritt
lektionspfaden die besten Tiere für die Zucht
pro Jahr in der Zuchtstufe in Euro für drei ver-
selektiert werden. Den größten Anteil am
ZAR-Seminar 2019 – Egger-Danner – Aspekte zur Optimierung von genomischen Zuchtprogrammen
20Sie können auch lesen

















































