Die Rolle von X-linked inhibitor of apoptosis protein (XIAP) in der Systemischen Sklerose - Ludwig Hallenberger
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Die Rolle von X-linked inhibitor of apoptosis protein
(XIAP) in der Systemischen Sklerose
Der Medizinischen Fakultät
der Friedrich-Alexander-Universität Erlangen-Nürnberg
zur
Erlangung des Doktorgrades Dr. med.
vorgelegt von
Ludwig Hallenberger
2021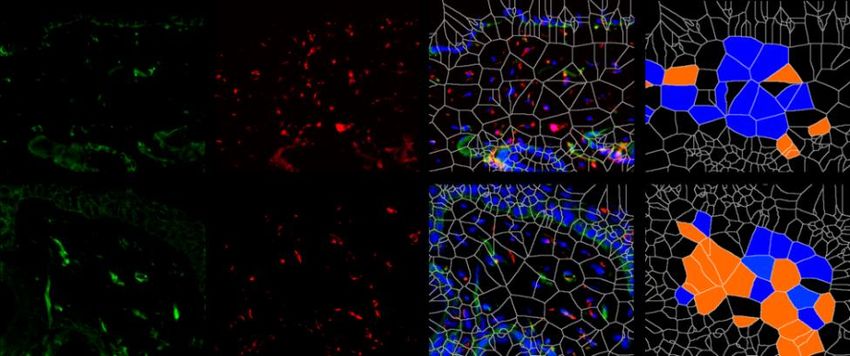
Als Dissertation genehmigt
von der Medizinischen Fakultät
der Friedrich-Alexander-Universität Erlangen-Nürnberg
Vorsitzender des Promotionsorgans: Prof. Dr. Markus Neurath
Gutachter: Prof. Dr. Jörg Distler
Gutachter: Prof. Dr. Georg Schett
Tag der mündlichen Prüfung: 31. August 2021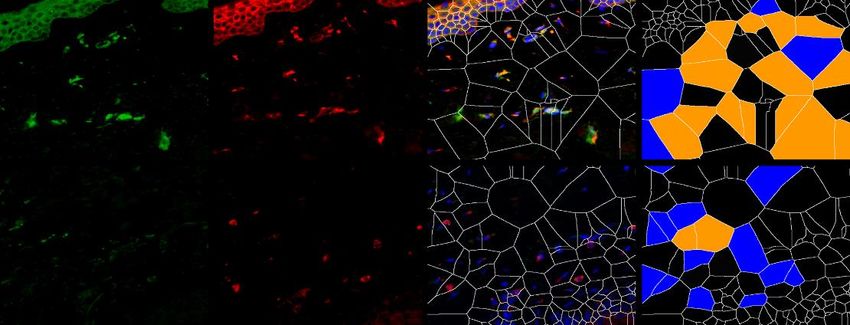
Meiner Familie in Liebe und Dankbarkeit gewidmet
Inhaltsverzeichnis
1 Abstract ................................................................................................................................... 1
2 Zusammenfassung ................................................................................................................... 3
3 Einleitung ................................................................................................................................ 5
3.1 Systemische Sklerose ....................................................................................................... 5
3.1.1 Epidemiologie ............................................................................................................ 5
3.1.2 Klinische Manifestation ............................................................................................. 6
3.2 Pathogenese der systemischen Sklerose ........................................................................... 7
3.2.1 Der TGF-β Signalweg ................................................................................................ 8
3.2.2 Der Wnt Signalweg .................................................................................................. 10
3.3 Das XIAP Protein ........................................................................................................... 11
4 Zielsetzung ............................................................................................................................ 12
5 Material und Methoden ......................................................................................................... 13
5.1 Mausmodelle .................................................................................................................. 13
5.1.1 Bleomycin-induzierte Fibrose .................................................................................. 13
5.1.2 cGvHD-induzierte Fibrose ....................................................................................... 13
5.1.3 TBRI/LacZ-induzierte Fibrose................................................................................. 13
5.1.4 Topoisomerase I-induzierte Fibrose ........................................................................ 14
5.1.5 Wnt-induzierte Fibrose ............................................................................................ 14
5.1.6 Weitere Mausbehandlungen..................................................................................... 14
5.1.7 Monitoring der Mäuse.............................................................................................. 14
5.2 Patienten und Fibroblasten ............................................................................................. 14
5.2.1 Stimulation von Fibroblasten ................................................................................... 15
5.2.2 Selektive Hemmung von XIAP durch Embelin ....................................................... 15
5.2.3 Nucleofection mit siRNA ........................................................................................ 15
5.3 Bestimmung von Apoptose ............................................................................................ 16
5.3.1 MTT Assay .............................................................................................................. 16
I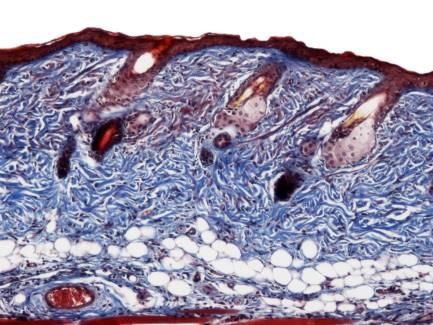
5.3.2 TUNEL Assay .......................................................................................................... 16
5.3.3 FACS........................................................................................................................ 16
5.4 Quantitative real-time PCR ............................................................................................ 17
5.5 Quantifizierung von Kollagen Protein ............................................................................ 17
5.5.1 SirCol Assay ............................................................................................................ 17
5.5.2 Hydroxyprolin Assay ............................................................................................... 18
5.6 Western Blot ................................................................................................................... 18
5.7 Histologische Analysen .................................................................................................. 18
5.7.1 HE Färbung .............................................................................................................. 19
5.7.2 Trichrom Färbung .................................................................................................... 19
5.7.3 Sirius Red Färbung .................................................................................................. 19
5.8 Immunhisto- und Immunzytochemie.............................................................................. 20
5.8.1 Immunhistochemie ................................................................................................... 20
5.8.1.1 α-SMA Färbung ................................................................................................ 20
5.8.2 Immunzytochemie.................................................................................................... 20
5.9 Statistik ........................................................................................................................... 21
6 Ergebnisse ............................................................................................................................. 22
6.1 Die Hochregulation von XIAP in SSc ............................................................................ 22
6.1.1 Die Expression von XIAP ist in SSc Fibroblasten erhöht ....................................... 22
6.1.2 Die Expression von XIAP ist in SSc Mausmodellen erhöht .................................... 25
6.1.2.1 Die Expression von XIAP ist in Modellen mit Bleomycin-induzierter
Hautfibrose erhöht ......................................................................................................... 25
6.1.2.2 Die Expression von XIAP ist in Modellen mit chronischer Graft-versus-Host
Erkrankung-induzierter Hautfibrose erhöht .................................................................. 27
6.1.2.3 Die Expression von XIAP ist in Modellen mit Topoisomerase I-induzierter
Hautfibrose erhöht ......................................................................................................... 28
6.1.2.4 Die Expression von XIAP ist in Modellen mit Wnt-induzierter Hautfibrose
erhöht ............................................................................................................................. 30
6.1.3 TGF-β induziert die Expression von XIAP ............................................................. 31
II
6.1.3.1 TGF-β induziert die Expression von XIAP in vitro .......................................... 31
6.1.3.2 TGF-β induziert die Expression von XIAP in vivo ........................................... 33
6.1.4 Die Inhibierung von XIAP verbessert die durch TGF-β-induzierte ........................ 36
Fibroblasten Aktivierung .................................................................................................. 36
6.1.5 Die Inhibierung von XIAP verbessert die durch Wnt-induzierte Fibroblasten
Aktivierung ....................................................................................................................... 40
6.1.6 Die Inhibierung von XIAP verbessert die in Mausmodellen induzierte Hautfibrose
........................................................................................................................................... 43
6.1.6.1 Die Inhibierung von XIAP verbessert Bleomycin-induzierte Hautfibrose ....... 43
6.1.6.2 Die Inhibierung von XIAP verbessert Topoisomerase I-induzierte Haut- und
Lungenfibrose................................................................................................................ 44
6.1.6.3 Die Hemmung von XIAP verbessert Wnt-induzierte Hautfibrose.................... 47
7 Diskussion ............................................................................................................................. 49
8 Literaturverzeichnis ............................................................................................................... 52
9 Abkürzungsverzeichnis ......................................................................................................... 57
10 Danksagung ......................................................................................................................... 63
III
1 Abstract
Background and Objectives:
Systemic Sclerosis (SSc) is a connective tissue disorder and has the highest morbidity and
mortality among rheumatic diseases. The underlying cause of this disease is linked to a
persistent fibroblast activation in connection with an increased production of extracellular
matrix, ultimately resulting in fibrosis. A key role in this process is the uncontrolled activation
of Wnt and transforming growth factor-β (TGF-β) signaling. The molecular mechanisms
underlying their interaction are not yet fully understood. The X-linked inhibitor of apoptosis
protein (XIAP) is a multifactorial protein, which is involved in a multitude of cellular functions
including immunological processes. In the current thesis, we evaluated XIAP as a potential
molecular link between Wnt and TGF-β signaling in the context of fibrotic diseases.
Methods:
Cultured dermal fibroblasts from SSc patients and matched healthy volunteers were stimulated
with Wnt-1 and TGF-β. XIAP was inactivated by siRNA-knockdown and pharmacologic
inhibition with Embelin. MTT-assay, TUNEL-assay, or FACS excluded potential toxic side
effects. XIAP expression was analyzed by qPCR, Western Blot, and immunofluorescence.
Collagen deposition was quantified by SirCol- and HP-assay. In vivo experiments were
performed using different mouse models such as Bleomycin-induced skin fibrosis,
Topoisomerase-induced fibrosis, a model of sclerodermatous chronic graft-versus-host-disease
(cGvHD), mice with overexpression of Wnt10b or overexpression of a constitutively active
TGF-β receptor type I (TBRI). Dermal and pulmonary fibrosis was analyzed via histologically
(HE, Trichrome, and Sirius Red staining), by immunofluorescence-based quantification of
myofibroblasts, by real-time PCR based assessment of Col1a1 mRNA and by quantification of
the hydroxyproline content.
Results:
The expression of XIAP was upregulated in fibroblasts of patients with SSc and cGvHD.
Additionally, an increased number of XIAP positive fibroblasts and of total XIAP protein was
observed in skin samples of Bleomycin-, cGvHD-, TopoI-, and Wnt10btg mice. TGF-β induced
XIAP expression in vitro and in vivo in a SMAD3-dependent manner. Inhibition of XIAP
reduced TGF-β and Wnt induced fibroblast activation. Col1a1 mRNA levels as well as the
amount of collagen and α-SMA protein were significantly reduced. Comparable effects were
1observed in vivo by inhibition of XIAP in Topoisomerase-, Wnt-, Bleomycin, and TBRI-
induced skin fibrosis. Inhibition of XIAP ameliorated fibrosis with reduced skin thickening,
myofibroblast counts, and hydroxyproline content.
Conclusions:
This research characterizes XIAP as a key regulator of TGF-β and Wnt mediated fibroblast
activation. XIAP is increased in SSc such as cGvHD patients and therefore might have an
important role in fibroblast activation in fibrotic diseases. Inhibition of XIAP ameliorates
fibrosis in complementary mouse models at well tolerated doses. These results suggest that
inhibition of XIAP might be a possible therapeutic approach in the treatment of fibrotic
diseases.
22 Zusammenfassung
Hintergrund und Ziele:
Die Systemische Sklerose zählt zu den Erkrankungen der Kollagenosen und hat unter den
rheumatischen Erkrankungen die höchste Mortalität und Morbidität. Die Ursache dieser
Erkrankung liegt in einer übermäßigen Fibroblastenaktivierung mit gesteigerter Produktion
extrazellulärer Matrix, was in der Entstehung von Fibrose endet. Eine Schlüsselrolle spielt
hierbei die unkontrollierte Aktivierung des Wnt und TGF-β Signalweges, deren genaue
Interaktionen miteinander nach wie vor nicht gänzlich verstanden sind. X-linked inhibitor of
apoptosis protein (XIAP), das als multifaktorielles Protein auch Einfluss auf viele
immunologische Prozesse hat, könnte eine Verbindung zwischen diesen Signalwegen
darstellen und wird in dieser Arbeit genauer untersucht.
Methoden:
Für in vitro Versuche wurden kultivierte dermale Fibroblasten von SSc Patienten und gesunden
Probanden verwendet. Zur weiteren Untersuchung wurden die Fibroblasten mit Wnt-1 oder
TGF-β behandelt. Mittels siRNA-Knockdown und pharmakologischer Hemmung durch
Embelin wurde XIAP inaktiviert. Toxische Effekte dieses Wirkstoffes wurden durch MTT-
Assay, TUNEL-Assay oder FACS geprüft. Die Expression von XIAP wurde mittels qPCR,
Western Blot und Immunfluoreszenz analysiert. Kollagen wurde durch SirCol- und HP-Assay
quantifiziert. In vivo Versuche wurden in Bleomycin, Topoisomerase, cGvHD, Wnt und TBRI
Modellen durchgeführt. Eine histologische Auswertung erfolgte durch HE-, Trichrom- und
Sirius Red Färbung.
Ergebnisse:
Die XIAP Expression war in Fibroblasten von SSc und cGvHD Patienten erhöht. Zusätzlich
ließ sich in Hautproben aus Mäusen mit Bleomycin-, cGvHD-, Topoisomerase- und Wnt-
induzierter Fibrose vermehrt XIAP positive Fibroblasten und XIAP Protein finden. TGF-β
induzierte die XIAP Expression SMAD3-abhängig in vitro und in vivo. Eine Hemmung von
XIAP verringerte die durch TGF-β und Wnt-induzierte Fibroblastenaktivierung. Die Col1a1
Expression sowie die Menge an Kollagen bzw. α-SMA waren signifikant reduziert. Der gleiche
Effekt zeigte sich in vivo durch eine Hemmung von XIAP in Topoisomerase-, Wnt-, Bleomycin
und TBRI-induzierter Hautfibrose. Hautdicke, Myofibroblastenzahl und Hydroxyprolingehalt
waren signifikant reduziert.
3Schlussfolgerungen:
In dieser Arbeit wird XIAP als zentraler Mediator der TGF-β und Wnt vermittelten
Fibroblastenaktivierung dargestellt. XIAP ist in SSc sowie cGvHD Patienten erhöht und spielt
somit eine wichtige Rolle in fibrotischen Erkrankungen. Eine Hemmung von XIAP verbessert
die Fibrose in Tiermodellen und reduziert die signalabhängige Fibroblastenaktivierung. Die
Ergebnisse dieser Studie stellen einen möglichen Therapieansatz in der Behandlung fibrotischer
Erkrankungen dar.
43 Einleitung
3.1 Systemische Sklerose
Bei der Systemischen Sklerose (SSc) handelt es sich um eine prototypische, multisystemische
Autoimmunerkrankung aus der Gruppe der Kollagenosen [1, 2]. Sie zeichnet sich durch
Gefäßveränderungen, der Bildung von Autoantikörpern gegen verschiedene zelluläre Antigene
und eine ausgeprägte Fibrose aus [3]. Die Fibrosierung des Gewebes ist dabei nicht
ausschließlich auf die Haut beschränkt und kann auch innere Organe wie Herz, Lunge oder
Nieren betreffen. Dies trägt maßgeblich dazu bei, dass die SSc die höchste fallbezogene
Mortalität unter den rheumatologischen Autoimmunerkrankungen aufweist [4, 5]. Da die SSc
multiple Organe im Körper befallen kann, weist die Erkrankung eine sehr hohe klinische
Heterogenität auf. Dieser Umstand stellt selbst für erfahrene Ärzte eine große Herausforderung
dar und führt nicht selten dazu, dass die Diagnose bei Patienten zu spät gestellt bzw. eine
adäquate Therapie verzögert wird [2, 6]. Obwohl sich das Outcome in den letzten vier Dekaden
aufgrund frühzeitiger Erkennung lebensbedrohlicher Komplikationen und konsequenterer
Nachsorge deutlich verbessert hat, gibt es nach wie vor keine adäquate antifibrotische Therapie
[6, 7].
3.1.1 Epidemiologie
Die SSc weist eine Prävalenz von 7-530/1.000.000 Einwohner und eine Inzidenz von 7-
489/1.000.000 Einwohner auf. Die große Spannweite der Ergebnisse hängt mit
unterschiedlichen geographischen Lagen, Methoden und Krankheitsdefinitionen zusammen.
Eine steigende Prävalenz in den letzten Jahrzehnten ist auf eine verbesserte Überlebensrate
zurückzuführen. Trotz dieser Steigerung ist die SSc nach wie vor selten und zählt zu den
seltenen Erkrankungen [7-9]. Der Beginn der Erkrankung liegt meist zwischen 30-50 Jahren
und Frauen sind 3-4 Mal häufiger betroffen als Männer [10-12]. Darüber hinaus gibt es
Hinweise darauf, dass farbige Menschen häufiger und schwerer von der Erkrankung betroffen
sind [3, 8]. Die kumulative Überlebensrate liegt bei 75% über fünf Jahre und bei 63% über zehn
Jahre. Eine pulmonale Manifestation ist hierbei die führende Todesursache und tritt in
Abhängigkeit des Subtyps bei 25-90% auf [7, 13]. Patienten mit SSc haben eine
durchschnittlich um 16-34 Jahre verringerte Lebenserwartung [5].
53.1.2 Klinische Manifestation
Die SSc lässt sich in Abhängigkeit von der Hautbeteiligung in zwei Subgruppen aufteilen: der
limitiert kutanen SSc (lcSSc) und der diffus kutanen SSc (dcSSc).
Die lokal kutane SSc macht rund 60% der Fälle aus und zeichnet sich durch eine distal betonte
Hautbeteiligung aus. Hierbei sind Hautareale proximal bis zu den Knien bzw. Ellenbogen
betroffen. Zusätzlich kann es zu einem Befall im Gesichts- und Nackenbereich kommen [2, 6].
Neben der Hautbeteiligung kann es auch zu einem fibrotischen Befall der Organe kommen.
Dieser ist aber limitiert, langsam progredient und tritt erst im späteren Verlauf der Erkrankung
auf [4, 10]. Ein Raynaud Phänomen tritt meist Jahre im Voraus auf, bevor es zu weiteren
Symptomen kommt [3, 14]. Weitere charakteristische Symptome sind Tabaksbeutelmund,
Sklerodaktylie, Teleangiektasien, dilatierte Nagelbettkapillaren oder Calcinosis Cutis [6]. Des
Weiteren kommt es bei der lcSSc häufig zu einer isolierten pulmonal arteriellen Hypertonie
(PAH). 50-90% der Patienten weisen positive Anti-Zentromer-Antikörper auf, die mit dem
erhöhten Risiko verbunden sind, im Verlauf eine PAH zu entwickeln [2, 3, 6].
Die diffus kutane Verlaufsform tritt bei 20-40% der SSc Patienten auf. Hierbei kommt es zu
einem Hautbefall proximal der Knie und Ellenbogen, des Stammes und der Gesichtspartien. Im
Vergleich zur lcSSc geht die Hautbeteiligung mit einem zeitgleichen Raynaud Phänomen und
der Fibrose innerer Organe einher. Häufig betroffen sind Ösophagus, Lunge, Herz, Nieren und
GI-Trakt, wenngleich prinzipiell jedes Organ betroffen sein kann [2, 10]. Die Fibrosierung
zeichnet sich zudem durch einen raschen Progress aus [4]. Bei der dcSSc kann es zu
verschiedenen unspezifischen Frühsymptomen kommen. Dazu zählen Hautspannen, Juckreiz,
muskuloskelettale Schmerzen und Schwäche, Fatigue, Belastungsdyspnoe und Gewichtsverlust
aufgrund verminderter Nahrungsmittelaufnahme [6]. Typisch für diese Untergruppe sind
positive Anti-Scl70-Antikörper. Hierbei handelt es sich um Antikörper, die gegen
Topoisomerase I gerichtet sind und mit einem erhöhten Risiko für schwere Lungenbeteiligung
assoziiert sind. Positive Anti-RNA Polymerase III-Antikörper sind mit einer schnell
progressiven Hautbeteiligung und erhöhtem Risiko für das Entwickeln einer renalen Krise
assoziiert. Diese Komplikation kommt bei rund 10% der Patienten vor und äußert sich durch
plötzliches Auftreten einer malignen Hypertonie. Unbehandelt kann dies schnell zum
Nierenversagen und zum Tode führen [2, 10].
Wie beschrieben zeigen die einzelnen Subgruppen Assoziationen zu bestimmten Symptomen
und Antikörperprofilen. Dennoch ist die Diagnosestellung und die Zuordnung zu den
6Subgruppen nicht immer eindeutig und stellt eine klinische Herausforderung dar. Daher ist die
Aktualisierung von Klassifikationskriterien fortlaufend Gegenstand aktueller Forschung.
Zuletzt wurden diese durch van den Hoogen et al. in den ACR/EULAR Kriterien von 2013
erneuert [15].
Die Therapie der SSc ist auch heute noch eine große Herausforderung für Ärzte und Patienten.
Ein Großteil der Therapie dreht sich um das Erkennen und Behandeln von Komplikationen.
Dazu zählen Lungenfibrose, PAH, kardiale Fibrose, digitale Kontrakturen, GI-Komplikationen
usw. [6]. Die antifibrotisch wirksame Therapie der SSc ist nach wie vor jedoch begrenzt und
nur unter bestimmten Bedingungen einsetzbar. So ist die autologe Stammzelltherapie nach
Hochdosischemotherapie, die in Studien gute Langzeitergebnisse hat, nur einem selektiven
Patientengut mit rapide fortschreitender SSc vorenthalten, da sie mit einem signifikanten
Letalitätsrisiko einhergeht [7].
3.2 Pathogenese der systemischen Sklerose
Die genaue Pathogenese der SSc ist nach wie vor nicht vollkommen verstanden. Man geht
davon aus, dass eine komplexe Wechselwirkung zwischen exogenen Faktoren und genetischer
Prädisposition zu einer Induktion von SSc spezifischen Gen-Programmen führt [1]. Mögliche
Umweltfaktoren, für die es bereits mehrere Hinweise in Studien gibt, sind Silikate,
Lösungsmittel, Vinylchlorid, Medikamente und Infektionen [1, 6, 10, 16]. Durch diese
exogenen Einflussfaktoren kommt es vermutlich auch zu einer direkten Beeinflussung von
zellulären Signalwegen und epigenetischen Mechanismen. Zu diesen zählen zum Beispiel die
Histonmodifikation oder die DNA-Methylierung [1]. Darüber hinaus stehen bestimmte Allele,
z.B. in den TGF-β bzw. ACE (angiotensin converting enzyme) Genen in Verdacht, eine erhöhte
Anfälligkeit für SSc zu verursachen [10].
Durch diese Prädispositionen vermittelt entstehen Schäden an den Endothelzellen der kleinen
und mittelgroßen Gefäße von Finger, Lunge, Herz und GI-Trakt [1, 10, 17]. Im Rahmen dieser
Schäden kommt es zu einer erhöhten Endothelin-1 (ET-1) Produktion, die mit einer
verminderten Prostazyklin Freisetzung und konsekutiver Vasokonstriktion einhergeht [10].
Zusätzlich kommt es zu einer erhöhten Produktion von Adhäsionsmolekülen und
Sauerstoffradikalen, die zu einer Intima Proliferation, Media Hyperplasie und Adventitia
Fibrose führen [2, 10, 17]. Diese Gefäßveränderungen stellen eine der drei Hauptcharakteristika
der SSc Pathogenese dar. Neben der Vaskulopathie kommt es zu einer
Autoimmunität/Entzündung und Fibrose [1, 2, 4, 18]. Infolge von Endothelschäden und
7Gefäßveränderungen kommt es zu einer Aktivierung des zellulären Immunsystems. CD4+ T-
Zell dominierte Zellinfiltrate führen zu einer erhöhten Freisetzung von Zytokinen (wie IL-2,
IL-4, IL-6) und Wachstumsfaktoren (z.B. TGF-β). Dies verursacht weitere Gefäßschäden und
verstärkt die Fibrose Entwicklung zusätzlich [10, 19]. Eine zeitgleiche Aktivierung von B-
Zellen führt zu einer vermehrten Produktion von Autoantikörpern, deren genauer Einfluss an
der Pathogenese jedoch noch nicht geklärt ist. Neben der Bildung von Autoantikörpern kommt
es auch zu einer Überexpression von B-Zell assoziierten Genen [2, 10].
Diese Veränderungen führen zu einer übermäßigen Fibroblastenaktivierung, wodurch es im
Verlauf zu einer erhöhten Produktion kollagenhaltiger Extrazellulärmatrix (ECM) kommt.
Insbesondere durch TGF-β kommt es zu einer Differenzierung von Fibroblasten zu
Myofibroblasten. Diese Zellen zeichnen sich durch eine vermehrte Expression von α-SMA (α-
smooth muscle actin), profibrotischer Zytokine und extrazellulärer Matrix aus. Zudem scheinen
Myofibroblasten in einem gewissen Umfang Apoptose-resistent zu sein [10, 18, 20]. Neben
Fibroblasten ist es auch anderen Zellen, wie den Endothelzellen, möglich, einen
Myofibroblasten ähnlichen Phänotyp anzunehmen und so zur Produktion von ECM beizutragen
[21]. Die exzessive Ansammlung der Extrazellulärmatrix ersetzt mit der Zeit zunehmend
gesundes Gewebe und zerstört nachhaltig die eigentliche Architektur [18]. Unter
physiologischen Umständen ist dieser Prozess auch bei der Wundheilung aktiv. Hier handelt es
sich jedoch um einen selbstlimitierenden Prozess, der durch ein Gleichgewicht aus pro- und
antifibrotischen Signalen reguliert wird. Bei der SSc hingegen ist diese Autoregulation gestört
und Myofibroblasten produzieren unabhängig exogener Stimuli extrazelluläre Matrix und
Zytokine [1, 4, 20, 21]. Diese Eigenschaften machen Myofibroblasten zu den zentralen
Schlüsselmediatoren in der Fibroseentstehung [18, 20].
3.2.1 Der TGF-β Signalweg
Bei TGF-β (transforming growth factor β) handelt es sich um eine Zytokin Superfamilie aus 33
Genen, die einen weitreichenden Einfluss auf viele physiologische Prozesse im menschlichen
Körper hat. Dazu zählen embryonale Entwicklung, Homöostase, Wundheilung, Chemotaxis
und Zellzykluskontrolle [20, 22-24]. Bisher sind mehr als 60 Mitglieder der TGF-β
Superfamilie bekannt. Darunter Bone Morphogenetic Proteins (BMPs), growth/differentiation
factors (GDFs), Anti Müller Hormone (AMHs) und die drei TGF-βs (TGF-β1, TGF-β2, TGF-
β3) [23, 25, 26].
8Wie alle anderen sezernierten Proteine wird auch TGF-β am endoplasmatischen Retikulum
gebildet und anschließend im Golgi-Apparat posttranslational modifiziert. Die entstandenen
Proteine werden in Vesikel verschlossen und mittels Exozytose in die extrazelluläre Matrix
(ECM) sezerniert. Dort liegt das inaktive TGF-β an Fibronectin und Fibrillin gebunden vor.
Durch weitere enzymatische Modifikation, u.a. durch Integrine vermittelt, wird das inaktive
TGF-β in seine aktive Form überführt und steht der weiteren Signaltransduktion zu Verfügung
[23, 27].
Das aktive TGF-β bindet für die weitere Signaltransduktion an TGF-β Typ I und II Rezeptoren
(TGFβI und TGFβII), die in der Plasmamembran lokalisiert sind [24]. Bisher wurden fünf Typ
II Rezeptoren und sieben Typ I Rezeptoren entdeckt, die sich in allen Zellen des Menschen
finden lassen. Initial bindet TGF-β an den TGFβII Rezeptor, der als Homodimer vorliegt und
eine höhere Affinität zu dem Liganden aufweist. Das führt dazu, dass zwei weitere Typ I
Rezeptoren an den Komplex binden und durch den Typ II Rezeptor phosphoryliert und aktiviert
werden. Der Typ I Rezeptor wiederum phosphoryliert SMAD Proteine (R-SMAD) als seine
Substrate. SMAD2 und SMAD3 werden phosphoryliert, dissoziieren von dem Rezeptor und
bilden einen Komplex mit SMAD4. Dieser transloziert in den Zellkern, in dem er an die DNA
bindet und die Transkription von Genen positiv oder negativ reguliert. Der TGF-β Signalweg
selbst ist auch durch viele positive und negative Mechanismen reguliert. So konkurriert zum
Beispiel SMAD7 mit SMAD2/3 an ihrem Rezeptor bzw. hemmt die Transkriptionsaktivität des
SMAD-Komplexes an der DNA [23-28].
Neben dem klassischen, kanonischen TGF-β Signalweg gibt es auch nicht-kanonische
Signalwege. Zu diesen gehören ERK, P38 und JNK Mitogen-aktivierte Proteinkinasen, Rho-
like GTPase und PI3K Signalwege [22, 27-29].
Außer dem Einfluss auf physiologische Prozesse kann eine Störung im TGF-β Signalweg zu
vielerlei Pathologien wie Krebserkrankungen, gefäß- oder immunbedingten Krankheiten und
Fibrose führen [24, 27, 30]. So reicht in Mäusen die alleinige Überexpression eines konstitutiv
aktiven TGF-β Rezeptor Typ I, um eine Fibrose der Haut zu erzeugen [29]. Zudem konnte in
SSc Fibroblasten eine erhöhte Expression an Integrinen festgestellt werden, die bei der
Aktivierung von latentem TGF-β eine Schlüsselrolle spielen [27]. Gleichzeitig konnte gezeigt
werden, dass die Hemmung des TGF-β Signalweges auf verschiedenen Ebenen antifibrotische
Effekte erzielt [20, 29]. Diese und viele andere Erkenntnisse machen deutlich, dass ein
fehlregulierter TGF-β Signalweg eine grundlegende Rolle in der Pathogenese der SSc spielt
[22, 30].
93.2.2 Der Wnt Signalweg
Der Wnt Signalweg ist ein weiterer Signalweg, der einen Einfluss auf die Entstehung
fibrotischer Erkrankungen hat. Er ist unterteilt in einen kanonischen, β-Catenin abhängigen,
und nicht-kanonische Signalwege, wobei bisher nur der kanonische Signalweg in direkten
Zusammenhang mit Fibrose Entstehung gebracht wurde [31]. Unter physiologischen
Bedingungen spielt der Wnt Signalweg eine Rolle bei der embryonalen Entwicklung und der
Gewebehomöostase [31, 32]. Defekte in diesem Signalweg sind dementsprechend mit
Geburtsfehlern, Krebs oder anderen Erkrankungen assoziiert [33]. Besonders in den letzten
Jahren häufen sich auch die Hinweise darauf, dass ein überaktiver Wnt Signalweg eine
wesentliche Rolle in der Pathogenese der SSc spielt und so ein potenzielles Ziel für zukünftige
Therapien darstellt [31, 34-36].
In Abwesenheit von Wnt Liganden wird β-Catenin kontinuierlich durch einen
Zerstörungskomplex bestehend aus Axin, APC (adenomatous polyposis coli gene product),
CK1 (casein kinase 1) und GSK3 (glycogen synthase kinase 3) degradiert. Dieser Komplex
phosphoryliert β-Catenin, was zu dessen Ubiquitinierung und anschließenden Degradierung im
Proteasom führt. Der kontinuierliche Abbau verhindert so die Anreicherung von β-Catenin im
Zellkern, wodurch die Expression von Wnt Zielgenen durch einen Komplex aus TCF (T cell
factor) und LEF (lymphoid enhancer factor) verhindert wird [32, 33, 37].
Bei der Wnt Familie handelt es sich um sezernierte Glykoproteine bestehend aus 19
verschiedenen Liganden. Diese Liganden binden zusammen mit den Korezeptoren LRP 5/6
(low-density lipoprotein receptor-related protein 5/6) an einen von sieben Frizzled
Transmembranrezeptoren (Fz) [38]. Dieser Ligand-Rezeptor Komplex bindet das
Strukturprotein Dishevelled und aktiviert durch Phosphorylierung LRP6. Zusätzlich wird der
Axin-Zerstörungskomplex an den Rezeptor Komplex gebunden. Diese Bindung verhindert die
Phosphorylierung und Ubiquitinierung von β-Catenin, wodurch dessen zytoplasmatische
Konzentration steigt und das Protein in den Zellkern transloziert. Dort bindet β-Catenin mit den
Kofaktoren p300 und CBP (CREB-binding protein) an TCF/LEF und aktiviert so die
Expression von Wnt Zielgenen [31, 33, 37].
Einen zusätzlichen Regulationsmechanismus stellen endogene Inhibitoren des Signalweges
dar. So zum Beispiel die Dickkopf Proteine (DKK), die an den LRP Korezeptor binden und so
die Formierung eines Komplexes mit dem Fz Rezeptor verhindern [36, 39].
103.3 Das XIAP Protein
X-linked inhibitor of apoptosis (XIAP) zählt zu der Familie der inhibitor of apoptosis (IAP)
Proteine. Durch ihre Hemmung von Caspasen zählen sie zu den intrinsischen Regulatoren des
programmierten Zelltods [40]. Neben dieser Funktion haben IAPs noch weitere Funktionen wie
den Einfluss auf Signalwege, den Zellzyklus oder Ubiquitinierung. So können sie auch Einfluss
auf Entzündungsprozesse, das Immunsystem oder Zellproliferation nehmen. Diese Fülle an
Eigenschaften macht IAPs zu multifaktoriellen Proteinen [40-42]. IAP Proteine sind definiert
durch ein ca. 80 Aminosäuren langes Baculoviral Inhibitor of apoptosis Repeat (BIR). Je nach
IAP liegen zwischen einer und drei dieser BIR Domänen vor [43]. Zusätzlich zu den BIR
Domänen enthalten einige IAPs eine RING Struktur. Diese RING Struktur ist ein Zink-Finger-
Protein mit E3 Ubiquitin Ligase Aktivität und ermöglicht es, andere Proteine durch
Ubiquitinierung zu degradieren [44, 45]. Bis heute sind in Säugetieren acht verschiedene IAPs
bekannt, die sich durch unterschiedliche Gewebespezifität und Funktionen voneinander
unterscheiden [41, 42].
XIAP zählt in der IAP Familie zu dem stärksten und vielseitigsten Caspase Inhibitor und wird
ubiquitär exprimiert [40, 46]. Das Protein besteht aus drei BIR Domänen und einer zusätzlichen
RING Struktur [43]. Neben den bereits genannten Funktionen konnte auch gezeigt werden, dass
XIAP ein Aktivator des TGF-β Signalweges ist und nach physiologischem Stress hochreguliert
wird [46]. So konnte bereits gezeigt werden, dass XIAP eine Rolle bei der Bleomycin
induzierten Lungenfibrose spielt [47]. Zusätzlich hat XIAP einen direkten Einfluss auf
Krebserkrankungen und steht in Zusammenhang mit Metastasierung, Tumorzell Motilität und
einem besonders aggressiven Krankheitsverlauf [44, 48, 49]. Zu erwähnen sei, dass bereits in
klinischen Studien eine Therapie mittels XIAP Inhibitoren untersucht wird [42, 50, 51].
Neben XIAP zählen noch cIAP1 und cIAP2 zu den klassischen anti-apoptotischen IAPs [41].
Diese ähneln sich nicht nur strukturell, sondern scheinen auch funktionell einen engen
Zusammenhang zu haben, da cIAP1 und 2 beide in ihren Konzentrationen kompensatorisch
steigen, sobald die Expression von XIAP vermindert wird [46].
114 Zielsetzung
Sowohl der TGF-β als auch der Wnt Signalweg spielen eine entscheidende Rolle bei der Fibrose
Entstehung generell und insbesondere bei der Systemischen Sklerose. Dennoch ist das genaue
Zusammenwirken dieser beiden Signalwege schlecht verstanden. Eine potenzielle Verbindung
zwischen diesen beiden Signalwegen könnte XIAP bilden. Es konnte bereits gezeigt werden,
dass XIAP als multifaktorielles Protein einen entscheidenden Einfluss auf immunologische
Prozesse und deren Mitspieler wie dem TGF-β Signalweg hat. Diesen Einfluss gilt es weiter zu
charakterisieren mit dem Ziel, dass aus diesen Erkenntnissen die Grundlage für neue
therapeutische Ansätze geschaffen werden können. Zweck dieser Arbeit ist es, den Einfluss von
XIAP auf diese Signalwege und somit der Pathogenese der Systemischen Sklerose weiter zu
verstehen.
125 Material und Methoden
5.1 Mausmodelle
5.1.1 Bleomycin-induzierte Fibrose
Sechs Wochen alte Mäuse wurden alle zwei Tage über vier Wochen durch eine Bleomycin
Injektion behandelt. Diese Injektion erfolgte in die Subkutanhaut des oberen Rückens.
Hierdurch kam es zu einer lokalen Hautfibrose und einer lokalen bzw. systemmischen
Inflammation. Es wurde jeweils eine Dosis von 0,5mg/ml injiziert. Randomisierte Kontrollen
wurden auf identische Weise mit 0,9% NaCl-Lösung behandelt.
5.1.2 cGvHD-induzierte Fibrose
Zur Induktion von sklerodermiformer Graft-versus-Host Erkrankung wurden [BALB/c (H-2d)]
Mäuse verwendet. Die tibialen und femoralen Knochenmarkskanäle wurden mit PBS von
Knochenmarkszellen freigespült. Daraufhin kam es zu erythrozytärer Hämolyse. Mit einem
100µm Nylon Zellsieb (Becton Dickinson, Franklin Lakes, USA) wurden die Splenocyten
separiert. Empfängermäuse ([BALB/c (H-2d)]) erhielten eine Komplettkörperbestrahlung mit
700Gy. 6h nach der Bestrahlung wurden 2x106 Splenocyten und 1x106 Knochenmarkszellen in
100µl PBS gelöst und den Mäusen durch venöse Injektion in den Schwanz transplantiert.
Hierbei erhielten die [BALB/c (H-2d)] Mäuse die Zellen entweder von [BALB/c (H-2d)]
Mäusen im Rahmen einer syngenen Transplantation oder von [B10.D2 (H-2d)] Mäusen als
allogene Transplantation.
5.1.3 TBRI/LacZ-induzierte Fibrose
Bei TBR handelt es sich um einen konstitutiv aktiven TGFβ Typ 1 Rezeptor (TBRact). Um
diesen Rezeptor in Mäusen zu überexprimieren, wurden attenuierte Adenoviren, die diesen
Rezeptor kodierten (adTBR), in den oberen Rücken der Mäuse injiziert. Attenuierte
Adenoviren, die für LacZ (β-Galaktosidase) kodierten, wurden als Kontrolle verwendet. Die
Behandlung der Mäuse erfolgte über einen Zeitraum von acht Wochen, wobei die Mäuse alle
zwei Wochen mit den jeweiligen Adenoviren injiziert wurden.
135.1.4 Topoisomerase I-induzierte Fibrose
Fünf Wochen alte C57Bl/6 Mäuse wurden durch eine subkutane Injektion mit 500U/ml
rekombinanter DNA Topoisomerase I behandelt. Der Wirkstoff wurde in einem Verhältnis von
3:2 in Freund’s Complete Adjuvant (CFA) gelöst und vier Mal über einen Zeitraum von acht
Wochen injiziert. Hieraus entwickelte sich sowohl eine lokale Hautfibrose an der
Injektionsstelle als auch eine pulmonale Fibrose. Randomisierte Kontrollen wurden auf
identische Weise mit CFA behandelt.
5.1.5 Wnt-induzierte Fibrose
Der Einfluss von XIAP auf den Wnt Signalweg wurde durch Mäuse untersucht, die transgen
eine Überexpression von Wnt-10b zeigten. Randomisierte Kontrollgruppen erhielten eine
Injektion mit Kochsalzlösung.
5.1.6 Weitere Mausbehandlungen
Mit Embelin therapierte Mäuse erhielten alle zwei Tage eine intraperitoneale Injektion mit
8mg/kg KG Embelin. Zusätzlich wurden Mäuse mit SD208 behandelt, einem spezifischen
Inhibitor der TGF-β Rezeptor I Kinase. Dieser wurde oral mit einer Dosis von 20mg/kg KG
verabreicht.
5.1.7 Monitoring der Mäuse
Die Mäuse wurden regelmäßig kontrolliert, um schädliche Effekte der Behandlung frühzeitig
zu erkennen. Dabei wurden Körpergewicht, Aktivität, Körperhaltung, Felltextur und Verhalten
beobachtet und beurteilt. Zusätzlich wurden die Mäuse bei der Sektion auf toxische Effekte
untersucht.
5.2 Patienten und Fibroblasten
Humane Fibroblasten wurden aus Hautbiopsien von 23 Patienten mit Systemischer Sklerose
(SSc) und 21 gesunden Probanden gewonnen. 16 dieser Patienten waren weiblich, sieben waren
männlich. Das mediane Erkrankungsalter lag bei 45 Jahren (Schwankung: 19-65 Jahre) und der
mediane Erkrankungszeitraum bei fünf (Schwankung: 0,5-10 Jahre) Jahren. 15 Patienten
(65,2%) hatten eine diffus kutane SSc und acht (34,7%) hatten eine limitiert kutane SSc. Alle
Patienten erfüllten die ACR/EULAR Kriterien für Systemische Sklerose von 2013. Eine aktive
SSc wurde durch den EUSTAR-Aktivitätsindex bestimmt. Die Biopsien wurden mit Dispase II
14(Invitrogen, Karlsruhe, Deutschland) und einem Medium aus RPMI, Collagenase D und
DNAse I verdaut. Die Zellen wurden anschließend mit DMEM-F12 (Thermo Fisher, Waltham,
USA), welchem 0,5% L-Glutamin, 0,2% Amphotericin B, 1% PIS und 10% FBS (fetales
Rinderserum) zugesetzt wurde, in 75cm2 großen Kulturflaschen bei 37°C mit 5% CO2 und 90%
Luftfeuchtigkeit kultiviert.
5.2.1 Stimulation von Fibroblasten
Fibroblasten für Experimente wurden aus Passagen zwischen vier und acht verwendet. Die
Fibroblasten wurden ab einer Dichte von ca. 90% mit 0,1% FBS haltigem DMEM-F12 und
0,1% Vitamin C für 24h vor den Experimenten gehungert. Anschließend wurden die Zellen mit
TGF-β1 (10ng/ml; R&D Systems, Minneapolis, USA) oder rekombinantem menschlichem
Wnt-1 (50ng/ml; Peprotech, Hamburg, Deutschland) zu verschiedenen Zeitpunkten stimuliert.
5.2.2 Selektive Hemmung von XIAP durch Embelin
Die Zellen wurden 24h vor der eigentlichen Stimulation mit Embelin (Sigma-Aldrich, St. Louis,
USA) und 0,1% FBS haltigem DMEM-F12 präinkubiert. Dabei wurden Konzentrationen
zwischen 5µM und 10µM gewählt. Anschließend wurden die Zellen mit dem jeweiligen Agens
(TGF-β1, Wnt-1) behandelt. Potenziell toxische Effekte durch Embelin wurden mittels MTT-
Assay, FACS-Färbung und TUNEL-Assay ausgeschlossen.
5.2.3 Nucleofection mit siRNA
Fibroblasten wurden mit einem Necleofector Kit (Amaxa GmbH, Köln, Deutschland)
transfiziert. Dabei wurden XIAP und SMAD3 mit 1,5µg siRNA ausgeknockt. Fibroblasten mit
non-target siRNA wurden hierbei als Kontrollen verwendet. Nach 6h wurde das Medium
gewechselt und nach 24h wurden die Zellen lysiert und gesammelt. Ein erfolgreicher
Knockdown wurde mittels real-time qPCR und Western Blot bestätigt.
Es wurden folgende siRNA Duplexe (Eurogentec, Seraing, Belgien) verwendet:
Duplex Sequenz (5‘ 3‘)
XIAP1 sense AAG UGG UAG UCC UGU UUC AGC UU
XIAP1 antisense AAG CUG AAA CAG GAC UAC CAC UU
XIAP2 sense GGU AAG AAC UAC UGA GAA AUU
XIAP2 antisense AAU UUC UCA GUA GUU CUU ACC
SMAD3 sense GCC UGG UCA AGA AAC UCA
SMAD3 antisense UUG AGU UUC UUG ACC AGG
155.3 Bestimmung von Apoptose
5.3.1 MTT Assay
Die metabolische Aktivität von Hautfibroblasten wurde mittels MTT-Assay ermittelt. Dabei
wurden Zellen in 96-Well Platten ausgesät (10.000 Zellen/Well) und mit Embelin (5µM, 10µM,
15µM) in 80µl Medium für 24h inkubiert. Die unbehandelte Gruppe diente dabei als Kontrolle.
Fibroblasten mit 50% DMSO (Dimethylsulfoxid) dienten als Positivkontrolle. 20µl MTT [MTT
3-(4,5-dimethylthiazol-2-yl) -2, 5-diphenyltetrazoliumbromid] Reagenz wurden 4h vor
Stimulationsende hinzugegeben. Nach Entfernung des Überstandes wurden 100µl Lysepuffer
hinzugegeben und die Aktivität mittels ELISA (BioTek, Winooski, USA) bei 570nm bzw.
630nm Wellenlänge ermittelt.
5.3.2 TUNEL Assay
Das Vorliegen von apoptotischen Zellen in vivo wurde mittels TACS 2 TdT Fluorescein Kit
(4812-30-K, Trevigen, Gaithersburg, USA) ermittelt. Nach Deparaffinisierung wurden die
Schnitte durch 50µl Cytonin permeabilisiert und mit Anti-Vimentin (Cell Signaling
Technology, Danvers, USA) bzw. Anti-CD31 (ab28364, Abcam, Cambridge, GB, 1:100)
Antikörpern über Nacht inkubiert. Als Zweitantikörper wurden AlexaFluor488 und
AlexaFluor594 (Invitrogen, Carlsbad, USA) verwendet. Anschließend wurden die Schnitte mit
50µl Labelling Puffer für 1h bei 37°C behandelt. Eine Positivkontrolle wurde mittels TACS-
Nuclease und eine unlabelled Kontrolle durch das Fehlen von TdT-Enzym erstellt. Nach dem
Labelling Prozess wurden die Schnitte mit Strep-Fluor Lösung und DAPI (Santa Cruz
Biotechnology, Dallas, USA) angefärbt.
5.3.3 FACS
Das Vorliegen von Apoptose bzw. Nekrose in Hautfibroblasten wurde mittels Annexin
V/Propidiumiodid Färbung und Durchflusszytometrie ermittelt. Fibroblasten wurden in 6-Well
Platten mit 10% FBS/DMEM-F12 ausgesät und nach 90% Konfluenz für 24h auf 0,1%
FBS/DMEM-12 herabgesetzt. Die Zellen wurden für weitere 24h mit Embelin inkubiert.
Positivkontrollen wurden durch die Zugabe von 50% DMSO 12h vor Behandlungsende erstellt.
1x106 Zellen wurden in 100µl Annexin V Binding Buffer (Thermo Fischer, Waltham, USA)
inkubiert und auf 96-Well-Platten transferiert. Die Färbung wurde mit Annexin V-APC 1:200
(Thermo Fischer, Waltham, USA) und Propidiumiodid 1:100 (Biolegend, San Diego,
California, USA) für 20min durchgeführt. Eine Färbekontrolle wurde durch das Fehlen von
16Annexin V/Propidiumiodid hergestellt. Die Zellen wurden mittels CytoFLEX (Beckman
Coulter, Brea, USA) detektiert.
5.4 Quantitative real-time PCR
Die RNA-Isolation wurde mittels NucleoSpin RNAII Kit (Macherey-Nagel, Düren,
Deutschland) durchgeführt. Fibroblasten wurden auf 6-Well Platten ausgesät und anschließend
mit 350µl Lysepuffer (RA1 + β-Mercaptoethanol) geerntet. Die RNA wurde durch MultiScribe
Reverse Transcriptase (Applied Biosystems, Foster City, USA) und Random Hexamers
(Thermo Fisher, Waltham, USA) in cDNA überschrieben. NonRT Kontrollen wurden durch
das Fehlen reverser Transkriptase erstellt. Die Genexpression wurde mittels SYBR Select
Master Mix (Applied Biosystems, Foster City, USA) und MX3005P Detection System (Agilent
Technologies, Santa Clara, USA) quantifiziert. Alle Proben bis auf nonRT Kontrollen wurden
in Doppelansätzen gemessen. Es wurden folgende Primer verwendet:
Primer Sequenz (5‘ 3‘)
Human XIAP forward GAG TGA AGA CCC TTG GGA ACA
Human XIAP reversed ACA GAT ATT TGC ACC CTG GAT ACC
Murine Xiap forward AGT TAT CTG CTG CCT CCA CAG
Murine Xiap reversed GGT GTA AGT GGT TAC CTG CCA
Human cIAP1 forward ATC GTG CGT CAG AGT GAG C
Human cIAP1 reversed GCC GAC AAG GAG ATA CGG C
Human cIAP2 forward AGT CAC TCC CAG ACT CTT TCC A
Human cIAP2 reversed TGA TGT GTT GAC CCC GTG TT
Human COL1A1 forward ACG AAG ACA TCC CAC CAA TC
Human COL1A1 reversed ATG GTA CCT GAG GCC GTT C
Human β-Actin forward AGA AAA TCT GGC ACC ACA CC
Human β-Actin reversed TAG CAC AGC CTG GAT AGC AA
5.5 Quantifizierung von Kollagen Protein
5.5.1 SirCol Assay
Die Menge an löslichem Kollagen wurde mittels Sircol Soluble Collagen Assay (#S1000,
Biocolor Ltd., Carrickfergus, United Kingdom) ermittelt. Dabei wurden Zellen in 6-Well-
Platten ausgesät und für 72h in 0,1% FBS/DMEM-F12 und 0,1% Vitamin C gehungert. 200µl
des Überstandes wurden mit 400µl DyeReagent für 30min inkubiert und anschließend für
10min bei 12.000g zentrifugiert. Der Überstand wurde verworfen, 250µl eiskaltes Acid-Salt-
Wash Reagent hinzugegeben und erneut für 10min bei 12.000g zentrifugiert. Der Überstand
17wurde erneut verworfen und 125µl Alkali Reagenz hinzugegeben. Die Proben wurden im
ELISA (BioTek, Winooski, USA) bei 555nm gemessen. Es wurden Standards mit
Konzentrationen von 0/2/5/10/15/30µg/ml erstellt.
5.5.2 Hydroxyprolin Assay
Die Menge an Kollagen in Gewebeproben wurde mittels Hydroxyprolin Assay ermittelt. Die
Hautproben wurden mit 500µl 6M Salzsäure für 3h bei 120°C verdaut. Der pH-Wert wurde
anschließend mit 6M Natriumhydroxid auf pH 6-7 eingestellt. 25µl der Proben wurden mit
225µl HP-Puffer vorbereitet. Nach der Zugabe von 125µl Chloramine T wurden die Proben
20min bei Raumtemperatur inkubiert. Eine Lösung aus 15% p-Dimethylaminobenzaldehyd,
60% Methoxyethanol und 3,15M Perchlorsäure wurde für 20min bei 60°C hinzugefügt. Die
Absorption wurde mittels ELISA (BioTek, Winooski, USA) bei 557nm Wellenlänge ermittelt.
Es wurden Standards mit Konzentrationen von 0/1/5/10/15/20/30/40µg/ml erstellt.
5.6 Western Blot
Proteinproben wurden mittels SDS-Polyacrylamid Gel separiert und auf Polyvinylidene Fluorid
Membranen (Millipore, Burlington, USA) transferiert. Nachdem die Membranen mit 2%
BSA/PBS blockiert worden sind, wurden sie mit folgenden Antikörpern bei 4°C über Nacht
inkubiert:
Antikörper Firma
Ziege gegen XIAP (AF8221) R&D Systems, Minneapolis, USA
Hase gegen GAPDH (ab181602) Abcam, Cambridge, GB
Maus gegen α-SMA (A5228) Sigma Aldrich, St. Louis, USA
Maus gegen Typ I Kollagen (ab6308) Abcam, Cambridge, GB
Hase gegen β-actin (ab8227) Abcam, Cambridge, GB
Hase gegen SMAD3 (9513S) Cell Signaling, Cambdrige, UK
Anschließend wurden die Membranen mit einem Zweitantikörper (Dako, Glostrup, Denmark),
der in Meerrettich Peroxidase konjugiert wurde, inkubiert.
5.7 Histologische Analysen
Hautproben wurden in 4% Formalin fixiert und in Paraffin eingebettet. Vor den Färbungen
wurden die Schnitte in einer Xylolreihe für zwei Mal 5min deparaffinisiert und in einer
Alkoholreihe mit 2x100% Ethanol für 5min, 1x96% Ethanol für 5min und 1x80% Ethanol für
5min rehydriert. Anschließend wurden die einzelnen Färbungen vorgenommen.
185.7.1 HE Färbung
Zur Bestimmung der dermalen Größe wurden 5µm große Hautschnitte mit Hämatoxylin und
Eosin angefärbt. Mit einem Nikon Eclipse 80i Mikroskop (Nikon, Badhoevedorp, Niederlande)
wurde der weiteste Abstand zwischen epidermalem/dermalem Übergang und
dermalem/subkutanem Übergang gemessen. Diese Auswertung wurde in Relation zur
jeweiligen Kontrolle gesetzt und als „x-facher Anstieg“ angegeben. Die Analyse der Schnitte
wurde verblindet und durch geübtes Personal bei einer Vergrößerung von 40- bzw. 100-fach
vorgenommen.
5.7.2 Trichrom Färbung
Die Trichrom Färbung wurde verwendet, um Kollagenfasern direkt sichtbar zu machen. Nach
Deparaffinisierung und Rehydrierung wurden die Schnitte für 15min bei 65°C in Bouin’s
Solution erhitzt und anschließend jeweils für 5min bei Raumtemperatur in Weigert’s Iron
Hematoxylin Solution und Biebrich Scarlet-Acid Fucshin inkubiert. Nach einer Behandlung
mit Phosphotungstic/Phosphomolybdic Säure für 5min wurde mit Anilinblau für 2min
gegengefärbt. Schließlich wurden die Schnitte dehydriert und mit Histokitt verschlossen. Alle
verwendeten Materialien stammten von Sigma-Aldrich, St. Louis, USA.
5.7.3 Sirius Red Färbung
Diese Färbung wurde verwendet, um Kollagen bzw. fibrotisches Areal in Lungengewebe
sichtbar zu machen. Hierzu wurden die Schnitte deparaffinisiert und rehydriert. Die Kerne
wurden für 8min mit Weigert’s Haematoxylin angefärbt und für 60min in Picro Sirius-red
(Sigma-Aldrich, St. Louis, USA) inkubiert. Anschließend wurden die Schnitte mehrmals in
angesäuertem Wasser gewaschen und in einer Ethanolreihe bzw. Xylol rehydriert. Die
fibrotischen Lungenareale wurden mittels Image J ausgemessen und mit einem abgewandelten
Ashcroft-Score ausgewertet [52]. Bei diesem Score werden Werte zwischen 0 und 8 vergeben.
Dabei sind die einzelnen Werte wie folgt definiert:
Wert Definition
0 normales Lungengewebe
1 minimale fibröse Verdickung der alveolären bzw. bronchialen
Wände
2-3 moderate Verdickung der Wände ohne sichtbare Schäden der
Lungenarchitektur
4-5 vermehrte Fibrose mit deutlichem Schaden der Lungenstruktur
und der Formation fibrinöser Bänder oder kleinfibröser Mengen
196-7 schwere Deformation der Struktur und große fibrinöse Areale
8 totale fibrinöse Obliteration des Feldes
5.8 Immunhisto- und Immunzytochemie
5.8.1 Immunhistochemie
Nach Deparaffinisierung und Rehydratation wurden die Epitope mittels Citratpuffer (pH = 6)
und Tris-EDTA-Tween (pH = 9) im Wechsel für 10min freigelegt. Die Schnitte wurden in
Citratpuffer für 20min abgekühlt und mit 5% BSA/PBS für 1h geblockt. Anschließend wurden
Primärantikörper über Nacht bei 4°C hinzugegeben. Dabei diente anti-prolyl-4-hydroxylase-β
Antikörper (P4Hβ) als Fibroblastenmarker und Vimentin als Cytoskelettmarker. Anti-IgG
Antikörper dienten als Färbekontrolle. Am folgenden Tag wurde nach mehrmaligem Waschen
der entsprechende Zweitantikörper für 1h bei Raumtemperatur inkubiert und mit DAPI für
10min gegengefärbt. Die Schnitte wurden mit fluorescent mounting medium verschlossen.
5.8.1.1 α-SMA Färbung
Nach Deparaffinisierung und Redhydratation wurden die Schnitte mit 5% Pferdeserum in 2%
BSA in PBS-Tween für 1h geblockt und anschließend mit monoklonalem Maus anti-α-SMA-1
(Sigma Aldrich, St. Louis, USA) in 2% BSA/PBS über Nacht bei 4°C inkubiert. Am
darauffolgenden Tag wurde die endogene Peroxidase durch 3% Wasserstoffperoxid (H2O2) für
10min geblockt und die Schnitte anschließend mit polyklonalem Hase Anti-Maus IgG/HRP
Antikörper (Vector Laboratories, Burlingame, USA) für 1h bei Raumtemperatur inkubiert. Die
α-SMA Expression wurde mittels Diaminobenzidin-Peroxidase-Substrat (Sigma-Aldrich, St.
Louis, USA) sichtbar gemacht. Die Anzahl von Myofibroblasten wurde verblindet von einem
Untersucher an sechs verschiedenen Orten der Hautschnitte pro Maus gezählt.
5.8.2 Immunzytochemie
Zur Färbung von Zellen wurden 5000 Zellen/Kammer in Chamber Slides in 10% FBS/DMEM-
F12 ausgesät. Daraufhin wurden die Zellen in 0,1% FBS/DMEM-F12 und Vitamin C (1:1000)
bzw. Embelin für 24h gehungert und mit TGF-β (10ng/ml) stimuliert. Nach einer Fixierung der
Zellen mit 4% Methanol-freiem PFA/PBS für 10min bei Raumtemperatur wurden sie mittels
0,25% Triton X-100/PBS für 5min bei Raumtemperatur permeabilisiert. Daraufhin wurden die
20Zellen mit 5% Pferdeserum in 2% BSA/PBS-T für 1h geblockt und mit folgenden
Primärantikörpern über Nacht bei 4°C inkubiert:
Antikörper Firma
Maus gegen P4Hβ (AF0910-1) Acris, Herfold, Deutschland
Maus gegen α-SMA (A5228) Sigma Aldrich, St. Louis, USA
Ziege gegen XIAP (AF8221) R&D Systems, Minneapolis, USA
Ziege gegen Vimentin (AB1620) Merck Millipore, Darmstadt, Germany
Hase gegen Vimentin (ab2365) Abcam, Cambridge, GB
Anschließend wurden die Zellen mit einem passenden Zweitantikörper (Vector Laboratories
Germany, Eching, Germany) für 1h bei Raumtemperatur inkubiert. Stressfasern wurden durch
eine zusätzliche Anfärbung mit Rhodamin-konjugiertem Phalloidin (#R415, Sigma Aldrich, St.
Louis, USA) für 20min bei Raumtemperatur sichtbar gemacht. Als Kernfärbung diente eine
Gegenfärbung mit DAPI (Santa Cruz Biotechnology, Heidelberg, Deutschland) für 10min bei
Raumtemperatur. Anschließend wurden die Zellen mit fluorescence mounting medium
abgedeckt. Die Färbungen wurden mit einem Nikon Eclipse 80i Mikroskop (Nikon,
Niederlande) untersucht.
5.9 Statistik
Alle Daten werden als Median mit Interquartilabstand gezeigt. Unterschiede zwischen den
einzelnen Gruppen wurden durch nicht-parametrischen Mann-Whitney-U-Test ermittelt. P-
Werte >0,05 wurden als signifikant bewertet. P-Werte wurden folgendermaßen dargestellt:
0,05>p>0,01 als *; 0,01>p>0,001 als ** und p6 Ergebnisse
6.1 Die Hochregulation von XIAP in SSc
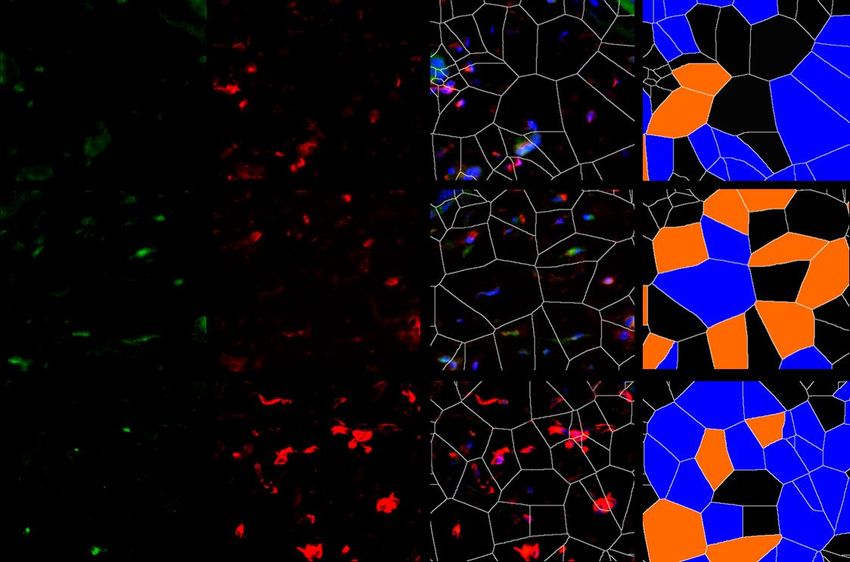
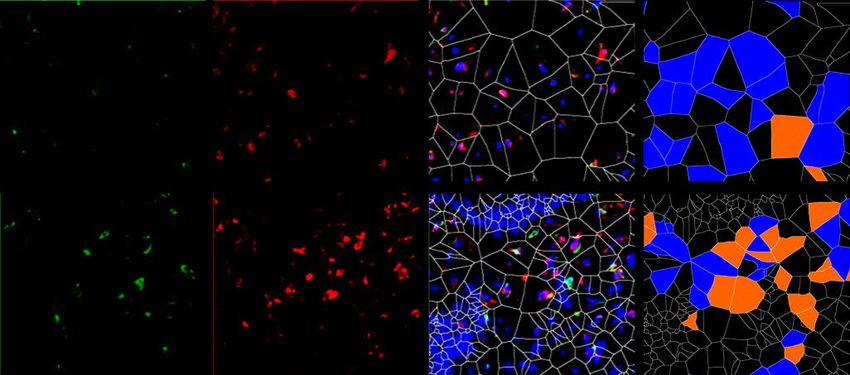
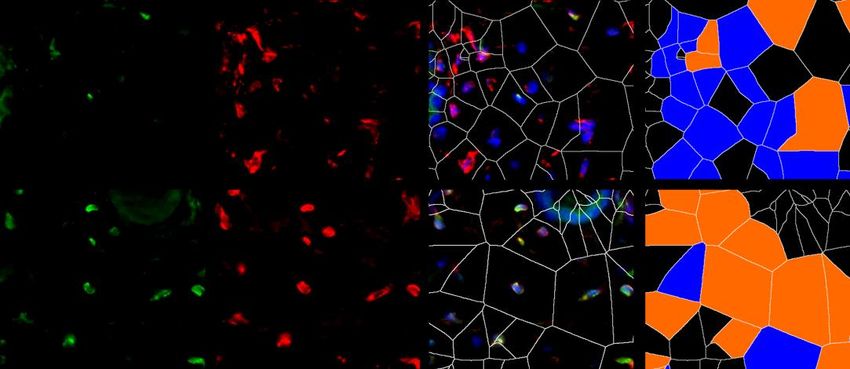
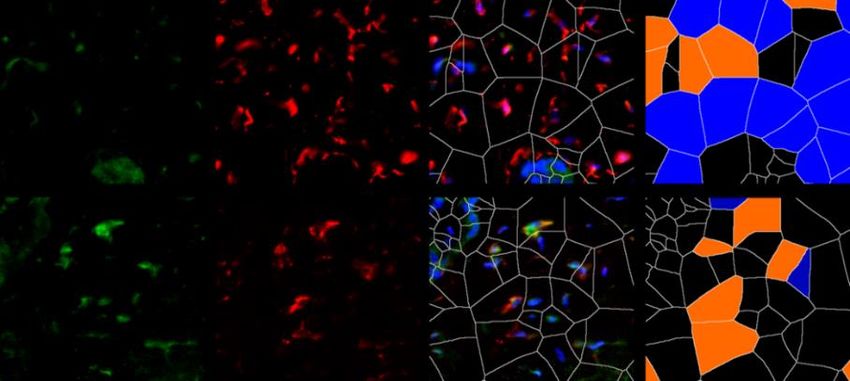
6.1.1 Die Expression von XIAP ist in SSc Fibroblasten erhöht
Um die Expression von XIAP im Menschen zu untersuchen, wurden Hautbiopsien von
Patienten mit systemischer Sklerose und gesunden Probanden verwendet. Dabei wurden
Hautproben von Patienten mit sowohl stabiler als auch progressiver systemischer Sklerose
analysiert. Die Größe beider Gruppen betrug n=7. Die Proben wurden immunhistochemisch
mit einem XIAP Antikörper angefärbt. Prolyl-4-Hydroxlase-β (P4Hβ) diente dabei als
Fibroblasten Marker. Mit 4‘,6-Diamidino-2-phenlyindol (DAPI) wurden Zellkerne angefärbt.
Zusätzlich wurde eine Hämatoxylin-Eosin (HE) Färbung durchgeführt. Es zeigten sich in den
Färbungen (Grafik 1.1 A) und der Auswertung der Bilder (Grafik 1.1 B) ein signifikanter
Anstieg von P4Hβ/XIAP positiven Zellen und eine signifikante Zunahme der Hautdicke bei
Patienten mit progressiver SSc (SScP) im Vergleich zu gesunden Probanden. Kein signifikanter
Unterschied zeigte sich bei Patienten mit stabiler SSc im Vergleich zu der Kontrolle. Die Bilder
wurden zusätzlich mittels Voronoi Technik ausgewertet (Grafik 1.1 A letzte Spalte). Dies
diente der Visualisierung XIAP positiver Zellen in der Dermis.
P4Hβ pos.; XIAP neg.
A
HE XIAP P4Hβ XIAP/P4Hβ/DAPI P4Hβ pos.; XIAP pos.
Gesund
SSc
22Sie können auch lesen

















































