Calciumantagonisten in der kardiovaskulären Therapie
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Diplomarbeit
Calciumantagonisten
in der kardiovaskulären Therapie
eingereicht von
Patrick Woschank
zur Erlangung des akademischen Grades
Doktor(in) der gesamten Heilkunde
(Dr. med. univ.)
an der
Medizinischen Universität Graz
ausgeführt am
Institut für Experimentelle und Klinische Pharmakologie
unter der Anleitung von
Univ.-Prof. Dr. med. univ. Josef Donnerer
Graz, am 17.03.2016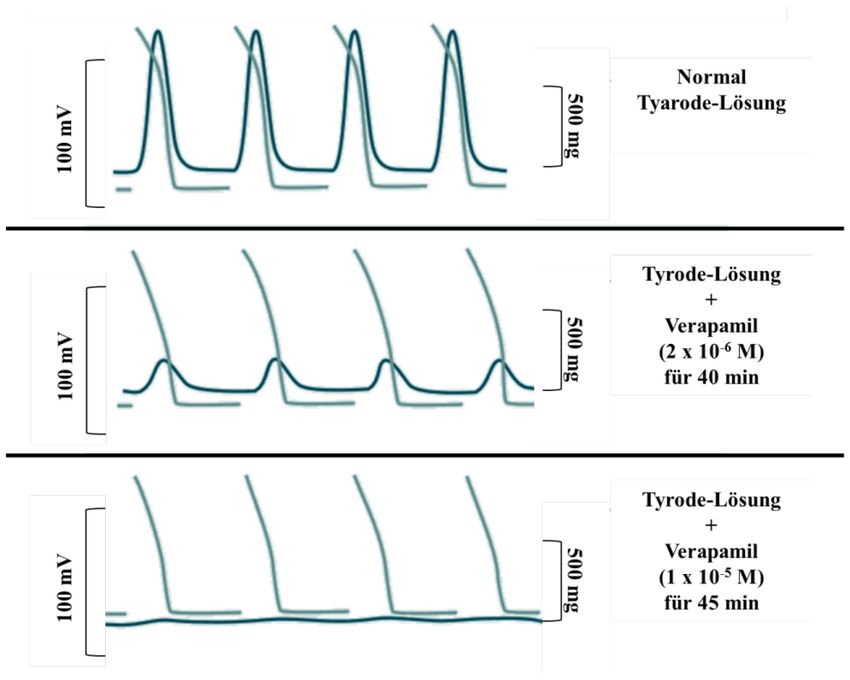
Eidesstattliche Erklärung
Ich erkläre ehrenwörtlich, dass ich die vorliegende Arbeit selbstständig und ohne fremde
Hilfe verfasst habe, andere als die angegebenen Quellen nicht verwendet habe und die den
benutzten Quellen wörtlich oder inhaltlich entnommenen Stellen als solche kenntlich
gemacht habe.
Graz, am 17.03.2016 Patrick Woschank e.h.
ii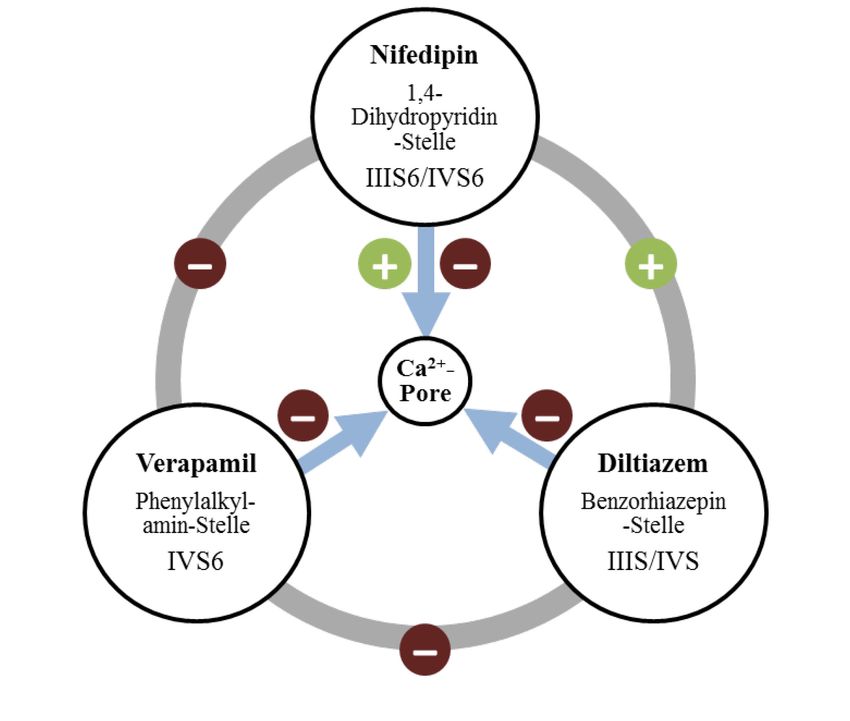
Danksagungen
Allen voran möchte ich mich bei meinem Betreuer Univ.-Prof. Dr. Donnerer für die
Bereitstellung dieses interessanten und auch für meine Zukunft essentiellen Themas und
seine Betreuung recht herzlich bedanken.
Ein besonderer Dank gilt meinem Bruder Manuel, welcher mir stets mit gutem Rat und
fachlichem Input zur Seite stand und dadurch essentiell zum Gelingen dieser Arbeit
beitrug.
Des Weiteren möchte ich meinen Studienkollegen und Freunden, insbesondere Andreas
Schleinzer und Patrick Fritzl, für ihre Freundschaft und ihren Beistand danken.
Zu guter Letzt möchte ich noch besonders meinen Eltern für ihre unermüdliche
Unterstützung und Motivation Dank aussprechen.
iii
Zusammenfassung
Calciumantagonisten (CCB) umfassen eine heterogene Gruppe von Wirkstoffen mit
typischer Struktur und pharmakologischen Charakteristika und finden Anwendung in einer
Vielzahl von kardiovaskulären Erkrankungen. Die kontinuierliche Weiterentwicklung von
Substanzen, Nutzen-Risiko-Analysen, neuen Erkenntnissen aus großangelegten, klinischen
Langzeit-Studien, sowie der Vergleich mit alternativen Herz-Kreislauf-Medikamenten
führten im Laufe der Zeit zu unterschiedlichen Präferenzen und auch zu inkohärenten
Ideologien bezüglich deren klinischen Gebrauchs. In dieser Diplomarbeit werden nun die
historische Entwicklung, das Wirkstoffprofil und klinische Anwendungsgebiete von CCB
in der Therapie kardiovaskulärer Erkrankungen veranschaulicht und in weiterer Folge ein
Rückschluss auf deren Stellenwert getroffen. Mittels einer Literaturrecherche erfolgte eine
Erhebung von pharmakologischen und klinischen Studien bezüglich der Pharmakokinetik/-
dynamik, Neben- und Wechselwirkungen, des Einsatzes und der Vergleichbarkeit mit
anderen kardiovaskulär wirksamen Substanzen, Wirkstoffkombinationen, sowie eine
Beurteilung der einzelnen Substanz-Subgruppen. Zusätzlich werden die, aufgrund der
häufig bei Herz-Kreislauf-Patienten/Patientinnen bestehenden Polypharmazie,
Arzneimittelinteraktionen mit anderen Substanzgruppen zusammengefasst. Das Ziel dieser
Diplomarbeit besteht darin, ein aktuell gültiges Profil der CCB zu erstellen, um die
Bedeutung dieser Substanzklasse und möglicher Anwendungsnischen in der modernen
Pharmakotherapie kardiovaskulärer Erkrankungen aufzuzeigen. CCB zeichnen sich durch
ihre pleiotropen Effekte und infolgedessen der Reduktion blutdruckabhängiger und
–unabhängiger kardiovaskulärer Risikofaktoren aus und finden dadurch zunehmend
Anerkennung in internationalen Guidelines. In der Therapie der arteriellen Hypertonie
bewähren sich langwirksame Formulierungen von Nifedipin, Amlodipin sowie späte
Generationen der Dihydropyridine und zeigen sich in der Reduktion des Insultrisikos und
bei Hochrisikohypertonie den übrigen Antihypertensiva überlegen. Diltiazem und
Verapamil finden Anwendung in der antiarrhythmischen Therapie, wobei sie sich im
Speziellen bei supraventrikulären Tachykardien auszeichnen. In der Therapie der stabilen
Angina pectoris erweisen sich CCB als gleichwirksam wie ihre Referenzsubstanzen und
zeigen sich auch besonders bei vasospatischer Angina Pectoris als vorteilhaft. Bei einem
Raynaud-Syndrom gilt Nifedipin weiterhin als besterforschter Wirkstoff und First-Line-
Medikament. Nimodipin reduziert die Wahrscheinlichkeit neurologischer Defizite nach
einer Subarachnoidalblutung und gilt hierbei weiterhin als Standardtherapie.
iv
Abstract
Calcium antagonists encompass a heterogeneous group of compounds with distinctive
structures and pharmacologic characteristics which are widely used in the treatment of
cardiovascular diseases. In course of time the continuous advancements of substances,
insights from novel, large-scale, clinical long-term studies as well as comparisons with
alternative cardiovascular drugs led to diverging preferences and incoherent ideologies
regarding their clinical application. In order to draw a conclusion about the significance of
calcium antagonists, in this thesis the historical development, drug profile and clinical
scopes of application are exemplified. An investigation of clinical and pharmacological
studies regarding pharmacokinetics/-dynamics, side effects, pharmacological interactions,
fields of application and comparability with other cardiovascular drugs, combination
therapies and assessments of the different substance sub-groups was conducted by means
of a literature research. Additionally, concerning the polypharmacy in patients with
cardiovascular diseases, pharmacological interactions with other drugs are summarized.
This serves the purpose to display the significance and potential therapeutic niches of
calcium antagonists in modern pharmacotherapy of cardiovascular diseases based on an
updated, valid profile. Calcium antagonists are characterized by their pleitropic effects and
therefore reduction of blood pressure dependent and blood pressure independent
cardiovascular risk factors. Moreover, an increasing number of positive recongitions in
international guidelines regarding calcium antagonists could be demonstrated. In the
therapy of arterial hypertension long-acting nifedipine, amlodipine and later generations of
the dihydropyridine calcium antagonists have been proven efficious and are superior in the
reduction of the risk of stroke and in the therapy of high-risk-hypertension in comparsion
to other antihypertensive agents. Diltiazem and verapamil are used in the treatment of
cardiac arrhythmias, especially supraventricular tachycardies. In the therapy of stable
angina pectoris, calcium antagonists have been proven as efficious as their reference
substances and in particular beneficial in the case of vasospatic angina. Nifedipine still
obtains the role of the best investigated substance and first-line treatment of Raynaud´s
Syndrome. Nimodipine reduces the probability of neurological deficits due to
subarachnoidal haemorrhage and therefore remains the standard therapy.
v
Inhaltsverzeichnis
Eidesstattliche Erklärung ....................................................................................................... ii
Danksagungen ...................................................................................................................... iii
Zusammenfassung ................................................................................................................ iv
Abstract .................................................................................................................................. v
Inhaltsverzeichnis ................................................................................................................. vi
Glossar und Abkürzungen .................................................................................................... ix
Abbildungsverzeichnis .......................................................................................................... x
Tabellenverzeichnis .............................................................................................................. xi
1 Einleitung ...................................................................................................................... 1
1.1 Problemdarstellung ................................................................................................. 1
1.2 Ziel der Arbeit ......................................................................................................... 2
1.3 Aufbau der Arbeit ................................................................................................... 2
1.4 Forschungsfragen .................................................................................................... 3
2 Grundlagen .................................................................................................................... 4
2.1 Historische Entwicklung der Calciumantagonisten ................................................ 4
2.2 Der Calciumkanal als Rezeptor der Calciumantagonisten ..................................... 8
2.2.1 Calcium in der Signaltransduktion .................................................................. 8
2.2.2 Spannungsabhängige Calciumkanäle .............................................................. 8
2.2.3 Struktur spannungsabhängiger Calciumkanäle ............................................. 10
2.2.4 L-Typ-Calciumkanäle.................................................................................... 11
2.3 Klassifizierung der Calciumantagonisten ............................................................. 12
2.3.1 Phenylalkylamine .......................................................................................... 14
2.3.2 Benzothiazepine............................................................................................. 15
2.3.3 1,4–Dihydropyridine ..................................................................................... 16
2.4 Nebenwirkungen und Wechselwirkungen von Calciumantagonisten .................. 18
2.4.1 Nebenwirkungen............................................................................................ 18
2.4.2 Wechselwirkungen ........................................................................................ 19
2.5 Pharmakologische Effekte der Calciumantagonisten ........................................... 21
2.5.1 Antiatheromatöse Effekte von Calciumantagonisten .................................... 22
2.5.2 Blutdruckunabhängige Effekte der Calciumantagonisten auf die vaskuläre
Schädigung .................................................................................................................. 24
2.5.3 Metabolische Effekte ..................................................................................... 25
vi
2.5.4 Effekte auf den Insulin- und Glukose-Metabolismus .................................... 25
2.5.5 Effekte auf den arteriellen Blutdruck ............................................................ 26
2.5.6 Effekte auf die zentrale Hämodynamik ......................................................... 27
2.5.7 Effekte auf die kardiale Reizleitung .............................................................. 28
2.5.8 Effekte auf die Herzfrequenz ......................................................................... 29
2.5.9 Effekte auf die linksventrikuläre Masse und Myokardfibrose ...................... 30
2.5.10 Effekte auf die kardiale Kontraktilität ........................................................... 31
3 Ergebnisse.................................................................................................................... 32
3.1 Calciumantagonisten zur Verbesserung kardiovaskulärer Ergebnisse ................. 35
3.1.1 Studien bezüglich der Verbesserung kardiovaskulärer Ergebnisse ............... 35
3.1.2 Meta-Analysen bezüglich der Verbesserung kardiovaskulärer Ergebnisse .. 36
3.1.3 Sicherheit der Anwendung von Calciumantagonisten................................... 37
3.1.4 Verbesserung der Prognose von Hypertonie-Patienten/-Patientinnen durch
Calciumantagonisten ................................................................................................... 38
3.1.5 Nicht-Unterlegenheit der Calciumantagonisten ............................................ 39
3.1.6 Überlegenheit der Calciumantagonisten ........................................................ 39
3.2 Calciumantagonisten in der Therapie der koronaren Herzkrankheit .................... 40
3.2.1 Stabile Angina Pectoris ................................................................................. 41
3.2.2 Therapie des akuten Koronarsyndromes und instabiler Angina Pectoris ...... 42
3.2.3 Vasospastische Angina .................................................................................. 43
3.2.4 Sekundärprävention bei myokardialen Re-Infarkt-Patienten/Patientinnen ... 44
3.3 Calciumantagonisten in der Therapie der arteriellen Hypertonie ......................... 45
3.3.1 Initiale Wirkstoffwahl zu Therapiebeginn ..................................................... 45
3.3.2 Status der β-Blocker in der Therapie der Hypertonie .................................... 46
3.3.3 Auswahl der antihypertensiven Therapie aufgrund von Begleiterkrankungen
und Kontraindikationen ............................................................................................... 47
3.3.4 Calciumantagonisten zur Erreichung eines Blutdruckzieles ......................... 49
3.4 Calciumantagonisten in Kombinationstherapien für das klinische Management
der Hypertonie ................................................................................................................. 50
3.4.1 Auswahl der Idealstrategie zur Blutdruckkontrolle ....................................... 50
3.4.2 Kombinationstherapie als Initialtherapie ....................................................... 50
3.4.3 Monotherapie versus Kombinationstherapie ................................................. 51
3.4.4 Komplementäre Effekte bei Kombinationstherapien .................................... 55
3.4.5 Vorteile für die kardiovaskuläre Protektion .................................................. 56
vii
3.4.6 Dualkombinationstherapie ............................................................................. 57
3.4.7 Trippelkombinationstherapie ......................................................................... 58
3.5 Calciumantagonisten in Therapie der Hochrisiko-Hypertonie ............................. 59
3.5.1 Anwendung bei Patienten/Patientinnen höheren Lebensalters ...................... 59
3.5.2 Diabetes und chronische Niereninsuffizienz ................................................. 60
3.6 Calciumantagonisten in der Therapie kardialer Arrhythmien .............................. 61
3.7 Calciumantagonisten in der Therapie hypertropher Kardiomyopathie................. 64
3.8 Calciumantagonisten in der Therapie des Raynaud-Syndroms ............................ 65
3.9 Calciumantagonisten in der Therapie klinischer Notfälle .................................... 66
3.9.1 Subarachnoidalblutung .................................................................................. 66
3.9.2 Ischämischer Insult ........................................................................................ 68
3.9.3 Hypertensive Enzephalopathie ...................................................................... 68
3.9.4 Akute Aortendissektion ................................................................................. 69
3.9.5 Hypertensiver Notfall aufgrund eines Catecholamin-Überschusses ............ 69
3.9.6 Hypertensiver Notfall während der Schwangerschaft ................................... 70
4 Diskussion ................................................................................................................... 71
Material und Methoden ....................................................................................................... 74
Literaturverzeichnis ............................................................................................................. 75
viiiGlossar und Abkürzungen
ACEI ACE-Hemmer
BB Beta-Rezeptoren-Blocker
BTZ Benzothiazepin
CCB Calciumantagonist
DHP Dihydropyridin
HCM hypertrophe Kardiomyopathie
KV kardiovaskulär
LV linksventrikulär
LVOT linksventrikulärer Ausflusstrakt
nDHP Nicht-Dihydropyridin-Calcium-Antagonisten; Calciumantagonisten,
welche nicht der Gruppe der 1,4-Dihydropyridine angehören
PPA Phenylalkylamin
ARB AT1-Antagonist
RAS Renin-Angiotensin-System
SAB Subarachnoidalblutung
TD Thiaziddiuretikum
WW Wechselwirkung
ixAbbildungsverzeichnis
Abbildung 1: Hemmung der Papillarmuskellkontraktilität durch Verapamil, nach [3] ........ 6
Abbildung 2: Calcium-Kanal-Subtypen, nach [4] ............................................................... 10
Abbildung 3: L-Typ-Calciumkanal, nach [4] ...................................................................... 11
Abbildung 4: Allosterische Wechselwirkungen, nach [4] ................................................... 12
Abbildung 5: Generationen der Calciumantagonisten, nach [28] ....................................... 13
Abbildung 6: Chemische Struktur der Calciumantagonisten, nach [4, 29] ......................... 15
Abbildung 7: Kombinationsmöglichkeiten der Antihypertensiva, nach [93]...................... 51
Abbildung 8: Therapie-Algorithmus für die arterielle Hypertonie, nach [91] .................... 54
xTabellenverzeichnis
Tabelle 1: Historische Entwicklung der Calciumantagonisten, nach [3, 5, 24] ................... 7
Tabelle 2: Calciumantagonisten für die orale Therapie der Hypertonie [4] ........................ 14
Tabelle 3: Weltweit erhältliche Calciumantagonisten [30] ................................................. 17
Tabelle 4: Nebenwirkungen der Calciumantagonisten nach Symptomen [24] ................... 18
Tabelle 5: Nebenwirkungen der Calciumantagonisten nach Organsystem [24] ................. 18
Tabelle 6: Wechselwirkungen der Calciumantagonisten [32]............................................. 19
Tabelle 7: Effekte der Calciumantagonisten-Subgruppen [28] ........................................... 21
Tabelle 8: Antiatheromatöse Effekte - Zusammenfassung der Studien, nach [28] ............. 24
Tabelle 9: Klinisch-elektrophysiologische Effekte [5]........................................................ 29
Tabelle 10: Calciumantagonisten - Klinische Studien, nach [30] ....................................... 33
Tabelle 11: Koronare Herzkrankheiten - Definitionen und Therapien [5] .......................... 41
Tabelle 12: Hypertonie – Initialtherapie, nach [92] ............................................................ 47
Tabelle 13: Auswahl der antihypertensiven Therapie nach Komorbiditäten [93]............... 48
Tabelle 14: Auswahl der antihypertensiven Therapie nach Kontraindikationen [93] ......... 48
Tabelle 15: Hypertonie - Kombinationstherapien [98]........................................................ 58
Tabelle 16: Effekte der Calciumantagonisten auf das Reiz-Leitungssystem, nach [5] ....... 63
xi1 Einleitung
1.1 Problemdarstellung
Kardiovaskuläre Erkrankungen stellen die weltweit häufigste Todesursache dar. Im Jahr
2012 wurden 31% der gesamten, globalen Todesursachen kardiovaskulären Erkrankungen
zugeschrieben, mit mehr als Dreiviertel an Todesfällen in Ländern mit niedrigem und
mittlerem Einkommen. Bei Patienten/Patientinnen mit einer kardiovaskulären Erkrankung
oder hohem kardiovaskulären Risiko aufgrund einer oder mehrerer Risikofaktoren
(Tabakkonsum, ungesunde Nahrungsmittel und Übergewicht, Diabetes, Hyperlipidämie
oder bereits bestehender Erkrankung) ist eine frühzeitige Erkennung, Management,
Beratung und Medikamententherapie vonnöten [1]. Calciumantagonisten stellen seit etwa
40 Jahren eine der wichtigsten Medikamente in der kardiovaskulären Therapie dar. Die
ständige Weiterentwicklung von Substanzen mit unterschiedlichen Wirkprofilen an den
verschiedenen Subtypen der spannungsabhängigen Calciumkanäle ergaben
unterschiedliche Anwendungsgebiete [2, 3, 4, 5]. Detaillierte Nutzen-Risiko-Analysen,
neue Erkenntnisse aus großen, klinischen Langzeit-Studien, Kontroversen bezüglich des
Einflusses auf Mortalität und Morbidität und der Sicherheit der Calciumantagonisten sowie
der Vergleich mit alternativen Herz-Kreislauf-Medikamenten führten im Laufe der Zeit zu
unterschiedlichen Präferenzen und auch zu inkohärenten Ideologien bezüglich deren
klinischen Gebrauchs. Hier soll nun der derzeitige Stand der Wissenschaft bezüglich des
Wirkprofiles der Calciumantagonisten und deren Anwendung im klinischen Bereich und
ihre Vergleichbarkeit und Kombinationsmöglichkeiten mit alternativen kardiovaskulär
wirksamen Pharmazeutika eruiert und ein strukturierter Überblick für eine aktuell gültige,
klinische Anwendung zur Verfügung gestellt werden.
.
11.2 Ziel der Arbeit
Ziel dieser Arbeit ist es, anhand einer Literaturrecherche neueste wissenschaftliche
Erkenntnisse aus klinischen Studien, Meta-Analysen und Guidelines betreffend
Calciumantagonisten in der kardiovaskulären Therapie zu sammeln und ein derzeit gültiges
Wirkstoffprofil zu erstellen, um daraus mögliche klinische Anwendungsgebiete ableiten zu
können. Anhand dieses Wirkstoffprofiles sollen nun die, für die Therapie mit
Calciumantagonisten in Frage kommenden Krankheiten aufgelistet, die Wirksamkeit und
Sicherheit bei den jeweiligen Krankheitsbildern widergespiegelt, ein Vergleich mit
alternativen Herz-Kreislauf-Medikamenten getroffen und mögliche Kombinationstherapien
bewertet werden. Eine prävalente Multimorbidität kardiovaskulärer Patienten/Patientinnen,
die mit kardiovaskulären Krankheiten vergesellschafteten Komorbiditäten und eine
Vielzahl an Pathogenesen erfordern oftmals eine, die Therapie erschwerende,
Polypharmazie. Diesbezüglich soll zusätzlich eine detaillierte Darstellung der
Wechselwirkungen erfolgen.
1.3 Aufbau der Arbeit
Kapitel 2 liefert die, für ein Verständnis der Einsatzgebiete notwendigen Grundlagen der
Entwicklung, Subtypen und pharmakologischen Eigenschaften und für die kardiovaskuläre
Therapie relevanten Effekte der Calciumantagonisten. Es erfolgt eine Darstellung der
geschichtlichen Entwicklung der verschiedenen Wirkstoffe, ihrer Entwicklungsprozesse in
der kardiovaskulären Therapie, ihres Wirkungsortes, des pharmakologischen Profils der
verschiedenen Calciumantagonisten und Erkenntnisse über ihre Effekte auf das
kardiovaskuläre System. Aufgrund der in der kardiovaskulären Therapie oftmals
prävalenten Polypharmazie wird hier zusätzlich auf Wechselwirkungen der
Calciumantagonisten eingegangen. Abgeleitet davon werden im Kapitel 3, basierend auf
neuen Erkenntnissen, Möglichkeiten der klinischen Anwendung der Calciumantagonisten
angeführt, ihre Wirksamkeit und Sicherheit bezüglich der diversen Einsatzgebiete bewertet
und Vergleiche mit alternativen Herz-Kreislauf-Medikamenten und möglicher
Kombinationstherapien eruiert.
21.4 Forschungsfragen
Ziel dieser Arbeit ist es, ein aktuell gültiges Wirkstoffprofil der Calciumantagonisten in der
kardiovaskulären Therapie zu erstellen. Weiters werden die nachfolgenden Forschungs-
fragen wissenschaftlich aufgearbeitet:
− Welche historische Entwicklung durchliefen Calciumantagonisten?
− Wie wirkt Calcium und wo greifen Calciumantagonisten an?
− Welche Calciumantagonisten stehen für die kardiovaskuläre Therapie zur
Verfügung und welches Wirkprofil weisen sie auf?
− Welche pharmakologischen Effekte weisen die verschiedenen Calciumantagonisten
auf und welche klinischen Anwendungsgebiete lassen sich daraus ableiten?
− Welche Wirksamkeit und Sicherheit weisen Calciumantagonisten bei den
verschiedenen kardiovaskulären Krankheiten und im jeweiligen Vergleich zu
alternativen, kardiovaskulären Medikamenten auf?
− Gibt es Kombinationstherapien und wie sicher bzw. wie effektiv sind sie?
− Welche Wechselwirkungen weisen Calciumantagonisten in der kardiovaskulären
Therapie auf?
32 Grundlagen
2.1 Historische Entwicklung der Calciumantagonisten
Beschreibungen kardioaktiver Substanzen konnten bereits in antiken Hochkulturen der
Ägypter und Griechen bestätigt werden, jedoch liegt kein Beweis vor, dass diese
Zivilisationen Einsatz von natürlich vorliegenden Calciumantagonisten (CCB) gemacht
hatten. Im Gegensatz dazu fand der, in der Natur vorkommende, CCB „Tashinone“ bereits
in den letzten 3000 Jahren in der chinesischen Medizin Anwendung. Das gegenwärtige
Interesse an dieser Wirkstoffgruppe, sowie ihr klinischer Gebrauch in der modernen
Medizin ist jedoch nicht auf antike, chinesische Medizin, sondern vielmehr auf
Experimente des Freiburger Physiologen Albrecht Fleckenstein im Jahre 1964, mittels den
damals neu entdeckten Substanzen Verapamil und Prenylamin, zurückzuführen [6].
Erstmalig konnte die Aufhebung der elektromechanischen Kopplung an elektrisch
gereizten Meerschweinchen-Papillarmuskeln im Organbad mittels dieser Substanzen, wie
in Abbildung 1 schematisch dargestellt, beschrieben werden: Die oberste Reihe zeigt den
gereizten Papillarmuskel in Tyrode-Lösung. Nach Hinzufügen von 2x10-6 M Verapamil
(mittlere Reihe) kann eine Senkung der Kontraktilität und nach einer Steigerung auf
1x10-5 M eine vollständige Blockade bei gleichbleibender Reizung beobachtet werden.
Somit wiesen Verapamil und Prenylamin in hohen Dosen eine bis dahin unerklärte,
negative Inotropie, ohne gleichzeitige Beeinflussung Na+-getragener Aktionspotential-
Parameter auf. Nur die Plateauphase und Repolarisation des Aktionspotentials wurden
gering modifiziert, während die maximale Depolarisationsgeschwindigkeit des
Aktionspotentials und infolgedessen die Erregbarkeit unverändert blieben [3, 7, 8]. Dieses
Ergebnis ähnelte Erkenntnissen aus, bereits zu Mitte des 19. Jahrhunderts beschriebenen,
Experimenten am Froschherzen durch Sydney Ringer, in denen er die Ca2+-Ionenanzahl im
Bad verminderte [9]. Auch Mines konnte bereits 1913 feststellen, dass ein Entzug von
Ca2+-Ionen die Kontraktion des Herzens stoppt, die elektrische Erregung aber fortbestehen
lässt [10]. Durch Maßnahmen, welche den Ca2+-Vorrat des kontraktilen Systems
wiederherstellen (Erhöhung extrazellulärer Ca2+-Ionen, Addition β-adrenerger
Katecholamine oder Herzglykoside) konnte dieser Effekt aufgehoben werden. Während
eine durch β-Blocker wie Pronethalol und Propranolol induzierte Herzinsuffizienz mittels
β-Adrenorezeptor-Agonisten nicht oder nur in hohen Dosen aufgehoben werden konnte,
4besserte sich die durch Verapamil und Prenylamin hervorgerufene Herzinsuffizienz bereits
nach Addition geringer Mengen und sprach somit für eine geringe oder nicht vorhandene
Wirkung auf kardiale β-Adrenozeptoren. Dies erlaubte eine Differenzierung zu β-Blockern
und führte zur Annahme der Entdeckung eines neuartigen Wirkmechanismus. Analoge
Effekte, welche in ihrer Wirkstärke jedoch diese von Verapamil übertrafen, konnten bei
Substanz D 600, einem Verapamil-Derivat welches später als Gallopamil bekannt werden
sollte, beobachtet werden. Ein Nachweis der Reduktion von isoproterenol-stimulierter,
45
myokardialer Ca-Inkorporation, Stabilisierung intrazellulärer Vorräte an energiereichen
Phosphaten, Senkung des 02-Verbrauches und Schutz von Myokardfasern gegenüber
deletären Konsequenzen einer intrazellulären Ca2+-Überladung konnte sowohl bei
Verapamil, als auch Gallopamil nachgewiesen werden. In den daraus resultierenden
Arbeiten erfolgte die erstmalige Verwendung des Terminus „Calciumantagonisten“ durch
Fleckenstein für diese Substanzen [11, 12]. 1969 konnte die selektive Blockade des
myokardialen Ca2+-Einstromes via potentialabhängige, „langsame“ Ca2+-Kanäle, ohne
gleichzeitige Beeinflussung „schneller“ Na+-Ströme der Membran mittels der damals
durch Reuter und Beeler introduzierten Voltage-Clamp-Methode konkretisiert werden
[13]. In den folgenden Jahren wurde durch intensive Forschung eine beträchtliche Anzahl
von Wirkstoffen identifiziert, wobei die Entdeckung der 1,4-Dihydropyridine (DHP) einen
weiteren Meilenstein der Geschichte der CCB darstellt. Der Chemiker Friedrich Bossert
und der Pharmakologe Wulf Vater forschten anhand von Carbochromen und Dipyridamol,
zwei damals bekannten, allerdings recht wirkungsschwachen Koronardilatatoren, als
Referenzsubstanzen, sowie in weiterer Folge Derivaten des Bischofskrautes (Ammi
visnaga) und konnten mit Hilfe der sogenannten Hantz'schen DHP-Synthese mehr als
2.000 DHP-Derivate synthetisieren, wobei ausgehend von 2-Nitrobenzaldehyd
schlussendlich auch die Synthese von Nifedipin erfolgte und als erster Wirkstoff dieser
Gruppe in den Markt eingeführt wurde. DHP zeigten eine große Nützlichkeit sowohl in der
Therapie als auch in der Wissenschaft. Diese im Vergleich zu Verapamil um
Größenordnungen vaskulär wirksameren Substanzen eröffneten neue Therapie-
möglichkeiten kardiovaskulärer Erkrankungen und trugen zur Charakterisierung der
spannungsabhängigen Calciumkanäle bei. Es erfolgten die Klassifizierung in Subgruppen,
die Erkenntnis über eine Selektivität der DHP für spannungsabhängige L-Typ-
Calciumkanäle, welche infolgedessen als DHP-Rezeptoren bezeichnet wurden und der
Nachweis dieser in glatter Muskulatur [3].
5Weitere Fortschritte in der Erkenntnisgewinnung bezüglich der spannungsabhängigen
Ca2+-Kanäle lieferte auch die Entdeckung der Calciumagonisten, ebenfalls der Gruppe der
DHP angehörigen Substanzen, welche die Öffnungswahrscheinlichkeit von Ca2+-Kanälen
erhöhen, im Jahr 1982 [3].
Abbildung 1: Hemmung der Papillarmuskellkontraktilität durch Verapamil, nach [3]
Elektrophyiologische Studie von Mehrschweinchenpapillarmuskeln unter steigenden Konzentrationen von
Verapamil in Tyrode-Lösung; Obere Reihe: normale Kontraktion ohne Verapamil, mittlere Reihe: Senkung
der Kontraktilität bei gleicher Reizung mit 2x10-6M Verapamil für 40 min, untere Reihe vollständige
Blockade bei konstanter Reizung mit 2x10-5M Verapamil für 45 min.
Die Bedeutung der CCB in der kardiovaskulären Therapie neben ihrem Nutzen als
antianginale Substanzen wurde, zumindest klinisch, erst spät anerkannt [2]. Im Jahr 1972
erfolgte die erste Erklärung der antiarrhythmischen Wirkung von Verapamil bei
paroxysmaler, supraventrikulärer Tachykardie [14]. 1975 wurde Verapamil zur Therapie
der hypertrophen Kardiomyopathie eingesetzt und Nifedipin für die Therapie der
koronaren Herzkrankheit zugelassen. Gleichzeitig wurden Studien zur Wirksamkeit von
Nifedipin in der Therapie vasospastischer Angina pectoris veröffentlicht und folgend 1979
klinische Ergebnisse der antianginösen, antiarrhythmischen und antihypertensiven Effekten
publiziert [15, 16, 17]. Im Zuge der im Jahr 1983 herausgegebenen, auf klinischen Studien
basierenden Veröffentlichungen zum Thema "Calcium Antagonists in the Treatment of
Hypertension" der American Heart Association wurden allmählich die Bedeutung dieser
6Pharmaka in der Therapie der Hypertonie anerkannt, für die Therapie der arteriellen
Hypertonie eingeführt und subsequentiell für den Einsatz einer zunehmenden Anzahl
kardiovaskulärer Indikationen erweitert. [18] Kontroversen betreffend der Sicherheit
(Einfluss auf Mortalität, Darmkrebsrisiko) kurzwirksamer CCB rückten diese Mitte der
1990er in ein schlechtes Licht, ein daraus resultierender WHO-Bericht und großangelegte
Studien konnten jedoch ihre Sicherheit und Wirksamkeit belegen [19, 20, 21, 22, 23].
Heutzutage gelten sie als etablierte Medikamente für die arterielle Hypertonie, koronare
Herzkrankheit, insbesondere vasospastischer Angina pectoris, supraventrikulärer
Tachyarrhythmien, dem Raynaud Syndrom und der Subarachnoidalblutung, wie in Kapitel
3 beschrieben. In Tabelle 1 erfolgt eine chronologische Darstellung der Geschichte der
Calciumantagonisten.
Tabelle 1: Historische Entwicklung der Calciumantagonisten, nach [3, 5, 24]
Jahr Entdeckung/Ereignis
1882 Abhängigkeit von Calciumionen (Ca2+) in myokardialer Kontraktion
Ca2+-Entzug verhindert die Kontraktionskraft im ventrikulären Myokard mehr als die
1913
bioelektrische Aktivität
1960 Pharmakologische Wirkung von Prenylamin
1962 Pharmakologische Wirkung von Verapamil
1963 Anwendung von Verapamil bei koronarer Herzkrankheit
Verapamil und Phenylamin verhindern die Erregungs-Kontaktionskopplung auf die
1964
gleiche Weise wie ein Ca2+-Defizit im Myokardium
1965 Verwendung von Verapamil als Antiarrhythmikum bei Vorhofflattern
1967 Einführung der Begriffe „Calciumantagonist“ und „calcium-antagonistisch“
Differenzierung der Calciumantagonisten Verapamil, Gallopamil und Prenylamin von
1966 –
β-Blockern. Definition des Calciumantagonismus und Calciumantagonisten als neue,
1969
pharmakologische Klasse
1976 Synthese von Gallopamil
1968 –
Kardioprotektive Wirkung von Calciumantagonisten im Tierexperiment
1969
1969 Therapie von arterieller Hypertonie mit Verapamil
1969 Pharmakologische Testung von Nifepidin
1969 –
Identifikation von Nifepidin als spezifischer Calciumantagonist
1970
1968-
Wirkung von Calciumantagonisten auf glatte Muskulatur
1972
1970 – Mechanismus und Angriffspunkt von Calciumantagonisten: Signifikanz der „slow
1972 calcium channels“ (Fleckenstein, publiziert in „Calcium and the heart“ 1970-1971)
1971 Pharmakologische Wirkung von Diltiazem (CRD 401)
1974 Behandlung paroxysmaler, supraventrikulärer Tachykardie mit Verapamil
1975 Identifikation von Diltiazem als Calciumantagonist
1976 Verwendung von Calciumantagonisten bei hypertropher Kardiomyopathie
1980 Editorial im American Journal of Cardiology
1995 Kontroverse bezüglich Sicherheit, Mortalität und Morbidität der Calciumantagonisten
1995 – Weiterentwicklung der Substanzen und Anwendungen,
dato wie in Kapiteln 2 und 3 beschrieben.
72.2 Der Calciumkanal als Rezeptor der Calciumantagonisten
2.2.1 Calcium in der Signaltransduktion
Calcium-Ionen (Ca2+) beeinflussen nahezu jeglichen Aspekt physiologischer Zellprozesse.
Eine Änderung der intrazellulären Calciumkonzentration ist Bestandteil der
elektromechanischen Kopplung bei Muskelkontraktion, Synthese und Sekretion von
Neurotransmittern und Hormonen, Genexpression, Enzymaktivität, sowie der
Aufrechterhaltung der Zellhomöostase. [4, 25] Im Ca2+-armen, intrazellulären Milieu
stellen Ca2+-Ionen weitverbreitete Botenstoffe („second messenger“) dar und ermöglichen
über eine streng regulierte Signalkaskade eine Informationsübertragung an den Zellkern
sowie dem kontraktilen Apparat. Eine zusätzliche Beeinflussung Ca2+-abhängiger
Enzyme, wie z.B. Proteinkinase C oder Ca2+-Calmodulinkinasen, und deren Effekte auf
weitere Kanäle, Rezeptoren und Enzyme tragen zur Komplexität der, einer Ca2+-
Kanalöffnung folgenden, Modulation der elektrischen und nicht elektrischen
Zellaktivitäten bei [5]. Im ruhenden Zustand verfügt die Zelle über einen
Calciumgradienten von 10-8M, welcher bei Erregung kurzzeitig durch den Einstrom
extrazellulärer Ca2+-Ionen um das 103-fache ansteigen kann. In kardialen und glatten
Muskelzellen wird durch die Aktivierung der Ca2+-Kanäle die Muskelkontraktion direkt
durch eine Erhöhung der cytoplasmatischen Ca2+-Konzentration und indirekt durch die
Aktivierung der calciumabhängigen Calciumfreisetzung mittels Ryanodin-Rezeptoren im
sarkoplasmatischen Retikulum eingeleitet [11].
2.2.2 Spannungsabhängige Calciumkanäle
Spannungsabhängige Calciumkanäle stellen das Schlüsselelement der Vermittlung einer
Membrandepolarisation zu einem Ca2+-Einstrom in die Zelle dar. Man unterscheidet
zwischen Low-Threshold-Kanälen, welche bei einem Membranpotential nahe dem
Ruhepotential (ca. -60 bis –70 mV) der Zelle aktiviert werden und High-Threshold-
Kanälen, welche eine stärkere Depolarisation (bei -40 mV) zur Aktivierung benötigen [26].
Basierend auf ihren biophysikalischen und pharmakologischen Eigenschaften sind weitere
8Differenzierungen möglich. High-Threshold-Kanäle können in L-Typ- (large conductance
bzw. long lasting), N-Typ- (neuronal bzw. neither T- nor L-Type), P/Q-Typ- (Purkinje-
Neuronen) und R-Typ-Kanälen (resistant oder residual) eingeteilt werden, während Low-
Threshold-Kanälen der T-Typ-Kanal (tiny conductance bzw. transient) aufgrund seiner
raschen, monoexponentiellen und nur vom Membranpotential abhängigen Inaktivierung
zugeordnet wird. Die pharmakologisch bedeutsamsten Typ-N-Kanäle sind für die
Erregungs-Kontraktions-Kopplung in Muskelzellen, die Erregungs-Transkriptions-
Kopplung in Nerven- und Muskelzellen, sowie die Erregungs-Sekretionskopplung in
endokrinen Zellen und spezialisierten Ribbon-Synapsen zuständig. Typ-P/Q, Typ-N- und
R-Kanäle sind an Zelloberflächen von Neuronen lokalisiert. Der Typ-T-Kanal sorgt für die
Generation rhythmischer Potenziale im Herzen und Thalamus. Die Relation der Typ-N-
Kanäle zu chronischen neuropathischen Schmerz dient als Grundlage der Erforschung
neuer Wirkstoffe. Den Typ-T-, Typ-P/Q- und Typ-N-Kanälen wird eine Rolle in der
Epilepsie, sowie auch in der Entstehung von Krebs und neuropathischem Schmerz bei
peripherer, diabetischer Neuropathie (Typ-N-Kanal) zugeordnet [4, 25, 27]. Die Typ-L-
Ca2+-Kanäle sind die Zielstruktur der Ca2+-Kanal-Blocker, weswegen der Begriff
„Calciumantagonist“ schließlich auf L-Typ-Kanal-Blocker ausgeweitet werden kann. Es
muss jedoch berücksichtigt werden, dass diese keinen irreversiblen Block, sondern eine
Hemmung der Kanalöffnung initiieren. Abbildung 2 zeigt die oben definierten
Einteilungen in die diversen Subkategorien, ihr Vorkommen, ihre selektiven Antagonisten
und die Überstimmung ihrer Aminosäuren der diversen spannungsabhängigen Calcium-
Kanäle [4, 5].
9selektiver
Schwelle Typ Klon überwiegendes Vorkommen
Antagonist
Cav 1.1(α1S) Skelettmuskel Dihydropyridine
Cav 1.2(α1C) Herz-/glatte Muskulatur, Endokrinium Dihydropyridine
L Cav 1.3(α1D) Gehirn, Ohr, Endokrinium Dihydropyridine
Cav 1.4(α1F) Retina Dihydropyridine
hoch Cav 2.1(α1A) Gehirn, Ohr, Hypophyse ω-Agatoxin
P/Q Cav 2.2(α1B) Gehirn, Neurone ω-Conotoxin-GVIA
N Cav 2.3(α1E) Gehirn, Ohr, Neurone -
R
Cav 3.1(α1G) Gehirn, Neurone Mibefradil
Cav 3.2(α1H) Gehirn, Herz Mibefradil
T Cav 3.3(α1I) Gehirn Mibefradil
niedrig
20 60 100
Anteil identischer
Aminosäuren [%]
Abbildung 2: Calcium-Kanal-Subtypen, nach [4]
Darstellung der Calcium-Kanal-Subtypen nach ihrer Reizschwelle, Ähnlichkeit in ihrer Aminosäure-
Komposition, ihr überwiegendes Vorkommen und ihre jeweiligen, selektiven Antagonisten.
2.2.3 Struktur spannungsabhängiger Calciumkanäle
Ähnlich den anderen, der Superfamilie zugehörigen, spannungsabhängigen Ionenkanälen
sind High-Treshhold-Calciumkanäle heteromere, aus multiplen Untereinheiten bestehende
Membrankomplexe. Die α1-Untereinheit stellt als das zentrale Transmembranprotein die
größte Untereinheit dar und dient als Kanalpore, Spannungssensor, sowie Schleuse und
beinhaltet die meisten Andockstellen für Second Messenger, Medikamente und Toxine zur
Kanalregulation. Sie besteht aus vier homologen Domänen (I-IV), wobei jede einzelne
Domäne wiederum durch sechs Transmembransegmente organisiert wird. Die α2-
Untereinheit ist ein extrazelluläres, extrinsisches Membranglykoprotein mit mehreren
Glykolysierungsstellen und hydrophoben Sequenzen, welches über eine Disulfidbrücke mit
der δ–Untereinheit und somit der Zellmembran verbunden ist. Die intrazellulär liegende β-
Untereinheit weist keine Transmembransegmente auf, wohingegen die γ-Untereinheit vier
Transmembransegmente besitzt. Die auxiliären α2δ-, β- und γ-Untereinheiten beeinflussen
die Kanalexpression, Spannungsabhängigkeit, Aktivierungskinetik und spielen eine Rolle
in der Verankerung innerhalb der Zellmembran7. In Abbildung 3 wird der L-Typ-
10Calciumkanal, seine Untereinheiten, deren Relation zur Zellmembran und untereinander
und die Andockstellen der Calciumantagonisten schematisch dargestellt [4, 27].
Ca2
α1-Untereinheit α2
S
S außen
II
I III δ
γ
IV
innen
β Domäne mit 6
transmembranalen
Segmenten
Abbildung 3: L-Typ-Calciumkanal, nach [4]
Abk.: BTZ = Benzothiazepine, DHP = Dihydropyridine, PAA = Phenylalkylamine
Die vier α1-Untereinheiten (I-IV) bilden die Kanalpore und sind direkt in der Zellmembran (blau-gelbe
Struktur) verankert. Die α2δ-, β- und γ-Untereinheiten beeinflussen die Kanaleigenschaften und Expression.
Die roten Markierungen zeigen die Andockstellen der Calciumantagonisten.
2.2.4 L-Typ-Calciumkanäle
Die, in kardialer, glatter und skelettaler Muskulatur prävalenten L-Typ-Calciumkanäle
werden durch eine langsame, spannungsabhängige Inaktivierung, dadurch langanhaltende
Aktivierungszeit und eine nicht-Ca2+-abhängige Inaktivierung und Hochregulation durch
cAMP-abhängige Proteinphosphorilisierungs-Singalwege charakterisiert. Sie zeigen eine
spezifische Inhibierung durch Dihydropyridine, Phenylalkylamine und Benzothiazepine.
Die chemisch divergenten Ca2+-Antagonisten binden hierbei direkt an verschiedene Stellen
der α1-Untereinheit: Dihydropyridine binden über Serin1115 in unmittelbarer Nähe zu dem
aus vier Glutaminsäuren bestehenden Ca2+-Selektivitätsfilter. Benzothiazepine an der
Brücke der Transmembrandomänen III und IV und Phenylalkylamine an einer weiteren
11Stelle der Transmembrandomäne IV [4, 27]. Wie in Abbildung 4 konnten sowohl zwischen
den diversen Calciumantagonisten, als auch zwischen Bindungsstellen und den funktionell
wichtigen Einheiten des Ca2+-Kanales positive (+) und negative (-) allosterische
Wechselwirkungen nachgewiesen werden.
Calciumantagonisten bewirken eine verminderte Bewegung der Ca2+-Ionen durch die
Kanalpore, ohne Veränderung der Leitfähigkeit des geöffneten Kanals, sondern durch eine
Veränderung der Öffnungswahrscheinlichkeit und Dauer [4, 5].
Abbildung 4: Allosterische Wechselwirkungen, nach [4]
Interaktionen der verschiedenenen Calciumantagonisten-Subgruppen bzw. ihrer Bindungsstellen
2+
untereinander und mit der Ca -Kanalpore; rotes Minus: negative allosterische Wirkungen; grünes Plus:
positive allosterische Wirkungen.
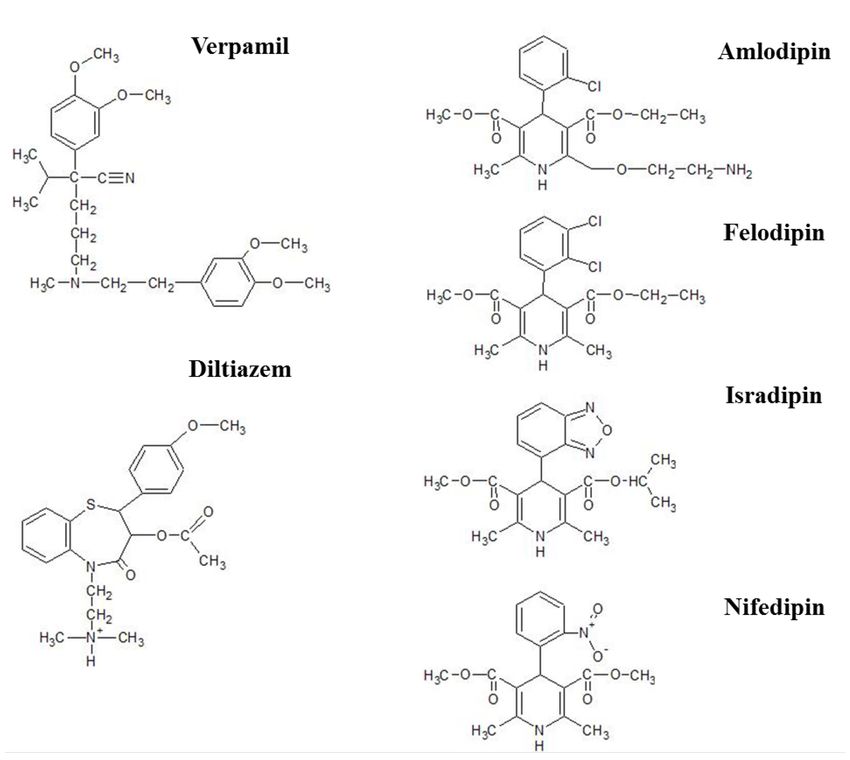
2.3 Klassifizierung der Calciumantagonisten
Calciumantagonisten (CCB) lassen sich, wie Abbildung 5 dargestellt, in drei heterogene
Gruppen einteilen: Phenylalkylamine, Benzothiazepine und Dihydropridine mit Verapamil,
Diltiazem und Nifedipin als Wirkstoffe der ersten Generation der jeweiligen Klasse. Die
zweite Generation umfasst CCB mit extended-release Darreichungsformen wie Verapamil
SR, Diltiazem CD, Nifedipin XL, Felodipine ER und Isradipin CR. Im Vergleich zu
12Nifedipin zeigen die 2. und 3. Generation der Dihydropyridine eine zunehmend höhere
vaskuläre Selektivität, geringere sympathische Aktivierung, geringere
Herzfrequenzzunahme und negative Inotropie. DHP können aufgrund ihres
Wirkungseintrittes und ihrer Wirkungsdauer in verschiedene Gruppen unterteilt werden. In
Kapselform verabreichtes Nifedipin wird rasch resorbiert und führt zu einer
unberechenbaren Zu- und Abnahme des Plasmaspiegels was einen starken, kurzwirkenden
hämodynamischen Effekt bedingt. Die retardierten Formulierungen wie Nifedipin GITS
(Gastro-Intestinal Therapeutic System) oder extended release-Formulierungen von
Felodipin besitzen langsamere Absorptionsraten mit langsamer Zu- und Abnahme des
Plasmaspiegels und 24-stündige Blutdruckregulation. Die langsam einsetzenden,
langwirksamen Formulierungen von Amlodipin, Lacidipin und Lercanidipin üben ihre
hämodynamischen Effekte über Tage aus und induzieren langwirksame und gestufte
Blutdruckänderungen [5, 28].
Abbildung 5: Generationen der Calciumantagonisten, nach [28]
Generationen der Calciumantagonisten geordnet nach ihrem Wirkungseintritt und ihrer Wirkungsdauer.
132.3.1 Phenylalkylamine
Verapamil und sein potenteres Derivat Gallopamil stellen die Prototypen der
Phenylalkylamine dar. Ihre hochaffine Bindung an der α1-Untereinheit inaktivierter Typ-L-
Ca2+-Kanäle führt zu einer Plateaudepression des monophasischen Aktionspotenzials im
Myokard. Verapamil ist chiral, besitzt somit zwei Enantiomere, die R-From und wirksame
S-Form, und wird klinisch als Razemat eingesetzt. Die Wirkung der Phenylalkylamine ist
frequenzabhängig. Im Ruhezustand des Ca2+-Kanales wird die Wirkstoffbindung gelöst,
bei einer Zunahme der Herzfrequenz wird die Blockade verstärkt. Obwohl
Phenylalkylamine einen vasodilatierenden Effekt aufweisen, finden sie primär aufgrund
ihrer kardialen Wirkungen (negative Ino-, Chrono und Dromotropie) Einsatz [4]. Tabelle 2
zeigt die verschiedenen Arzneiformen, ihre zeitlichen Verläufe und vorgeschlagene,
tägliche Verabreichungsdosen für die Therapie der arteriellen Hypertonie.
Tabelle 2: Calciumantagonisten für die orale Therapie der Hypertonie [4]
Antihypertensive Tagesdosis Verabreichungen
Subklasse/
Formulierung Wirkung [mg] pro Tag
Wirkstoff
Einsatz [h] Dauer [h]
Phenylalkylamine
Verapamil klassisch 0,5 6–8 80 – 320 2–3
Verapamil slow release 1,5 – 5 8 – 24 120 – 240 1–2
Verapamil retard >5 6–8 120 – 240 1
Benzothiazepine
Diltiazem klassisch 0,5 6–8 120 – 360 2–3
Diltiazem slow release >5 120 – 240 1
Dihydropyridine
Amlodipin klassisch >5 ≥ 24 2,5 – 10 1
Felodipin slow release 1,5 – 5 8 – 24 2,5 – 10 1
Isradipin klassisch 0,5 ≥ 24 2,5 – 10 2
Isradipin slow release 1,5 – 5 ≥ 24 5 – 10 1
Lacidipin klassisch 0,5 ≥ 24 2–8 1
Manidipin klassisch 0,5 ≥ 24 10 – 20 1
Nifedipin klassisch 0,5 6–8 30 – 40 3
Nifedipin slow release 1,5 – 5 8 – 24 10 – 40 2
Nifedipin GITS >5 ≥ 24 20 – 60 1
Nisoldipin slow release 1,5 – 5 ≥ 24 10 – 40 1
Nitrendipin klassisch 0,5 8 – 24 20 – 40 2
Abk.: GITS = Gastrointestinal Therapeutic System = Osmotische Minipumpe
142.3.2 Benzothiazepine
Diltiazem ist der einzige klinisch eingesetzte Vertreter dieser Klasse und weist eine
vornehmlich frequenzabhängige Interaktion auf. Diltiazem führt neben einer
vasodilatorischen Wirkung zu einer Plateausenkung des Aktionspotentials im Myokard
und wirkt dadurch kardiodepressiv. Die pharmakologische Wirkung der Benzothiazepine
ähnelt denen der Phenylalkylamine, weist jedoch eine höhere vaskuläre Selektivität auf
und ist somit in ihrer Wirksamkeit zwischen den beiden anderen Gruppen der
Calciumantagonisten einzugliedern [4]. Abbildung 6 zeigt die chemische Struktur der
Prototypen der verschiedenen Substanzklassen der Calciumantagonisten und weitere
Beispiele für verschiedene Generationen der Dihydropyridine.
Abbildung 6: Chemische Struktur der Calciumantagonisten, nach [4, 29]
Prototypen der Calciumantagonisten-Subgruppen:
Verapamil – Phenylalkylamine; Diltiazem - Benzothiazepine; Nifedipin als Prototyp und weitere Vertreter
der 1,4-Dihydropyridine.
152.3.3 1,4–Dihydropyridine
In Relation zu Verapamil und Diltiazem verfügt Nifedipin über ein zehnfach stärkeres
Potential der Inhibierung des Ca2+-Einstromes. Im Tierversuch führen sie zu einer initial
starken Erhöhung der koronaren Sauerstoffsättigung, einer wesentlichen Reduktion des
systemischen Gefäßwiderstandes, Blutdrucksenkung, sowie reflektorischer Tachykardie.
Diese Erhöhung der Herzfrequenz ist besonders nach intravenöser Verabreichung oder
nach Verabreichung schnellwirksamer Formulierungen prägnant, wobei neuere
Formulierungen und Wirkstoffe dieser Klasse beim Menschen keine oder nur noch eine
geringe Reflextachykardie aufweisen. Im Gegensatz zu den übrigen Substanzklassen
weisen Dihydropyridine eine hohe vaskuläre Selektivität und geringe kardiale Wirkungen
auf. Die allen Calciumantagonisten eigene negative Ino- und Chronotropie ist hier so
schwach, dass diese im gesunden Herzen vollständig reflektorisch kompensiert werden.
Bei bereits reduzierter Rechtsherzfunktion kann es zu einer Minderung der Kontraktilität
kommen. Die vaskuläre Selektivität beruht auf Unterschieden der Affinität zu den
organspezifischen Splice-Varianten, sowie dem Status der Ca2+-Kanäle und einer
wirkstoffspezifischen Abhängigkeit vom Membranpotential. Während die meisten 1,4-
Dihydropyridine weitgehend im inaktivierten Zustand des Kanales blockieren, weisen
Substanzen wie Nisoldipin auch eine Blockade im Ruhezustand auf. Des Weiteren zeigen
sie eine Zunahme in ihrer Wirkung in Abhängigkeit des Depolarisationszustandes der
Zellen und weisen somit einen stärkeren Effekt in glatter Gefäßmuskulatur mit relativ
geringerem Membranpotential im Vergleich zum Herzmuskel auf. Die Splice-Variante b
des IS6-Segments der α1c-Untereinheit, mit welcher DHP niederaffin interagieren, trägt
auch zur vaskulären Selektivität bei [4]. In Tabelle 3 werden alle derzeit weltweit, für die
kardiovaskuläre Therapie erhältlichen, Calciumantagonisten, ihre Hauptindikationen,
Metabolisierungswege, Erhältlichkeiten und Applikation aufgelistet.
16Tabelle 3: Weltweit erhältliche Calciumantagonisten [30]
Metabol
t1/2a [h] Zugelassene Indikationen Erhältlichkeit Appl.
-isierung
Dihydropyridine
Amlodipin 30-50 Angina pectoris, Hypertonie hepatisch weltweit oral
Aranidipin 3 Hypertonie hepatisch Japan (Sapresta®) oral
®
Azelnidipin 19,2 Hypertonie hepatisch Japan (Calblock ) oral
Barnidipin 10 Hypertonie hepatisch Eur, Asien oral
Benidipin 3 Angina pectoris, Hypertonie hepatisch Asien oral
Cilnidipin 0,5 Hypertonie hepatisch Asien oral
®
Clevidipin 0,02 Hypertonie Esterase US (Cleviprex ) i.v.
Isradipin 8 Hypertonie hepatisch weltweit oral
b ®
Efonidipin 2 Hypertonie hepatisch Japan (Landel ) oral
Angina pectoris, Hypertonie,
Felodipin 11-16 hepatisch weltweit oral
Raynaud-Syndrom
Lacidipin 13-19 Hypertonie hepatisch Eur, Asien oral
Lercanidipin 8-10 Hypertonie hepatisch Eur, Asien, SA oral
Manidipin 5-9 Hypertonie hepatisch Eur, Asien oral
Nicardipin 8 Angina pectoris, Hypertonie hepatisch weltweit oral/i.v.
Angina pectoris, Hypertonie,
Nifedipin 4 hepatisch weltweit oral/i.v.
Raynaud-Syndrom
Nilvadipin 15-20 Hypertonie hepatisch Eur, Japan oral
c
Nimodipin 9 zerebrovaskuläre Störungen hepatisch weltweit oral/i.v.
Nisoldipin 6-12 Angina pectoris, hepatisch Eur, US oral
Nitrendipin 10-22 Hypertonie hepatisch Eur, Asien oral
Phenylalkylamine
Angina pectoris, Hypertonie,
Verapamil 4,5-12 hepatisch weltweit oral/i.v.
Arrhythmien
Angina pectoris, Hypertonie,
Gallopamil 3-6 hepatisch Europa, Asien oral
Arrhythmien
Benzothiazepine
Angina pectoris, Hypertonie,
Diltiazem 3-5 hepatisch weltweit oral/i.v.
Arrhythmien
Abk.: Appl. = Applikation; Eur = Europa
a
Halbwertszeit
b
L/T-Typ-Calciumantagonist
c
überschreitet Blut-Hirn-Schranke
172.4 Nebenwirkungen und Wechselwirkungen von
Calciumantagonisten
2.4.1 Nebenwirkungen
Die Calciumantagonisten zeigen nur geringe Nebenwirkungen und sind besonders bei
langsam einsetzenden Formulierungen gut verträglich. Häufige, wichtige Nebenwirkungen
umfassen Beinödeme, Flush, Vertigo, Konstipation, Nausea, Ausschläge und Müdigkeit.
Eine Zusammenfassung der Nebenwirkungen ist in Tabelle 4 nach Symptom und Inzidenz
des Symptoms und Therapieabbruches, und in Tabelle 5 nach Organ oder Wirkung und der
jeweilig zuordenbaren unerwünschten Nebenwirkung angeführt [24, 28, 31].
Tabelle 4: Nebenwirkungen der Calciumantagonisten nach Symptomen [24]
Symptom Verapamil Gallopamil Diltiazem Nifepidin
Vertigo/Cephalgie + + + ++
Hypotonie +/- +/- +/- ++
Periphere Ödeme ++ + + ++
Konstipation ++ + - +/-
Gastrointestinale
+ + +/- +/-
Nebenwirkungen/Nausea
Bradykardie + + + -
Tachykardie - - - +
Angina Pectoris - - - +
Herzinsuffizienz + + +/- -
Allergische Reaktionen +/- +/- +/- +/-
Inzidenz der Neben-
5-20 5-15 4-10 15-50
wirkungen (in %)
Inzidenz des Therapie-
1 3 1-5 5
abbruches (in %)
Bei oraler Verabreichung
Abk.: ++ = häufig, + = selten, +/- = sehr selten, - = beobachtet
Tabelle 5: Nebenwirkungen der Calciumantagonisten nach Organsystem [24]
Organ/Wirkung Nebenwirkung prädominant bei
Negative Inotropie Herzinsuffizienz V (D) (N)
Negative Chronotropie Bradykardie V D
Arterielle Vasodilation
Blutdruckabfall, Hypotension, Vertigo, Flush,
(Reduktion des peripheren (V) (D) N
Ödeme
Widerstandes)
Gesicht Flushing N
Zentralnervensystem Cephalgie N
Nase Nasale Verstopfung N
Ventilations-Perfusions-Mismatch (mit einer
Lunge N
vorbestehenden Ventilationsstörung)
Verschlechterung der renalen Funktion (bei
Niere vorbestehender Einschränkung der N
Nierenfunktion)
18Fortsetzung von Tabelle 5: Nebenwirkungen der Calciumantagonisten nach Organsystem [24]
Organ/Wirkung Nebenwirkung prädominant bei
Uterus Menorrhagien N
Autonome Gegenregulation
(Aktivierung des sympathischen Tachykardie, Palpitationen, Angina Pectoris N
Nervensystems)
V
Konstipation (N)
N
Inhibierung der glatten hiatale Insuffizienz
Muskulatur gastroösophagialer Reflux Miktionsstörungen
N
Menorrhagien (V)
N
Aktivierung des Renin-
Natriumretention, Ödeme N
Angiotensin-Aldosteron-Systems
Inhibierung der Insulinsekretion Verschlechterung der Glukosetoleranz N (V)
Abk.: V = Verapamil-Gruppe, D = Diltiazem, N = Nifepidin (Dihydropyridin-Gruppe)
2.4.2 Wechselwirkungen
Calciumantagonisten weisen eine Vielzahl an wichtigen Interaktionen mit anderen
Medikamenten auf. Allen Calciumantagonisten ist eine verstärkt hypotone Wirkung bei
gleichzeitiger Verabreichung mit anderen Antihypertensiva und eine hauptsächliche,
hepatische Metabolisierung durch das Enzym Cytochrom P450 3A4, wodurch
Plasmakonzentrationen von anderen Wirkstoffen mit gleichem Metabolisierungsweg
beeinflusst werden, gemein. Von einem Konsum von Grapefruitsaft während einer
Nifedipin-Therapie wird abgeraten. Eine Übersicht der Wechselwirkungen wird in Tabelle
6 angeführt [28, 31, 32, 33].
Tabelle 6: Wechselwirkungen der Calciumantagonisten [32]
Kombination CCB Effekt Anmerkungen
mit
Alpha-1- vermehrte bzw. verstärkte First-
Rezeptor- V, D Dose-Effekte der Alpha-
Antagonisten Blocker können auftreten.
V,D, N Additive kardiodepressive Die gleichzeitige Behandlung mit Amiodaron und
Amiodaron
Wirkung möglich CCB vom Verapamil-Typ wird nicht empfohlen.
Reduktion der oralen Dosis von Midazolam um 50-
Verstärkte zentral dämpfende
75% empfohlen. Lorazepam, Lormetazepam,
Benzodiazepine V, D Wirkungen der betroffenen
Oxazepam und Temazepam sind wahrscheinlich
Benzodiazepine möglich
nicht von der Interaktion betroffen.
In Einzelfällen Bradykardie, Die gleichzeitige parenterale Anwendung von N und
Beta-Blocker N
Hypotonie, Herzinsuffizienz Beta-Blockern soll unterbleiben.
Verstärkte kardiodepressive Die i.v.-Gabe eines oder beider Arzneistoffe ist
Beta-Blocker V, D
Wirkung kontraindiziert - außer in der Intensivmedizin.
Cholesterol- Amlodipin Verstärkte Wirkungen von Höchstdosen: Simvastatin 20 mg/die, Lovastatin 40
Synthese- Simvastatin nicht mg/die; Alternativen: Fluvastatin oder Pravastatin
Hemmer auszuschließen bzw. DHP; Die Creatinkinase-Aktivität soll dann
(CSE-Hemmer) bestimmt werden; bei einem Anstieg auf mehr als
Lovastatin, V, D Erhöhtes Risiko von Myo- das 10fache des oberen Normwertes (etwa 2000 U/l)
Simvastatin, pathien, Rhabdomyolysen mit oder intolerablen Muskelbeschwerden muss der
Atorvastatin Myoglobinurie und NV CSE-Hemmer abgesetzt werden.
19Sie können auch lesen

















































