Biochemische und molekularbiologische Charakterisierung des neuartigen Influenza-A-Virus Proteins PB1-F2
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Biochemische und molekularbiologische Charakterisierung des neuartigen
Influenza-A-Virus Proteins PB1-F2
Der Naturwissenschaftlichen Fakultät der
Friedrich-Alexander-Universität Erlangen-Nürnberg
zur Erlangung des Doktorgrades
Vorgelegt von
David Mitzner
aus Lichtenfels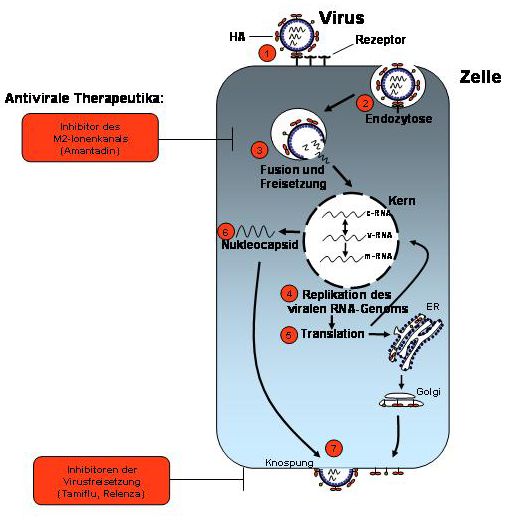
Als Dissertation genehmigt von der Naturwissenschaftlichen Fakultät der Universität
Erlangen-Nürnberg
Tag der mündlichen Prüfung: 12. März 2009
Vorsitzender der Prüfungskommision: Prof. Dr. Bänsch
Erstberichterstatter: Prof. Dr. Ulrich Schubert
Zweitberichterstatter: Prof. Dr. Andreas Burkovski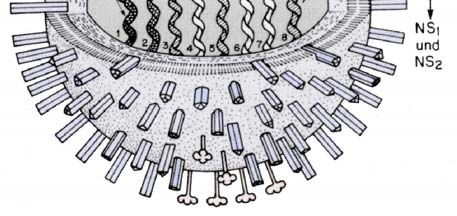
Inhaltsverzeichnis
Inhaltsverzeichnis
1. Zusammenfassung........................................................................................................ 1
1.1 Summary................................................................................................................... 2
2. Einleitung....................................................................................................................... 4
2.1 Die Familie der Orthomyxoviren............................................................................. 4
2.2 Epidemiologie und Tropismus des Influenza-A-Virus............................................ 5
2.3 Morphologie und Replikation des Influenza-A-Virus............................................. 7
2.4 Influenza und die virusinduzierte Apoptose............................................................ 10
2.5 Die influenzainduzierte Signaltransduktion und die Rolle der PKC-Aktivierung.. 14
2.6 PB1-F2, das 11. Influenzaprotein............................................................................ 17
3. Zielsetzung..................................................................................................................... 23
4. Material und Methoden................................................................................................ 24
4.1 Material.................................................................................................................... 24
4.1.1 Verbrauchsmaterial.......................................................................................... 24
4.1.2 Chemikalien und Reagenzien........................................................................... 24
4.1.3 Standard Puffer und Lösungen......................................................................... 25
4.1.4 Synthetische Peptide......................................................................................... 26
4.1.5 Antikörper......................................................................................................... 26
4.1.6 Plasmide und Oligonukleotide.......................................................................... 27
4.1.7 Bakterien und Bakterienkulturreagenzien......................................................... 27
4.1.8 Eukaryotische Zellen und Medien.................................................................... 28
4.1.9 Virus-Stocklösungen......................................................................................... 29
4.2 Methoden.................................................................................................................. 29
4.2.1 Standardmethoden............................................................................................. 29
4.2.2 Kultivierung eukaryotischer Zellen................................................................... 30
4.2.2.1 Inkubation und Passagieren von eukaryotischen Zellen................................ 30
4.2.2.2 Kultivierung von primären Zellen.................................................................. 30
4.2.2.3 Einfrieren und Auftauen von Zellen.............................................................. 31
4.2.2.4 Virusherstellung in Zellkultur........................................................................ 31
4.2.2.5 Virusinfektionen von Zellen........................................................................... 31
4.2.2.6 Plaque-Assay und Virustiter-Bestimmung.................................................... 32
4.2.3 Apoptosemessungen......................................................................................... 32
4.2.3.1 Annexin-V-Färbung zur FACS-Analyse....................................................... 32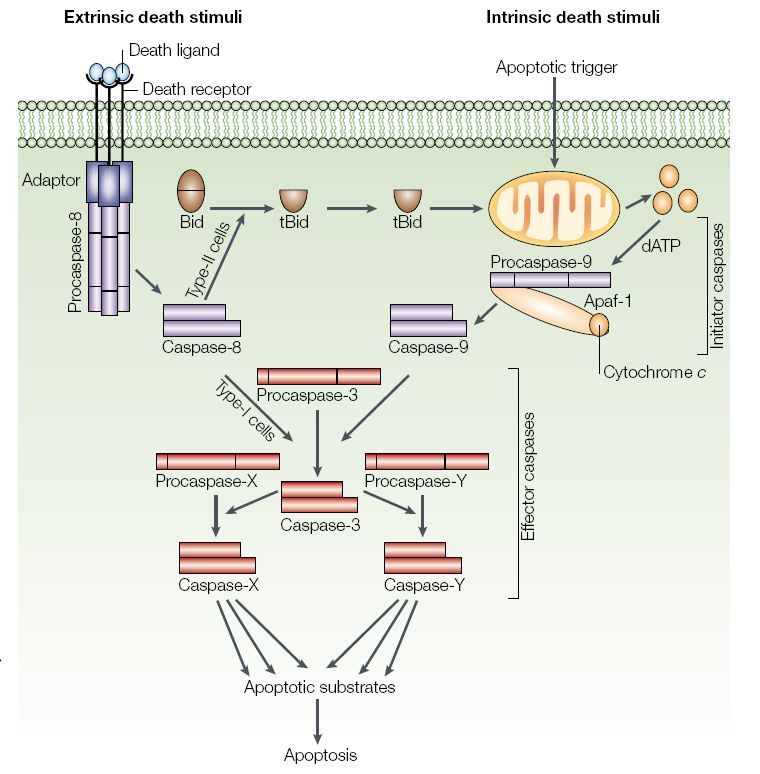
Inhaltsverzeichnis
4.2.3.2 Caspase-3-Aktivitätsmessung........................................................................ 33
4.2.3.3 JC1-Färbung.................................................................................................. 33
4.2.4 Phosphorylierungsexperimente........................................................................ 33
4.2.4.1 Herstellung von S10-Extrakten..................................................................... 33
4.2.4.2 In-vitro-Phosphorylierung............................................................................. 34
4.2.4.3 In-vivo-Phosphorylierung.............................................................................. 34
4.2.4.4 Immunopräzipitation der radioaktiven Proben.............................................. 35
4.2.5 Chemisches Crosslinking.................................................................................. 35
5. Ergebnisse
5.1 Kinetik der PB1-F2 Expression in IAV infizierten Zellen.................................. 37
5.2 Untersuchung der pro-apoptotischen Wirkung von exogenem und endogenem
PB1-F2 in Zellkultur............................................................................................... 38
5.2.1 Inkubation von Epithelzelllinien mit synthetischem PB1-F2 und Analyse
mittels Annexin-V-Färbung............................................................................. 38
5.2.2 Exogenes sVpr, aber nicht sPB1-F2, induziert Apoptose in T-Lymphozyten. 40
5.2.3 Die Expression von endogenem PB1-F2 in 293T-Zellen hat keinen Einfluss
auf die Caspase-3-Aktivität.............................................................................. 41
5.2.4 Exogenes sPB1-F2 hat keinen Einfluss auf die apoptotische Sensibilisierung
verschiedener Zelllinien................................................................................... 43
5.2.5 PB1-F2 verstärkt die virusinduzierte Apoptose in humanen Monozyten......... 44
5.3 Das IAV-Protein PB1-F2 wird von der Proteinkinase C phosphoryliert.......... 45
5.3.1 PB1-F2 enthält mehrere potentielle Phosphorylierungsstellen mit PKC- und
CK2-Konsensussequenzen............................................................................... 46
5.3.2 sPB1-F2 wird von der Protein-Kinase-C (PKC) in-vitro-phosphoryliert......... 47
5.3.3 PB1-F2 wird in transfizierten 293T-Zellen phosphoryliert.............................. 49
5.3.4 PB1-F2 wird an den PKC-Phosphorylierungsstellen Serin-35 und
Threonin-27 in vitro phosphoryliert................................................................. 51
5.3.5 Mutationsanalysen zur Identifikation der relevanten Phosphorylierungs-
stellen von PB1-F2 in vivo............................................................................... 52
5.3.6 Die PB1-F2-Phosphorylierungsstelle Serin-35 ist in zahlreichen human-
pathogenen Isolaten konserviert....................................................................... 54
5.3.7 Die Mutation der PB1-F2-Phosphorylierungsstellen resultiert in einer
reduzierten Caspase-3-Aktivierung in primären humanen Monozyten............ 54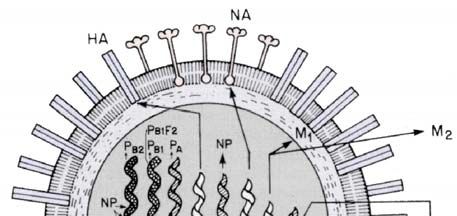
Inhaltsverzeichnis
5.4 Biochemische Charakterisierung der Oligomerisierung des PB1-F2-Proteins. 56
5.4.1 Das PB1-F2-Protein zeigt eine deutliche Tendenz zur Ausbildung
von Oligomeren................................................................................................ 56
5.4.2 Das N-terminale PB-(1-40)- und C-terminale PB-(50-87)-Fragment von
PB1-F2 besitzt die inhärente Fähigkeit zu oligomerisieren.............................. 57
5.4.3 Das Cystein in Position 42 ist durch die Ausbildung von Disulfidbrücken
an der Oligomerisierung von PB1-F2 beteiligt................................................. 60
6. Diskussion
6.1 PB1-F2, ein viraler Pathogenitätsfaktor und die Frage nach den molekularen
Mechanismen............................................................................................................ 62
6.2 Die Expression von PB1-F2 ist zeitlich reguliert..................................................... 64
6.3 Exogenes sPB1-F2 zeigt keine zytotoxischen Effekte in Zellkultur........................ 65
6.4 PB1-F2 ist ein Phosphoprotein und wird von der Proteinkinase C phosphoryliert. 67
6.5 PB1-F2 besitzt die intrinsische Eigenschaft, Oligomere auszubilden..................... 70
7. Abkürzungsverzeichnis................................................................................................ 73
8. Literaturverzeichnis...................................................................................................... 76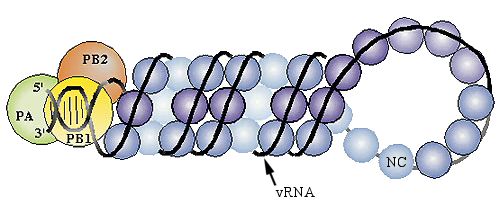
Zusammenfassung/Summary
1. Zusammenfassung
Diese Arbeit beschäftigt sich mit der funktionellen und biochemischen Charakterisierung des
im Jahre 2001 entdeckten 11. Influenza-A-Virus-(IAV)-Proteins PB1-F2. Vor kurzem wurde
im Mausmodell gezeigt, dass PB1-F2 die Virulenz hochpathogener IAV-Isolate verstärkt.
Weiterhin besitzt PB1-F2 funktionelle Ähnlichkeit zum pro-apoptotischen Vpr-Protein von
HIV-1, weshalb zunächst untersucht wurde, ob extrazelluläres PB1-F2, ähnlich dem
transduzierenden Vpr-Protein in der Lage ist, apoptotische Prozesse einzuleiten. Grundlage
für diese Arbeitshypothese war die bekannte Tatsache, dass PB1-F2 in den Mitochondrien
lokalisiert und pro-apoptotische Funktionen in einem noch nicht verstandenen Mechanismus
vermittelt. In dieser Arbeit wurde gezeigt, dass weder extrazelluläres synthetisches (s)PB1-F2
noch intrazelluläres, durch Plasmid exprimiertes PB1-F2 ausreichend ist, um in verschiedenen
Zelllinien Apoptose zu induzieren. Des Weiteren konnte hier gezeigt werden, dass nach der
Infektion von primären humanen Monozyten ein apoptoseverstärkender Effekt in Gegenwart
von PB1-F2 gemessen werden kann. Wurden dieselben Zellen mit einem PB1-F2-defizienten
Virus infiziert, war die Apoptoseinduktion deutlich reduziert. Diese Befunde legen nahe, dass
PB1-F2 nur im Kontext einer Virusinfektion pro-apoptotisch wirkt und dieser Effekt
zellspezifisch ist, was in späteren Publikationen anderer Labore bestätigt wurde.
Ein weiterer Schwerpunkt war die Untersuchung einer möglichen posttranslationalen
Modifikation von PB1-F2. Es konnte hier erstmals gezeigt werden, dass PB1-F2 ein
Phosphoprotein ist und direkt mit der Proteinkinase C (PKC) interagiert. Das virale Protein
wurde von einem Extrakt aus verschiedenen konventionellen PKC-Isoformen sowie den
32
Zellextrakten verschiedener Zelllinien effizient in vitro phosphoryliert. Die radioaktive P-
Markierung von transfizierten 293-T-Zellen und infizierten MDCK-Zellen ergab, dass PB1-
F2 in vivo phopshoryliert wird. Die in vitro und in vivo Phosphorylierung konnte in
Gegenwart PKC-spezifischer Inhibitioren dosisabhängig inhibiert und durch den Aktivator
PMA verstärkt werden. Es wurde mehrfach berichtet, dass die PKC nach einer IAV-Infektion
aktiviert vorliegt, jedoch ist nur wenig über die Funktion dieser Aktivierung im Kontext der
Virusreplikation bekannt. Als PB1-F2-relevante Phosphorylierungsstellen wurden in dieser
Arbeit die Aminosäuren Threonin in Poistion 27 und Serin in Position 35 ermittelt, wobei die
letztere in verschiedenen Isolaten konserviert ist, was eine Grundvoraussetzung für eine
mögliche funktionelle Bedeutung der PB1-F2-Phosphorylierung wäre. Ein rekombinantes
Virus, das für die Phosphorylierungsstellen in PB1-F2 mutiert wurde, zeigte nach der
Infektion von primären Monozyten einen vergleichbaren Phänotyp wie eine Mutante, die
durch gezielte Austauschmutationen nicht mehr in der Lage ist, PB1-F2 zu exprimieren.
1Zusammenfassung/Summary
Beide Virusmutanten waren im Vergleich zum Wildtyp-Virus in ihrer Fähigkeit
eingeschränkt, effektiv Apoptose zu induzierten. Ob die Phosphorylierung von PB1-F2 auch
für die kürzlich beschriebenen immunmodulatorischen Effekte des Proteins im Tiermodell
von Bedeutung ist und in welchem Zusammenhang sie zur IAV-aktivierten, zellulären
Signaltransduktion steht, bleibt noch Gegenstand zukünftiger Untersuchungen.
PB1-F2 besitzt die inhärente Fähigkeit zu oligomerisieren. Dieses Phänomen wurde durch die
Behandlung von sPB1-F2 mit chemischen Crosslinkern und anschließenden Westernblot-
Analysen untersucht. Es zeigte sich, dass sowohl ein N-terminales als auch C-terminales
Fragment von PB1-F2 Oligomere ausbildet, was mit dem in silico Befund übereinstimmt,
dass das Protein je eine Oligomerisierungsdomäne im N- und C-terminalen Bereich besitzt,
wobei die C-terminale Domäne ein deutlich größeres Potential zur Multimerisierung aufweist.
Unter chemisch reduzierenden Bedingungen konnte gezeigt werden, dass das Cystein in
Position 42 maßgeblich an der Ausbildung der dimeren Spezies von PB1-F2 durch
Disulfidbindung beteiligt ist, wohingegen die PB1-F2-Multimerisierung ausschließlich auf die
Integrität der Oligomerisierungsdomänen zurückgeführt werden kann. Über die funktionelle
Relevanz der Oligomerisierung von PB1-F2 wurde bisher viel spekuliert. Die prominenteste
Hypothese ist, dass PB1-F2 Membranporen ausbildet und möglicherweise zur
Destabilisierung des mitochondrialen Membranpotentials beiträgt und damit funktionell der
Gruppe der Viroporine zugeordnet werden kann.
1.1 Summary
This study is focused on the functional and biochemical characterization of the novel 11th
influenza A virus (IAV) protein PB1-F2. Recently, it was shown that PB1-F2 contributes to
the increased virulence of highly pathogenic IAV strains and enhances the effects of
secondary bacterial coinfection in mice. This results in severe pneumonia which is the major
cause of influenza related mortality. PB1-F2 shares functional similarities to the pro-apoptotic
Vpr protein of HIV-1. Initial experiments in this work are based on the hypothesis that
extracellular synthetic (s)PB1-F2 might be able to induce apoptosis in cell culture, similar to
the membrane transducing Vpr protein. PB1-F2 is predominantely located in the mitochondria
of transfected or infected cells and mediates pro-apoptotic functions by a so far unknown
mechanism. No cytotoxic effects were detected after incubation of different cell lines with
extracellular PB1-F2. Also the plasmid mediated expression of PB1-F2 after transfection of
293T cells showed no influence on apoptosis induction. Additional experiments revealed that
PB1-F2 is an enhancer of virus induced apoptosis after infection of primary human
2Zusammenfassung/Summary
monocytes, measured by caspase 3 acitivity. Therefore it was concluded that the pro-apoptotic
effects of PB1-F2 are predominantly mediated after virus infection and in a cell-type-specific
manner.
Further studies shown in this work describe the discovery of PB1-F2 phopshorylation, which
is a totally new finding in the field. PB1-F2 is phopshorylated in vitro by an extract
containing the conventional isoforms of the protein kinase C (PKC) and by cellular S10
extract of different cell lines. 32P radiolabelling of transfected 293T cells and infected MDCK
cells revealed that PB1-F2 is phosphorylated after expression in cell culture. The in vitro and
in vivo phosphorylation of PB1-F2 is sensitive to inhibitors of PKC and could be increased by
the PKC activator PMA. Several studies have shown that activation of the PKC occurs
subsequent to influenza virus infection. However, the function of the PKC activation in the
context of virus replication is unknown. The threonine in position 27 and the serine in
position 35 have been identified as the relevant PKC phopshorylation sites of PB1-F2 with the
latter being conserved in different influenza isolates which is a prerequisite for a general
function of the phosphorylation phenomenon. A recombinant virus which is mutated for the
PB1-F2 phopshorylation sites was impaired in its ability to induce apoptosis, similar to a
mutant virus which is knocked out for PB1-F2 expression. If PB1-F2 phopshorylation is also
important for the recently described immunomodulatory effects and pathogenicity remains to
be investigated. PB1-F2 displays the inherent ability to undergo oligomerization. This
phenomenon was characterized by the use of sPB1-F2 with chemical crosslinkers and western
blot analysis. In silico analysis revealed that the protein displays two oligomerization domains
in the C-terminus and a much weaker domain in the N-terminus, which is consistent with the
finding that both, a N-terminal and a C-terminal fragment of PB1-F2 are able to form
multimers, while the C-terminal fragment has a much higher propensity for oligomerization.
Under chemically reducing conditions it was shown that the cystein in position 42 mediates
the dimerization of PB1-F2 by disulfid-bridges whereas the protein multimerization depends
mainly on the integrity of the oligomerization sites.
There has been much speculation about the functional relevance of the oligomerization
phenomenon. The most prominent hypothesis suggests that PB1-F2 oligomerization results in
the formation of membrane-pores and therefore contributes to the destabilization of the
mitochondrial membrane potential, suggesting that PB1-F2 mediates a viroporin-like
function.
3Einleitung
2. Einleitung
2.1 Die Familie der Orthomyxoviren
Die Familie der Orthomyxoviren zeichnet sich durch ein einzelsträngiges RNA-Genom mit
negativer Polarität aus, das linear segmentiert vorliegt, wodurch sie sich von anderen RNA-
Viren deutlich abgrenzen und ein hohes Maß an genetischer Flexibilität besitzen. Die
Bezeichnung leitet sich vom griechischen „myxa“ (Schleim) ab, da die Vertreter dieser
Familie eine hohe Affinität zu Schleimhäuten besitzen. Orthomyxoviren werden in vier
Gattungen unterteilt (Leahy, Dessens et al. 1997; Pringle 1998). Zu ihnen gehören, wie in
Tabelle 1 dargestellt, die Influenza-Viren A, B und C, das Thogotovirus, zusammen mit dem
Dhorivirus und das Isavirus (Infectious Salmon Anemia Virus), das jedoch keine eigene
Gattung bildet (Klenk 1995; Falk, Namork et al. 1997; Regenmortel 2000). Die Aufteilung
der Influenza-Viren in die Genera A, B und C erfolgt aufgrund der serologischen
Unterschiede in ihrem Nukleoprotein (NP) bzw. Matrixprotein (M). Die Anzahl der RNA-
Segmente variiert zwischen den verschiedenen Gattungen: acht Segmente bei Influenza A und
B und dem Isavirus, sieben Segmente bei Influenza C und dem Dhorivirus und sechs für das
Thogotovirus. Eine weitere Unterteilung erfolgt durch die Hüllproteine der Viren. Nur
Influenza-A und -B-Viren besitzen die Oberflächenproteine Hämagglutinin (HA) und
Neuraminidase (NA). Bei Influenza-C ist die NA durch ein glykosyliertes Hämagglutinin-
Esterase-Fusionsprotein (HEF) ersetzt (Herrler and Klenk 1991). Das Thogoto- und
Dhorivirus haben nur ein einziges Glykoprotein auf der Oberfläche, welches bei anderen
Influenza-Viren nicht zu finden ist (Morse, Marriott et al. 1992). Die Influenza-A-Viren
werden je nach Antigenität ihrer HA- und NA-Proteine in mehrere Subtypen klassifiziert. Im
Augenblick sind 16 HA (H1, H2, usw.) und 9 NA (N1, N2, usw.) antigene Subtypen
beschrieben (Fouchier, Munster et al. 2005), für die Influenza-B und -C-Viren sind bisher
keine Subtypen bekannt. Durch die WHO (World Health Organisation) wurde eine
einheitliche Nomenklatur für Influenza-A-Viren eingeführt. Sie bezeichnet den Typ, den Ort
der Isolierung, die Nummer des Isolates und das Jahr, in dem das Virus isoliert wurde, z.B.
der Influenza-A-Virus (IAV) Stamm A/Puerto Rico/8/34 (H1N1).
Der Krankheitsverlauf des Influenza-C-Virus ist beim Menschen von geringer Bedeutung,
und auch das Influenza-B-Virus verursacht eher selten ernsthafte Erkrankungen (Chen and
Holmes 2008). Einzig die Gattung der Influenza-A-Viren stellt eine alljährliche und globale
Bedrohung dar, die neben extremen ökonomischen und gesundheitspolitischen Problemen
auch die aktuelle Diskussion einer möglichen bevorstehenden Pandemie durch das
Vogelgrippevirus entfacht hat (Korteweg and Gu 2008; Lee and Saif 2008).
4Einleitung
Gattung Wirte Segmentzahl Oberflächenproteine
Influenza-A-Virus Mensch,Vogel, HA, NA
Schwein, Pferd, 8
Robbe
Influenza-B-Virus Mensch, Robbe 8 HA, NA
Influenza-C-Virus Mensch, Schwein, HEF
7
Hund
Isavirus Lachs 8 HA, Esterase
Thogotovirus Zecke, Rinder, 6 1 Glykoprotein
Schafe, Nagetiere
Tab. 1 Die Familie der Orthomyxoviren mit ihren natürlichen Wirten und taxonomischen
Unterscheidungsmerkmalen.
2.2 Epidemiologie und Tropismus des Influenza-A-Virus
Die Evolution humaner Influenza-A-Viren erfolgt durch zwei primäre Mechanismen
(Webster, Bean et al. 1992; Wright 2001). Beim sogenannten „antigenic drift“ bilden sich in
einem langsam fortschreitenden Prozeß von Punktmutationen subtile Varianten von aktuell
zirkulierenden Viren. Diese Mutanten sind für die Influenza-A-Virus Epidemien
verantwortlich, wie sie in den meisten Ländern jährlich vorkommen. Im Gegensatz dazu steht
der „antigenic shift“, der sich auf den Austausch ganzer RNA-Segmente zwischen
verschiedenen Influenza-A-Viren innerhalb eines Wirtes zurückführen lässt.
Der primäre Wirt der Influenza-A-Viren sind Wasservögel, die ein riesiges, permanent
präsentes und globales Reservoir an Influenzaviren darstellen (Webster, Bean et al. 1992;
Webby and Webster 2001). Alle 16-HA und 9-NA Subtypen konnten bisher aus ihnen isoliert
werden, wohingegen beim Menschen nur H1, H2, H3 und N1 sowie N2 nachgewiesen
wurden (Webster, Bean et al. 1992). Dabei besteht die eher seltene Möglichkeit, dass es
entweder zu einer direkten Übertragung einer aviären Variante auf den Menschen kommt,
oder dass im sogenannten „genetic reassortment“ ein Austausch von Segmenten aktuell
zirkulierender humaner Viren mit einem Vogelgrippevirus zur Bildung eines neuen,
hochpathogenen und auf den Menschen übertragbaren Virus führt. Dieser Prozess wird als
„reassortment“ bezeichnet und findet in einem Zwischenwirt, wie z.B. dem Schwein statt, das
als „mixing vessel“ fungiert, in dem neue Viren mit heterogenen Oberflächenantigenen
gebildet werden.
Besonders hervorzuheben sind die vier großen Pandemien des vergangenen Jahrhunderts,
bekannt als die „Asiatische Grippe“ von 1957, die „Hongkong Grippe“ von 1968 und die
5Einleitung
„Russische Grippe“ von 1977, mit jeweils einer Million Opfer weltweit (Cox and Subbarao
2000) und die berüchtigte „Spanische Grippe“ von 1918, die nach heutigen Schätzungen etwa
25% der damaligen Weltbevölkerung infiziert und zwischen 20 und 40 Millionen Menschen
getötet hat (Taubenberger, Reid et al. 2000; Taubenberger, Reid et al. 2001). Es gilt
inzwischen als wahrscheinlich, dass sich die Spanische Grippe auf einen aviären Ursprung
zurückführen lässt (Taubenberger, Reid et al. 2005). Dabei wächst die Befürchtung, dass die
Bildung solcher hochpathogener Viren jederzeit wieder erfolgen kann, wobei die größten
Bedenken in der Mensch zu Mensch Übertragung eines reassortierten Vogelgrippevirus
liegen, da sich in keiner humanen Population Antikörper gegen H5, H7 oder H9 befinden
(Webby and Webster 2001) (Abb. 2.2).
Eine entscheidende biologische Barriere hierfür ist der unterschiedliche Tropismus zwischen
Vogelgrippeviren und humanen Viren. Zur Bindung an die Zelloberfläche nutzen Influenza-
Viren Sialinsäure, welche über die Galactose mit Zuckerresten verknüpft ist. Aviäre
Influenza-Viren binden bevorzugt an Sialylsäureoligosaccharide mit einer α2,3-
galactosidischen Bindung, wie sie vor allem auf Zellen des Darmtraktes von Vögeln zu finden
ist, wohingegen humane Influenza-Viren eine α2,6-Verknüpfung präferieren, eine Variante,
die für tracheale Epithelzellen typisch ist (Suzuki, Ito et al. 2000; Wright 2001).
Abb. 2.2 Weltkarte mit allen Ländern, in denen das H5N1 Virus nachgewiesen werden konnte.
(Nach dem U.S. Department of Health and Human Services, www.hhs.gov).
6Einleitung
Jedoch befinden sich im menschlichen Respirationstrakt Bereiche von zilienbildenden Zellen,
welche α2,3-verknüpfte Sialinsäurerezeptoren exponieren und ausreichend sind, um eine
Vogelgrippevirus-Infektion zu ermöglichen (Matrosovich, Matrosovich et al. 2004). Die
Tatsache, dass es bisher nur 385 belegte Fälle einer solchen Übertragung auf den Menschen
gibt, verdeutlicht, dass es sich hier um ein eher seltenes Ereignis handelt.
2.3 Morphologie und Replikation des Influenza-A-Virus
Das Influenza-Viruspartikel ist vielgestaltig (pleiomorph), da die Partikelgröße und -form
stark variieren kann. Hauptsächlich lassen sich sphärische Formen mit einem Durchmesser
von etwa 80-120 nm vorfinden (Abb. 2.3.1A), aber auch filamentöse Partikel mit einer Länge
von bis zu 300 nm sind bekannt. Die Virushülle besteht aus einer Lipidmembran, die von
Glykoproteinen durchsetzt ist. Bei diesen handelt es sich um vereinzelte Oberflächenprotein-
Cluster, die in einem HA/NA-Verhältnis von etwa 5:1 stehen. Die Innenseite der Hülle ist
durch das Matrixprotein (M1) ausgekleidet und von einigen Ionenkanälen, bestehend aus dem
Tetramer des M2-Proteins, durchsetzt.
Das Genom des Influenza-A-Virus besteht aus einzelsträngiger RNA mit negativer
Orientierung (-RNA). Die Genomgröße beträgt 13,5 Kilobasen (kB), welche sich auf acht
Segmente, die viralen Ribonukleoproteinkomplexe (vRNP`s) aufteilen. Diese Komplexe sind
zwischen 50 und 130 nm lang, besitzen eine helikale Symmetrie und bestehen aus
Nukleoproteinen (NP’s) mit gebundener RNA und einem assoziierten RNA-
Polymerasekomplex, der sich aus den „basischen“ viralen Polymerasen PB1 und PB2 sowie
der „sauren“ PA-Untereinheit zusammensetzt (Abb. 2.3.1B).
Jedes funktionsfähige Virusparitkel benötigt zur effizienten Infektion einen zellulären
Rezeptor. Dieser wurde für Influenza bereits vor 50 Jahren als Sialinsäure beschrieben
(Gottschalk 1959). Seitdem ist bekannt, dass das Virus über sein HA- Glykoprotein an die
Sialinsäurereste der Zelloberfläche bindet (Abb. 2.3.2), wie sie auf zellulären Glykoproteinen
oder Lipiden zu finden sind (Skehel and Wiley 2000). Als typische Wirtszelle der humanen
Influenza gilt die Lungenepithelzelle des Respirationstraktes mit einer α2,6-verknüpften
Sialinsäure auf der apikalen Plasmamembran (Baum and Paulson 1990). Die Aufnahme des
Viruspartikels erfolgt durch rezeptorvermittelte Endozytose (Patterson, Oxford et al. 1979;
Matlin, Reggio et al. 1981), wobei das HA-Protein auch als Fusionsprotein von Bedeutung ist
(Skehel and Wiley 2000). Nach der Ausbildung eines sogenannten späten endosomalen
Kompartimentes erfolgt die Freisetzung (uncoating) in das Zytoplasma (Bui, Whittaker et al.
7Einleitung
1996). Diesem Prozess geht eine vorhergehende Ansäuerung des Viruspartikels voraus,
welche über den M2-Ionenkanal vermittelt wird (Pinto, Holsinger et al. 1992).
A B
NP
Abb. 2.3.1 Aufbauf des Influenza-A-Virions und RNP-Komplexes.
(A) Das Influenza-A-Virus besitzt 11 Proteine, welche durch 8 einzelsträngige RNA-Segmente in
negativer Orientierung kodiert werden und ist von einer Lipiddoppelschicht umhüllt, in welche die
Hüllproteine eingelagert sind (Abb. nach Webster et al. 1992). Zu den Influenzaproteinen zählen die
Neuraminidase (NA), Hämagglutinin (HA), der virale Polymerase-Komplex mit dem basischen
Protein 1, 2 (PB1, PB2) und dem sauren Protein (PA), das RNA bindende Nukleoprotein (NP), Matrix
1 und 2 (M1, 2), die Nicht-Strukturproteine (NS 1, 2) und das akzessorische PB1-F2-Protein aus dem
offenen Leserahmen 2 des PB1-Gens.
(B) Modell der Ribonukleoproteinstruktur. Die einzelsträngige virale RNA (vRNA) umwickelt die
NP-Monomere und bildet eine Haarnadelstruktur aus. Die 5’- und 3’-Enden bilden eine kurze
doppelsträngige Region aus, welche an die heterotrimere RNA-Polymerase (PA, PB1, PB2) bindet
(Abb. nach Portela und Digard, 2002).
Dies ist auch der Angriffspunkt für Amantadin, eine antivirale Sustanz, welche den
Ionenkanal und somit die Nukleokapsidfreisetzung blockiert (Bukrinskaya, Vorkunova et al.
1982; Martin and Helenius 1991). Durch den niedrigen pH-Wert ändert sich die
Konformation der aminoterminalen Region des HA-Proteins, so dass die endosomale und
virale Membran miteinander fusionieren. Die zunehmende Ansäuerung des Partikels führt zu
einer erneuten Konformationsänderung des HA und löst die Interaktion zwischen den
Nukleoproteinen und RNP-Partikeln mit den M1-Hüllstrukturproteinen, was die
Voraussetzung für die Freisetzung der viralen RNA in das Zytoplasma ist (Bui, Whittaker et
al. 1996). Die RNPs gelangen durch einen Mechanismus, der bisher nicht eindeutig geklärt
ist, in den Zellkern, den Ort der viralen Transkription (Herz, Stavnezer et al. 1981).
Voraussetzung hierfür ist die Passage des RNPs durch die Kernmembran. Für einen passiven
8Einleitung
Diffusionsprozess sind die Komplexe zu groß (Paine 1975). Alle Proteine des RNP,
einschließlich der Polymeraseproteine besitzen eine NLS (nuclear localization sequence)
(Nath and Nayak 1990; Nieto, de la Luna et al. 1994). Die aktive Aufnahme in den Kern
erfolgt über das NP, welches hierfür an das Kernporenprotein Importin-α bindet, einer
wichtigen Komponente des Kernimport-Systems (Gorlich, Prehn et al. 1994).
Abb. 2.3.2 Der IAV-Replikationszyklus. Nach der rezeptorspezifischen Bindung des viralen HA an
die Zelloberfläche (1) erfolgt die endozytotische Aufnahme des Viruspartikels (2) und die Freisetzung
des Nukleokapsides in das Zytosol der Wirtszelle (3). Dieses wandert in den Kern, welcher der Ort der
viralen RNA-Replikation ist (4). Die Translation der viralen Oberflächenproteine erfolgt in das
endoplasmatische Retikulum (ER), und ein Teil der für die RNA-Replikation notwendigen
Virusproteine gelangt wieder in den Kern (5). Am Ende der Replikation steht die Generierung und der
Kernexport eines Ribonukleoproteinkomplexes (6), welcher zur Zelloberfläche gelangt und mit den
viralen Oberflächenproteinen unter Ausbildung eines neuen Viruspartikels abgeschnürt wird (Abb. der
ViroLogik GmbH Erlangen).
Die virale RNA hat im Zellkern zwei wesentliche Funktionen: Sie dient als Matrize zur
Synthese viraler mRNA und neuer genomischer RNA. Zu den früh translatierten
Virusproteinen zählen PB1, PB2, PA, NP, NS1 und NS2, die nach der Synthese wieder in den
9Einleitung
Kern zurück gelangen. Im Kern erfolgt die Initiation der RNA-Synthese durch die virale
RNA-abhängige RNA-Polymerase, die sich aus den PB1-, PB2- und PA-Proteinen
zusammensetzt. Zur Replikation der viralen RNA (vRNA) wird zunächst copy-RNA (cRNA)
synthetisiert, die einen Anteil von nur etwa 5-10% der gesamten viralen RNA ausmacht (Hay,
Lomniczi et al. 1977; Herz, Stavnezer et al. 1981) und weder eine Cap-Struktur aufweist,
noch polyadenyliert ist. Ein weiterer bedeutender Faktor der Replikation ist das NP. Es wird
spekuliert, dass seine Bindung an vRNA den Übergang der viralen mRNA- zur cRNA-
Synthese reguliert (Klumpp, Ruigrok et al. 1997). Nach der Synthese viraler RNA bilden sich
neue RNPs, die sich aus den drei Untereinheiten des viralen Polymerasekomplexes und der
einzelsträngigen v-RNA, die unspezifisch an NP gebunden ist, zusammensetzt (Huang, Palese
et al. 1990). Nach dem Kernexport der RNPs assembliert das Influenza-A-Virus in
Epithelzellen an der apikalen Plasmamembran (Bachi, Gerhard et al. 1969), wobei die viralen
Proteine HA, NA und M2 unabhängig voneinander vom endoplasmatischen Retikulum über
den Golgiapparat zur Zellmembran transportiert werden (Gottlieb, Gonzalez et al. 1986). Die
HA- und NA- Glykoproteine werden zu cholesterinreichen Mikrodomänen der
Zelloberfläche, den sogenannten „lipid rafts“, gesteuert (Barman and Nayak 2000), von denen
aus die Viruspartikel assembliert werden (Scheiffele, Rietveld et al. 1999; Takeda, Leser et al.
2003). Ob der Transport der RNP-Komplexe und des viralen M1 zur Zellmembran
ungerichtet (Patterson, Gross et al. 1988) oder, wie neuere Daten andeuten, gerichtet ist (Ali,
Avalos et al. 2000), bleibt kontrovers. Das virale M1-Protein dient als Adaptermolekül
zwischen der Zellmembran und dem RNP als auch zwischen dem RNP und dem
zytosolischen Teil der viralen Glykoproteine HA, NA, M2 (Barman, Ali et al. 2001), und das
so assoziierte neue Viruspartikel wird durch Abknospung von der Membran freigesetzt
(Webster, Bean et al. 1992).
2.4 Influenza und die virusinduzierte Apoptose
Die Untersuchung der Apoptoseinduktion durch das Influenza-Virus ist von zunehmender
Bedeutung für das Verständnis der Mechanismen der viralen Pathogenität (Benedict, Norris et
al. 2002; Yewdell and Garcia-Sastre 2002; Lowy 2003).
Unter Apoptose versteht man eine biochemisch und morphologisch definierte Form des
programmierten Zelltodes, welche von einer Zelle aktiv durchgeführt wird (Kerr, Wyllie et al.
1972) und für eine Vielzahl von Viruserkrankungen von Bedeutung ist (Razvi and Welsh
1995). Dabei können die komplexen und verzweigten apoptotischen Prozesse in eine
mitochondriale (intrinsische) und eine Todesrezeptor-abhängige (extrinsische) Signalkaskade
unterteilt werden (Abb. 2.4), welche beide in der Aktivierung der spezifischen Caspasen
10Einleitung
(Cystein Aspartasen) konvergieren (Cohen 1997). Apoptose-induzierende Caspasen werden
hauptsächlich in Initiatorcaspasen, wie die Procaspasen-8 und -9, und in die Effektorcaspasen
wie zum Beispiel Caspase-3, -6 und -7, unterteilt. Die Procaspasen dienen als Vermittler
zwischen apoptotischen, zellulären Signalen und der proteolytischen Aktivierung der
Effektorcaspasen. Sind letztere erst einmal aktiviert, erfolgt der gezielte und irreversible
proteolytische Abbau von zellulären Proteinen und Strukturen sowie die Fragmentierung der
Kern-DNA (Kroemer, Dallaporta et al. 1998; Thornberry and Lazebnik 1998).
Es gibt eine Vielzahl biochemischer und morphologischer Apoptosemarker, welche in der
Vergangenheit zur Detektion des IAV-induzierten Zelltodes verwendet wurden. Zu den
wichtigsten biochemischen Markern zählen die Detektion von extrazellulär exponiertem
Phosphatidylserin durch Annexin-V, die Messung des mitochondrialen Membranpotentials
und einsetzender Depolarisation, die Freisetzung von Cytochrom-c aus dem mitochondrialen
Membranzwischenraum und die spezifische Caspasen-Aktivierung, im Besonderen die der
Caspasen-8 und -3 (Lowy 2003). Die Zeiosis, eine spezielle und apoptosetypische
morphologische Erscheinung, die sich durch Zytoplasmaschrumpfung und Abschnürung
kleiner membranumhüllter Bläschen (membrane blebbing) äußert, ist im Kontext einer IAV-
Infektion nur schwer und in bestimmten Zellen nachweisbar (Wyllie, Kerr et al. 1980; Lowy
and Dimitrov 1997; Fujimoto, Takizawa et al. 1998). Apoptotische Prozesse werden durch
Influenza-A- und -B-Viren induziert und können sowohl in Zellkultur als auch in vivo
nachgewiesen werden (Fesq, Bacher et al. 1994; Hinshaw, Olsen et al. 1994; Mori, Komatsu
et al. 1995; Ito, Kobayashi et al. 2002). Neben den Lungenepithelzellen wurde die IAV-
induzierte Apoptose in verschiedenen Tiermodellen auch in Milz, Thymus, Leber, Nieren und
sogar dem Gehirn festgestellt (Mori, Komatsu et al. 1995; Ito, Kobayashi et al. 2002; Lowy
2003). Auch in Zellen, welche die IAV-Replikation nicht produktiv unterstützen, z.B. T-
Lymphozyten oder Monozyten, sind apoptotische Prozesse deutlich nachweisbar (Fesq,
Bacher et al. 1994). Besonders schwere IAV-Infektionen werden meist im Kontext einer
virusbedingten Lymphopenie beschrieben (Suarez and Schultz-Cherry 2000; Tumpey, Lu et
al. 2000; Zitzow, Rowe et al. 2002). Für die Depletion von T-, B- und dendritischen Zellen
werden in verschiedenen Studien vor allem extrinsische Faktoren wie Interferone oder die
Liganden der TNF- (tumor necrosis factor) Familie verantwortlich gemacht (Toth, Szegezdi
et al. 1999; Oh and Eichelberger 2000; Sedger, Hou et al. 2002). Die Depletion von
Immunzellen verläuft zudem häufig synergistisch mit einer bakteriellen Koinfektion, weshalb
schwere Krankheitsverläufe in einer durch bakterielle Superinfektion induzierten Pneumonie
resultieren (Sweet and Smith 1980; Colamussi, White et al. 1999; Engelich, White et al.
11Einleitung
2002). Die IAV-induzierte Apoptose verläuft überwiegend nach extrinsischen Mechanismen
in einer autokrinen oder parakrinen Stimulation, wobei die Interferone-α/β und -γ sowie der
TNF und TGF-β (transforming growth factor) als relevante pro-apoptotische Faktoren
identifiziert wurden (Lowy 2003).
Extrinsic pathway Intrinsic pathway
2.4 Schematische Darstellung der extrinsischen und intrinsischen apoptotischen Signalkasksade.
Der extrinsische Pfad wird durch eine spezifische Ligand-Rezeptor-Bindung (TNF-Rezeptor,
Fas/CD95) induziert, was zur Ausbildung eines DISC-Komplexes (death inducing signalling
complex) führt. Durch die membranständige DISC-Formation werden mehrere Caspase 8 Moleküle
rekrutiert, aktiviert und in das Zytosol freigesetzt. Die Caspase 8 ist je nach Zelltyp (Typ I+II) in der
Lage, entweder direkt die zentrale Effektorcaspase-3 zu aktivieren oder durch die proteolytische
Spaltung des pro-apoptotischen Bid-Proteins die mitochondriale Apoptose zu induzieren. Der
intrinsische Pfad kann durch zelluläre Stresssignale oder nach einer Infektion durch pathogene
Faktoren gezielt aktiviert werden, wie es beispielsweise für die akzessorischen Virusproteine Vpr
(HIV), p13 (HTLV) oder PB1-F2 (IAV) bekannt ist. Im weiteren Verlauf erfolgt die Freisetzung von
Cytochrom-c, dem AIF (apoptosis inducing factor) und SMAC (second mitochondria-derived
activator of caspase) aus dem mitochondrialen Membranzwischenraum. Cytochrom-c bildet mit
Apaf-1 (apoptotic protease activating factor) einen Komplex, der zunächst die Procaspase 9 und
daraus folgend die Effektorcaspase-3 aktiviert. (Abbildung aus V.Jesenberger und S.Jensch, nature
reviews, 2002).
12Einleitung
Weiterhin gibt es Befunde, die schwere, virusbedingte Gewebsschäden in der Lunge mit der
Generierung von ROS-Radikalen (reactive oxigen species) in Verbindung bringen (Oda,
Akaike et al. 1989).
Gegenstand neuerer Untersuchungen ist die Rolle der Transaktivatoren NF-κB (nuclear
factor kappa B) und AP-1 (activator protein 1), die zumindest indirekt durch die Modulation
der Zytokin- und Interferonantwort sowie Aktivierung von Stress-Kinasekaskaden die
apoptotischen Prozesse nach einer IAV-Infektion gleichsam regulieren (Pahl and Baeuerle
1995; Abraham, Stojdl et al. 1999; Balachandran, Roberts et al. 2000; Lin, Zimmer et al.
2001; Ludwig, Ehrhardt et al. 2001).
Von zentraler Bedeutung ist das Fas-Rezeptor-System (Takizawa 93, Fujimoto 98), welches
auch mit der Aktivierung der RNA-abhängigen Proteinkinase R (PKR) nach einer IAV-
Infektion in Zusammenhang gebracht wurde (Takizawa, Ohashi et al. 1996), wodurch der
Zelltod letztlich über den FADD/Caspase8-Pfad (Fas-associated death domain protein)
vermittelt wird (Balachandran, Roberts et al. 2000). Das Fas-FasL-System scheint weiterhin
an der virusinduzierten humanen Lymphopenie beteiligt zu sein, da die CD95 Expression in
CD 8+, 4+ und 3+ T-Zellen sowie in mononuklearen Leukozyten nach einer Infektion deutlich
gesteigert ist (Nichols, Niles et al. 2001). Von den Virusproteinen wurden das NS1-Protein
und das M1-Protein im Kontext pro-apoptotischer und anti-apoptotischer Prozesse
beschrieben (Schultz-Cherry, Dybdahl-Sissoko et al. 2001; Zhirnov, Konakova et al. 2002;
Zhirnov, Ksenofontov et al. 2002). Weiterhin ist bekannt, dass auch die virale NA pro-
apoptotisch wirkt, da sie die latente Form des exogenen Apoptoseinduktors TGF-β aktiviert
(Schultz-Cherry and Hinshaw 1996).
Die Regulierung apoptotischer Prozesse nach einer IAV-Infektion wird durch die zellulären
Caspasen vermittelt, wobei die zentrale Stellung der Effektorcaspase-3 bereits beschrieben
wurde (Zhirnov, Konakova et al. 1999; Lin, Holland et al. 2002; Wurzer, Planz et al. 2003).
Die Caspase-3 wird in einem Zeitraum von 12 h bis 36 h nach der IAV-Infektion aktiviert,
wobei ihre Funktion für die Virusreplikation noch umstritten ist (Fesq, Bacher et al. 1994;
Ludwig, Pleschka et al. 1999). Grundsätzlich wird die Apoptose als zellulärer
Abwehrmechanismus gegen eine Vielzahl von Pathogenen betrachtet. Es konnte jedoch
gezeigt werden, dass die Überexpression des anti-apoptotischen Proteins Bcl-2 in einer
reduzierten Virusproduktion und HA-Fehlglykosylierung resultiert (Hinshaw, Olsen et al.
1994; Olsen, Kehren et al. 1996). Die Virusvermehrung ist in der Gegenwart von Caspase-3-
Inhibitoren ebenfalls stark eingeschränkt, was auch in Zellen gezeigt werden konnte, die
shRNA gegen die Caspase-3 exprimierten (Wurzer, Planz et al. 2003). Dieser Effekt wird mit
13Einleitung
einer Retention des viralen RNP-Komplexes im Nukleus in Verbindung gebracht, die letztlich
die Ausbildung von Viruspartikeln verhindert (Wurzer, Planz et al. 2003). Die Tatsache, dass
das überexprimierte, virale NS1-Protein pro-apoptotisch wirkt, wurde als zusätzlicher Beleg
dafür betrachtet, das die virusinduzierte Apoptose im Kontext einer produktiven Replikation
steht (Schultz-Cherry, Dybdahl-Sissoko et al. 2001). Im Gegensatz hierzu konnte gezeigt
werden, dass ein NS1-defizientes Virus in einer verstärkten Apoptoseinduktion resultiert und
NS1 demnach anti-apoptotische Funktion besitzt (Zhirnov, Konakova et al. 2002), wofür auch
die IFN-α/β-antagonistische Funktion des Proteins sprechen würde (Balachandran, Roberts et
al. 2000). Weiterhin ist bekannt, dass das virale NP ein Substrat des Caspase-vermittelten
proteolytischen Abbaus ist, was als ein zusätzliches Argument der antiviralen Funktion
zellulärer Apoptose gilt (Zhirnov, Konakova et al. 1999).
Ob die virusinduzierte Apoptose letztlich replikationsfördernde Elemente besitzt oder ein
reiner zellulärer Abwehrmechanismus ist, bleibt Gegenstand weiterer Untersuchungen
(Ludwig, Pleschka et al. 2006).
2.5 Die influenzainduzierte Signaltransduktion und die Rolle der PKC-Aktivierung
Virale Infektionen resultieren in der unmittelbaren Aktivierung einer Vielzahl zellulärer
Signale und Faktoren (Ludwig, Pleschka et al. 2006). In Zellkultur konnte gezeigt werden,
dass nach einer IAV-Infektion alle bekannten Mitglieder der sogenannten MAPK-Familie
(Mitogen-activated protein kinase) durch Phosphorylierungskaskaden aktiviert werden
(Kujime, Hashimoto et al. 2000; Ludwig, Ehrhardt et al. 2001; Pleschka, Wolff et al. 2001).
Zu ihnen zählen als Kinase-Prototypen ERK (extracellular signal regulated kinase), JNK
(Jun-N-terminal kinase, BMK-1/ERK5 (Big MAP kinase) und die p38-MAPK (Pearson,
Robinson et al. 2001). Durch die MAPK-Kaskade werden zelluläre Prozesse der
Differenzierung und Proliferation wie auch die Aktivierung der Immunantwort reguliert
(Dong, Davis et al. 2002). Beispielsweise resultiert die p38-MAPK-Aktivierung in der
Expression von IL-8 (Interleukin-8) und des Chemokins RANTES (Regulated on Activation,
Normal T Expressesd and Secreted), welches für das gezielte Rekrutieren von T-
Lymphozyten und Makrophagen sowie von Eosinophilen und Neutrophilen an den
Entzündungsherd von Bedeutung ist (Kujime, Hashimoto et al. 2000).
Die von doppelsträngiger RNA (dsRNA) abhängige Aktivierung der Kinase-JNK hat einen
deutlich antiviralen Effekt, da sie Prozesse in Gang setzt, welche die Expression des Zytokins
IFNβ (Interferon β) induziert (Ludwig, Ehrhardt et al. 2001; Samuel 2001). Im Gegensatz
hierzu scheint die Aktivierung der Raf/MEK/ERK-Kaskade für die Virus-Replikation von
14Einleitung
Vorteil zu sein (Pleschka, Wolff et al. 2001). Die Inhibition der Kinase-MEK oder Expression
dominant-negativer ERK- oder Raf-Mutanten führten zur reduzierten Virusfreisetzung nach
der Infektion durch das Influenza-A und -B-Virus (Ludwig, Wolff et al. 2004). Mechanistisch
resultierte die ERK-Inhibition in der Retention der viralen RNP-Komplexe im Zellkern, was
sich unmittelbar auf die Replikation auswirkt. Da die Inhibition der Kinase MEK
virusspezifisch und effektiv gegen beide Virusgattungen wirkt, werden MEK-Inhibitoren als
antivirale Substanzen für eine eventuelle klinische Anwendung zur Zeit untersucht (Planz,
Pleschka et al. 2001; Ludwig, Pleschka et al. 2006).
Ein weiterer wichtiger Faktor, der nach einer IAV-Infektion durch die Akkumulation viraler
RNA und anschließender IKKβ-Stimulation aktiviert wird, ist der IκB/NFκB Kinasekomplex
(Julkunen, Melen et al. 2000; Hiscott, Kwon et al. 2001). Durch Phosphorylierung des NFκB-
Inhibitors IκB (inhibitor of NFκB) erfolgt dessen proteasomaler Abbau und die Translokation
des NFκB-Dimers in den Zellkern (Ghosh, May et al. 1998; Karin and Delhase 2000). Die
Transkriptionsfaktoren, welche zur NF-κB-Familie zählen, regulieren über 150 verschiedene
Gene, welche hauptsächlich immunologische und inflammatorische Prozesse steuern, unter
anderem die Expression von Zytokinen, Chemokinen sowie von pro- und anti-apoptotischen
Proteinen (Pahl 1999). Gleich der Kinase JNK reguliert auch die NFκB-Aktivierung die
Expression der bedeutenden antiviralen Faktoren IFNβ und TNFα (Maniatis, Falvo et al.
1998; Chu, Ostertag et al. 1999; Taniguchi and Takaoka 2002).
Eine weitere Kinase, die häufig im Kontext viraler Infektionen beschrieben wurde und eine
Vielzahl von Effektor-Signalkaskaden reguliert, ist die Proteinkinase C (PKC)
(Constantinescu, Cernescu et al. 1991; Toker 1998; Warrilow, Gardner et al. 2006). Die PKC-
Familie besteht aus 13 bekannten Isoformen (Tab. 2), welche in 3 Klassen unterteilt werden
können, basierend auf der Abhängigkeit von Ca2+ und Phorbolesteraktiverung (Abb. 2.5.1) (Li
and Gobe 2006).
PKC Subfamilie Isoforme
cPKC α, βI, βII, γ
nPKC Δ, ε, η, θ, ζ
aPKC ξ, ι/λ, ν, μ
Tab.2 Die Protein-Kinase-C-(PKC)-Subfamilien und ihre Isoformen.
Die PKC Familie wird in die konventionellen (cPKC), neuartigen (nPKC) und atypischen (aPKC)
unterteilt. Zum aktuellen Zeitpunkt sind 13 Isoforme bekannt.
15Einleitung
Abb. 2.5.1 Schematischer Aufbau der PKC-Subfamilien. Die verschiedenen Subtypen lassen sich
in vier konservierte (C 1-4) und fünf isoenzym-spezifische, variable (V 1-5) Regionen unterteilen. Die
Mitglieder der konventionellen PKC Isoformen besitzen neben der C-terminalen katalytischen
Domäne auch eine Ca2+-, Phosphatidylserin (PS)- und Diacylglycerin (DAG)-Bindestelle, welche alle
zur vollständigen Aktivierung benötigt werden. Die neuartigen und atypischen PKC Isoformen sind
Ca2+-unabhängig.
Die Bedeutung der PKC hinsichtlich der Regulation des Viruseintritts in die Zellen konnte
durch die PKC-Inhibitoren Staurosporin und H7 für verschiedene umhüllte Viren gezeigt
werden (Vesicular stomatitis-virus, herpes simplex-I, vaccinia virus, poliomyelitis virus)
(Constantinescu, Cernescu et al. 1991). Nach einer IAV-Infektion oder HA-Inkubation von
Zellen konnte eine sofortige Aktivierung der konventionellen PKC Isoformen detektiert
werden (Arora and Gasse 1998; Kunzelmann, Beesley et al. 2000; Root, Wills et al. 2000).
Der PKC spezifische Inhibitor Bisindolylmaleimid (BIM), sowie eine Reihe hochselektiver
Inhibitoren (CalphostinC, Gö6976) waren in der Lage, den frühen IAV-Eintrittsprozess in die
Zelle dosisabhängig und reversibel zu inhibieren (Abb. 2.5.2) (Root, Wills et al. 2000).
Durch die Expression einer phosphorylierungs-defizienten Mutante und den Einsatz selektiver
PKC-Inhibitoren, konnte eine Funktion der PKCβII-Isoform im Kontext der Regulation der
späten Endosomen und blockierter RNP-Freisetzung identifiziert werden (Sieczkarski, Brown
et al. 2003). Die PKC-Aktivierung betrifft jedoch auch Ereignisse des späten
Replikationszyklusses durch die indirekte Regulation von Effektormolekülen in der
Signaltransduktion. Beispielsweise wird die Phosphorylierung der Raf-Kinase ebenfalls durch
verschiedene PKC-Isoforme reguliert (Kolch, Heidecker et al. 1993; Cai, Smola et al. 1997),
was letztlich in der Aktivierung der mitogenen Raf/MEK/ERK-Kaskade resultiert (siehe
oben). Die Aktivierung der PKCα durch HA-Akkumulation an den lipid rafts in späten
Stadien des Replikationszyklusses und die daraus resultierende Raf-Phosphorylierung konnte
sogar direkt bestätigt werden (Abb. 2.5.2), (Marjuki, Alam et al. 2006). Auch wenn die
genauen Mechanismen der PKC-Aktivierung durch das Influenza-A-Virus und deren
Interaktion mit der zellulären Signaltransduktion noch nicht geklärt sind, zeichnet sich
16Einleitung
dennoch die zentrale Rolle der konventionellen-α/β-Isoformen ab, welche ubiquitär in allen
bisher untersuchten Zellen vorhanden sind (Li and Gobe 2006).
Abb. 2.5.2 Die Rolle der PKC-Aktivierung im Replikationszyklus von IAV. Nach der Infektion
bzw. HA-Inkubation von Zellen erfolgt die Aktivierung der konventionellen PKCßII-Kinase, die vom
Zytosol zur Zellmembran transloziert und über den N-Terminus der regulatorischen Domäne (R) im
Zytoplasma verankert ist. Die katalytische Domäne (C) wird durch diesen Vorgang durch eine
Konformationsänderung freigesetzt und aktiviert. Die spezifische und selektive Inhibition der PKC
oder die Expression einer funktionell depletierten PKC-Mutante (dnPKCβII) resultiert in der Retention
der viralen RNPs im späten Endosom. Im Gegensatz hierzu wird erst spät im Replikationszyklus die
Raf/MEK/ERK-Kaskade durch die Einlagerung von HA-Proteinen in lipid-rafts während der Virus-
Assemblierung aktiviert. Die gezielte Inhibition der PKCα resultierte in einem analogen
Aktivitätsverlust der ERK-Kinase und der RNP-Retention im Nukleus.
2.6 PB1-F2, das 11. Influenzaprotein
Im Jahr 2001 wurde ein elftes IAV-Genprodukt mit der Bezeichnung PB1-F2 beschrieben
(Chen, Calvo et al. 2001). Entdeckt wurde das Protein bei einem Screening nach MHC-I
Epitopen, die sich von viralen Polypeptiden ableiten lassen, die außerhalb des Standard-
Leserahmens der Influenza-Virusproteine exprimiert werden. PB1-F2 ist das einzige IAV-
Protein, welches in einem zweiten, alternativen +1-Leserahmen (F2, frame 2) kodiert wird,
der innerhalb des pb1-Gens der viralen RNA-Polymerase (PB1) liegt (Abb. 2.6.1). Dies wird
17Einleitung
durch das sogenannte „ribosomale Scanning“ ermöglicht, einem Vorgang, bei dem die 40S-
Untereinheit des Ribosoms nach weiteren AUG Startcodons innerhalb eines Leserahmens
„scannt“, um die Translation zu initiieren (Jackson 2005). Voraussetzung hierfür ist eine
ideale Initiationssequenz, welche auch als „Kozaksequenz“ (Kozak 1991) bezeichnet wird
und am Startcodon von PB1-F2 vorhanden ist.
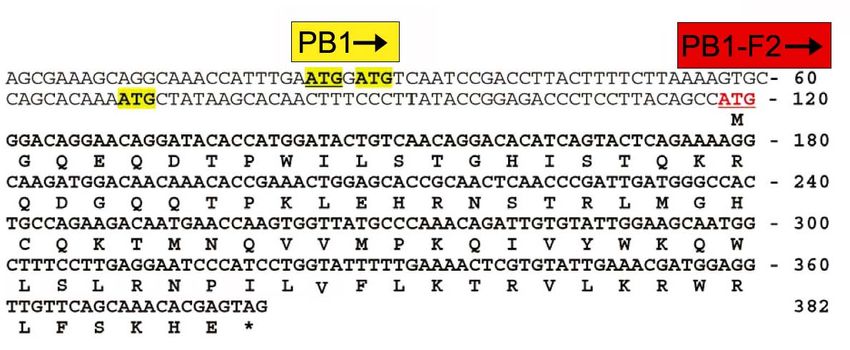
Abb. 2.6.1 DNA und Aminosäuresequenz von PB1-F2 im Leserahmen von PB1. Das PB1-F2
Startcodon (rot) befindet sich downstream vom bekannten ATG Initiations-Triplett (gelb,
unterstrichen) und zwei weiteren potentiellen ATG-Abfolgen (gelb) (Abbildung verändert nach Chen
et al. 2001).
Das vom IAV-Laborstamm A/PuertoRico/8/34(H1N1) (IAVPR8) exprimierte Protein, welches
in anfänglichen Studien untersucht wurde, hat ein Molekulargewicht von 11 kDa und eine
Länge von 87 A.s. (Chen, Calvo et al. 2001). Die neuesten Sequenzanalysen zeigen, dass
PB1-F2 in den meisten humanen und Vogelgrippe-Isolaten als funktionelles Protein mit einer
Länge von 78 bis 90 A.s. vorliegt. In Influenza-B-Virusisolaten ist PB1-F2 nicht präsent
(Chen, Calvo et al. 2001; Zell, Krumbholz et al. 2007). Interessanterweise exprimiert ein
Großteil der seit 1950 zirkulierenden humanen H1N1-Isolate ein C-terminal verkürztes PB1-
F2 mit einer Länge von nur 57 A.s. (Zell, Krumbholz et al. 2007). Strukturanalysen
vorangegangener Arbeiten haben gezeigt, dass PB1-F2 unter hydrophoben Bedingungen zwei
kleinere α-Helices im N-Terminus und eine große α-Helix von Position Ile-53 bis Lys-85 im
C-Terminus ausbildet (Abb. 2.6.2) (Bruns, Studtrucker et al. 2007). Der strukturierte C-
Terminus umfasst alle bis jetzt beschriebenen funktionellen Regionen des Proteins. Zu ihnen
zählen die Oligomerisierungsdomänen (OD), welche für die intrinsische Fähigkeit von PB1-
F2 verantwortlich sind, Dimere und Multimere auszubilden, und die mitochondriale
Zielseqeunz (MTS) (Gibbs, Malide et al. 2003; Bruns, Studtrucker et al. 2007). Es wird
spekuliert, dass die Oligomerisierung von PB1-F2 der Funktion dient, Poren in biologischen
Membranen auszubilden, was für eine mögliche Destabilisierung der mitochondrialen
18Einleitung
Membran und anschließender Depolarisierung des Membranpotentials von Bedeutung sein
könnte (Chanturiya, Basanez et al. 2004; Zamarin, Garcia-Sastre et al. 2005). Weiterhin
Abb. 2.6.2 Sekundärstruktur des PB1-F2-Proteins vom IAV-Stamm A/Puerto Rico/8/34 (H1N1)
mit allen bisher bekannten funktionellen Domänen und C-terminaler α-Helix.
konnte gezeigt werden, dass virales PB1-F2 nach radioaktiver 35S Markierung von infizierten
MDBK-Zellen in drei kurz aufeinanderfolgenden Banden im SDS-Gel migriert, was auf einer
möglichen posttranslationalen Modifikation des Proteins beruhen könnte (Chen and Holmes
2008). Erste in-silico-Sequenzanalysen gaben Hinweise darauf, dass PB1-F2 mehrere
potentielle Phosphorylierungsstellen besitzt, was im Rahmen dieser Arbeit experimentell
bestätigt werden konnte (Henklein, Bruns et al. 2005).
Die exakte biologische Funktion von PB1-F2 und seine Bedeutung im Kontext der zellulären
IAV-Replikation sind bis heute ungeklärt. Ursprünglich wurde PB1-F2 als pro-apoptotisch
und als ein Verstärker der IAV-induzierten Apoptose in infizierten Monozyten beschrieben
(Chen, Calvo et al. 2001). Das Protein ist sowohl im Zytoplasma als auch in den
Mitochondrien von transfizierten und infizierten Zellen lokalisiert und ist bei Letzteren
partiell auch im Nukleus vorzufinden (Chen, Calvo et al. 2001). Die mitochondriale
Lokalisation von PB1-F2 und der Befund, dass die Präsenz von nanomolaren Mengen an
synthetischem sPB1-F2 im Zellkulturmedium zur Cytochrom-c Freisetzung in HeLa-Zellen
führt, gaben Hinweise darauf, dass PB1-F2 das erste IAV-Protein ist, das den intrinsischen
Apoptosepfad induziert (Chen, Calvo et al. 2001; Lowy 2003). Andererseits wurde berichtet,
dass Plasmid-exprimiertes PB1-F2 nicht ausreichend ist, um in Epithelzellen Apoptose
auszulösen. In der Gegenwart von apoptotischen Stimuli wie UV-Strahlung oder Cisplatin
konnte jedoch eine verstärkte Apoptoseinduktion in PB1-F2 transfizierten 293T-Zellen im
Vergleich zu nicht transfizierten Zellen beobachtet werden (Zamarin, Garcia-Sastre et al.
2005). In Koimmunopräzipitationsstudien wurden die beiden Untereinheiten des
mitochondrialen PTPC (Permeabiltiy transition Pore complex) als zelluläre Bindungspartner
von PB1-F2 identifiziert (Zamarin, Garcia-Sastre et al. 2005). Ob PB1-F2 durch diese
Interaktion pro-apoptotische Prozesse vermittelt oder durch seine Fähigkeit zu
multimerisieren und Membranporen zu bilden, ist bis heute nicht geklärt.
19Sie können auch lesen

















































