Entwicklung eines Assays für die quantitative und qualitative Einschätzung humaner Mitochondrien-DNA bei biologischen Minimalspuren
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Universitätsklinikum Ulm
Institut für Rechtsmedizin
Ärztlicher Direktor: Prof. Dr. Erich Miltner
Entwicklung eines Assays für die quantitative
und qualitative Einschätzung humaner
Mitochondrien-DNA bei biologischen
Minimalspuren
Dissertation
zur Erlangung des Doktorgrades der Medizin an der
Medizinischen Fakultät der Universität Ulm
vorgelegt von
Eva Franke
geboren in Breisach am Rhein
2019
1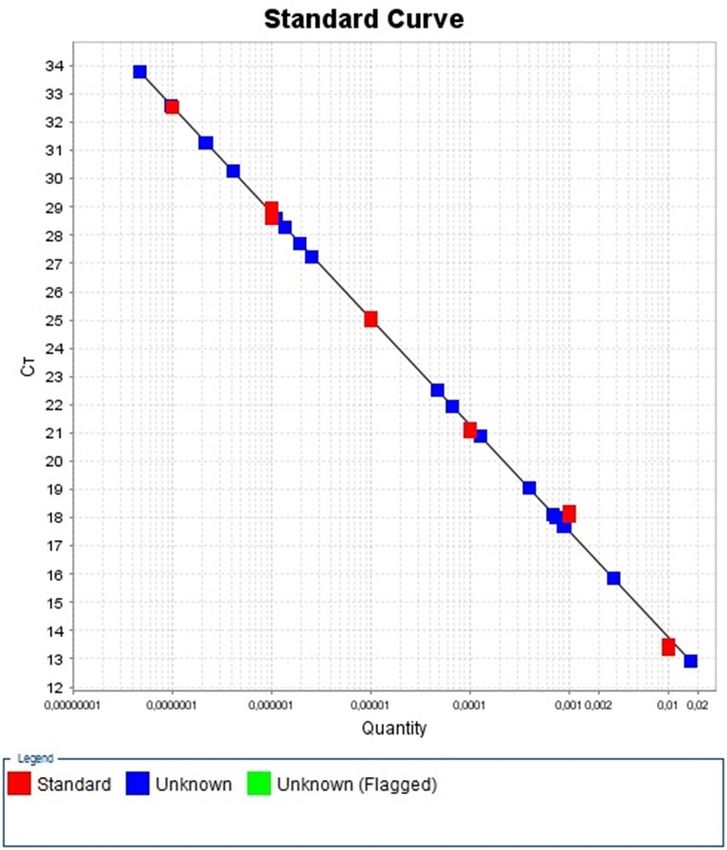
Amtierender Dekan: Prof. Dr. Thomas Wirth
1. Berichterstatter: Prof. Dr. Peter Wiegand
2. Berichterstatter: PD Dr. Bernd Baumann
Tag der Promotion: 22.10.2020
i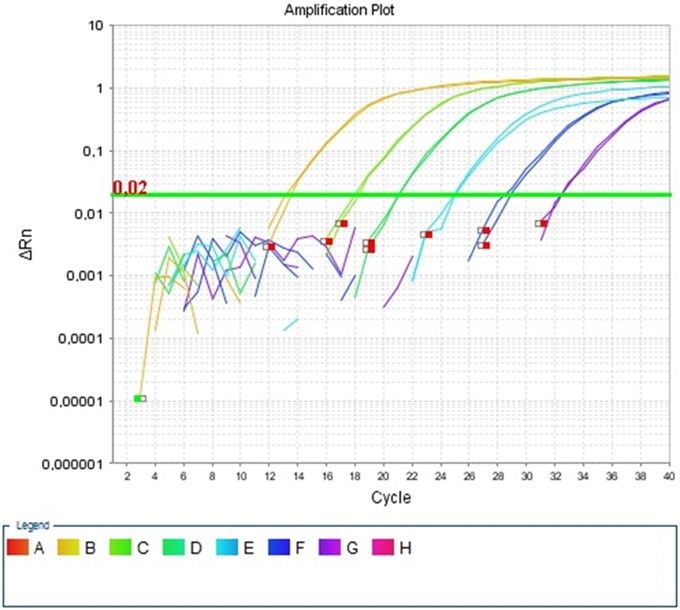
INHALTSVERZEICHNIS
Abkürzungsverzeichnis ...................................................................................................................... iii
1. Einleitung ................................................................................................................................... 1
1.1. Mitochondriale Humane DNA ................................................................................................ 1
1.2. mtDNA Sequenzierung und deren Nutzung in der forensischen Spurenanalyse ................. 4
1.3. Bedeutung der mtDNA Quantifizierung ................................................................................ 7
2. Material und Methoden .......................................................................................................... 10
2.1. Material ................................................................................................................................ 10
2.2. Geräte ................................................................................................................................... 11
2.3. Proben ................................................................................................................................... 11
2.4. Versuchsaufbau .................................................................................................................... 12
2.5. DNA-Extraktion ..................................................................................................................... 13
2.6. PCR-Amplifikation und Agarose Kontrolle .......................................................................... 14
2.7. Aufreinigung PCR-Amplifikat ............................................................................................... 18
2.8. Klonierung............................................................................................................................. 18
2.9. Real Time PCR ....................................................................................................................... 20
2.10. Umrechnung der Messungen der Standardkurve von Kopienzahl in ng/l ..................... 27
2.11. Schmelzkurvenanalyse ....................................................................................................... 29
2.12. Sequenzierung nach Sanger und Kapillarelektrophorese ................................................. 29
3. Ergebnisse ................................................................................................................................ 34
3.1. Klonierung............................................................................................................................. 34
3.2. Real Time PCR ....................................................................................................................... 34
3.3. Schmelzkurven-Analyse ....................................................................................................... 41
3.4. Ergebnis Sequenzierung ....................................................................................................... 45
4. Diskussion ................................................................................................................................ 51
4.1. Probengewinnung und Material .......................................................................................... 51
4.1.I.DNA-Extraktion................................................................................................................ 51
4.1.II. PCR und Klonierung ....................................................................................................... 52
4.2. Real Time PCR ....................................................................................................................... 53
4.3. Standardkurve ...................................................................................................................... 58
4.4. Sequenzierung ...................................................................................................................... 60
5. Zusammenfassung ....................................................................................................................... 65
6.Literaturverzeichnis....................................................................................................................... 67
Anhang ............................................................................................................................................. 71
Danksagung ................................................................................................................................... 80
Lebenslauf...................................................................................................................................... 81
ii
Abkürzungsverzeichnis
BLAST: Basic Local Alignment Search Tool (Sammlung von DNA- und Protein-
Sequenzdaten zum Vergleich von Mutationen)
bp: Basenpaare
BRCA-Gen: Breast-Cancer-Gen (Brustkrebsgen)
Ct: Treshold cycle (theoretische Größe für den Beginn eines expotentiellen
Wachstums einer Kurve)
D-Loop: displacement loop (Verdrängungsschleife, spezifische Sekundärstruktur der
DNA)
DNA: deoxyribonucleic acid (Desoxyribonukleinsäure)
dNTP: Desoxyribonukleosidtriphosphat
dUTP: Deoxyuridintriphosphat
GE: Genome equivalent
HVR: Hypervariable Region
LB-Medium: Lysogeny Broth-Medium
LF: Long Forward
LR: Long Reverse
MSH: Mundschleimhaut
mtDNA: Mitochondriale DNA
ng: Nanogramm
NCBI: National Center for Biotechnology Information
PCR: Polymerase Chain Reaktion (Polymerasekettenreaktion)
pg: Picogramm
rpm: rounds per minute (Runden pro Minute, Umdrehungszahl der Zentrifuge)
iii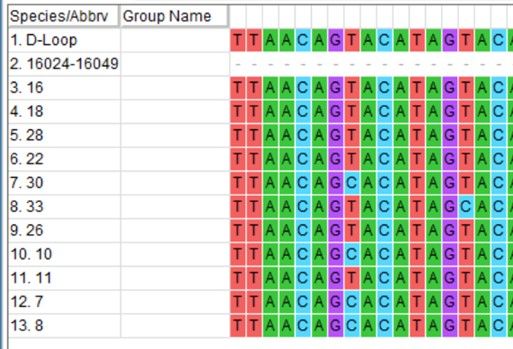
RNA: Ribonukleinsäure
rRNA: ribosomale-Ribonukleinsäure
SF: Short Forward
SNP: Single Nucleotide Polymorphism (Einzelnukleotidpolymorphismus)
SR: Short Reverse
STR: Short tandem repeat (Mikrosatelliten)
tRNA: transfer-Ribonukleinsäure
qPCR: quantitative Echtzeit-PCR, Real Time PCR
UDG: Uracil-DNA-Glykosidase
iv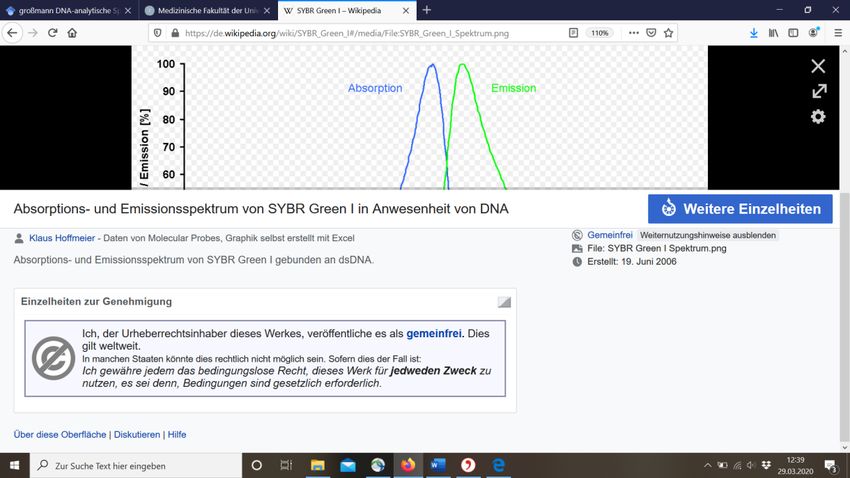
1. EINLEITUNG
1.1. MITOCHONDRIALE HUMANE DNA
Nahezu die gesamte menschliche DNA liegt durch Proteine verpackt, als 22 gepaarte
homologe autosomale Chromosomen und 2 Geschlechtschromosomen, X und Y, vor.
Man nennt dies auch diploider Chromosomensatz, wobei jeweils ein Chromosom von
einem Elternteil vererbt wird. Diese DNA nennt man Kern-DNA, da sie sich im Zellkern
befindet. Auf molekularer Ebene ist die doppelsträngige Desoxyribonukleinsäure auf
Nukleosomen aufgewickelt und je nach Phase, in der sich die Zelle befindet mehr oder
weniger verdichtet. Diese genomische DNA hat Abschnitte mit für die Zelle wichtigen
Informationen, die als Gen bezeichnet werden. Ein Gen ist „eine lokalisierbare Region
genomischer DNA-Sequenz, die einer Erbeinheit entspricht und mit regulatorischen,
transkribierten und/oder funktionellen Sequenzregionen assoziiert ist“ (Pearson H.
2006). Das Gen wird dann, während der sogenannten Transkription (Nordheim A. et
al. 2015) , abgelesen und es entsteht ein RNA-Strang, den man nach seiner Funktion in
tRNA, mRNA und rRNA einteilt. Das transkribierte Gen enthält jedoch auch Abschnitte
ohne relevante Information, die sogenannten Introns, welche bei der Bearbeitung der
prä-RNA zur RNA herausgeschnitten werden. Übrig bleiben die Exons mit codierender
Sequenz. Das Verhältnis von Introns zu Exons ist in jedem Gen unterschiedlich und
schwankt zwischen über 10% Exons beispielsweise im BRCA2-Gen (Górski B. u.
Debniak T, Jakubowska A, Cybulski C, Huzarski T, Byrski T, Złowocka E, Lubiński J.
2003) und 0,4% im Dystophin-Gen (Shiga N. et al. 1997). Insgesamt codiert nur etwa
1,5% der gesamten Kern-DNA für Proteine während 98,5% sogenannte „Junk-DNA“
(Lander E. S. et. al. 2001) aus Introns und anderen nicht-kodierenden
Desoxyribonukleinsäuren bestehen. Kodierende DNA ist bei Individuen größtenteils
identisch, nicht-kodierende DNA jedoch enthält auf bestimmten Abschnitten
Wiederholungen bestimmter Basenpaare, welche interindividuell verschieden bei
einem Menschen sind. Diese Wiederholungen von Basen, beispielsweise
TAGTAGTAG… nennt man Mikrosatelliten oder auch short tandem repeats (STRs). Dies
sind Sequenzlängenpolymorphismen, welche in der forensischen Untersuchung von
biologischen Spuren standardmäßig genutzt wird (Butler J. M. 2005).
1
Ein Prozent der genetischen Information jedoch ist in einer anderen Zellorganelle
vorhanden, dem Mitochondrium. 1963 wurde die mitochondriale DNA, abgekürzt
mtDNA, von Margit M. K. Nass und Sylvan Nass elektronenmikroskopisch entdeckt
(Nass M. M. K. u. Nass S. 1968). Der Endosymbiontentheorie (Sagan L. 1967) zufolge
waren Mitochondrien ursprünglich Prokaryonten, welche sich in die tierische
Vorläuferzellen eingeschleust haben. Hinweise darauf sind die zirkulär angeordnete
Form der DNA, die Ähnlichkeit der Proteinbiosynthese, die Lagerung von DNA in
Nukleoid und der Aufbau der DNA, der größtenteils aus Exons besteht. Der größte Teil
der mitochondrialen DNA codiert für Proteine der Atmungskette einer Zelle. Die
Atmungskette ist eine Abfolge von molekularchemischen Reaktionen, bei denen es
durch verschiedene Reduktions- und Oxidationsvorgängen zur Bildung von ATP
(Adenosintriphosphat), einem Hauptenergieträger von Zellen, kommt. Am Ende dieser
Atmungskette steht die Reduktion von Sauerstoff zu Wasser, welches bei der
physiologischen Atmung in der Lunge wieder ausgetauscht wird. Da die mtDNA also
einen wichtigen Teil der Proteine für die Atmungskette codiert und diese im
Mitochondrium liegt, wird das Mitochondrium auch als „Kraftwerk der Zelle“ (Jakobs
S. 2004) bezeichnet.
Ein weiterer Unterschied von mtDNA zur Kern-DNA ist die Art der Vererbung. MtDNA
wird ausschließlich von der Mutter vererbt. Die paternale mtDNA befindet sich im Hals
des Spermiums, der kein Teil der Zygote wird, und wird daher von der Eizelle mit
Ubiquitin markiert, sodass sie anschließend abgebaut wird (Nordheim A. et al. 2015).
Im Tierversuch seien jedoch auch kleine Anteile väterlicher mtDNA nachgewiesen
worden. (Schwartz M. u. Vissing J. 2002)
Das komplette mitochondriale Genom beinhält 37 Gene für Proteine, Transfer-RNAs
(tRNA) und ribosomale-RNAs (rRNA). Zwei rRNAs und 14 tRNAs, sowie 12 von 13
proteinkodierenden Genen kodieren auf dem schwereren, Purin-reichen Strang oder
auch heavy (H-)strand. Acht tRNA-Gene und ein proteinkodierendes Gen kodieren auf
dem leichteren, Pyrimidin-reichen Strang oder light (L-)strand (Anderson et al. 1981).
Des Weiteren gibt es eine Kontrollregion, auch D-Loop genannt, die wiederum 3
Hypervariable Regionen (HVR I-III) (van Oven M. u. Kayser M. 2009) enthält. Im
Gegensatz zur restlichen mtDNA ist der D-Loop, mit circa 1100 Basenpaaren, nicht
kodierend für Proteine und beinhaltet daher mehr inter- sowie intraspezifische
Unterschiede. Hier liegt die größte interindividuelle Varianz (Budowle B. et al. 1999).
2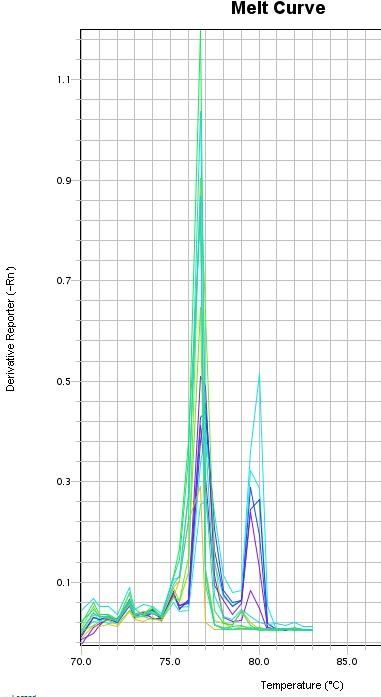
Innerhalb dieser Region liegt zwischen 16024-16365bp die Hypervariable Region I,
zwischen 73-340bp die HVR II, sowie die HVR III zwischen 438-574bp
ABBILDUNG 1) HYPERVARIABLE REGIONEN AUF DEM D-LOOP DER MITOCHONDRIELLEN
DESOXYRIBONUKLEINSÄURE, QUELLE: VERENA THIAS, EINE FORENSISCHE METHODE ZUR
EINZELZELLUNTERSUCHUNG MIT SEQUENZIERUNG DES D-LOOPS DER MITOCHONDRIALEN
DESOXYRIBONUKLEINSÄURE, 2008
HYPERVARIABLE REGIONEN I, II UND III UND DEREN POSITION AUF DEM D-LOOP
Die zirkuläre Mitochondrien-DNA besitzt zwei Stellen, eine auf jedem Strang, als Start
der Replikation. Der „Origin of replication“ des H-Strangs, sowie die Promoter-Region
für beide Stränge sind in der D-Loop Region enthalten. Am „Origin of replication“
beginnt die DNA Replikation also die Vervielfachung der Gen-Sequenzen. Diese
Promoter-Region ist der Beginn der Transkription für die Bildung der verschiedenen
RNAs.
ABBILDUNG 2) AUFBAU DER MENSCHLICHEN MITOCHONDRIALEN DESOXYRIBONUKLEINSÄURE
(QUELLE: WILDLIFE DNA ANALYSIS, 2013, DNA, GENOMES AND GENETIC VARIATION. IN A. LINACRE,
& S. S. TOBE, WILDLIFE DNA ANALYSIS (1ST ED.). WEST SUSSEX: WILEY-BLACKWELL
MITOCHONDRIELLER DESOXYRIBONUKLEINSÄURE-RING MIT 37 GENEN, ÄUßERER HEAVY-STRANG
UND INNERER LIGHT-STRANG
3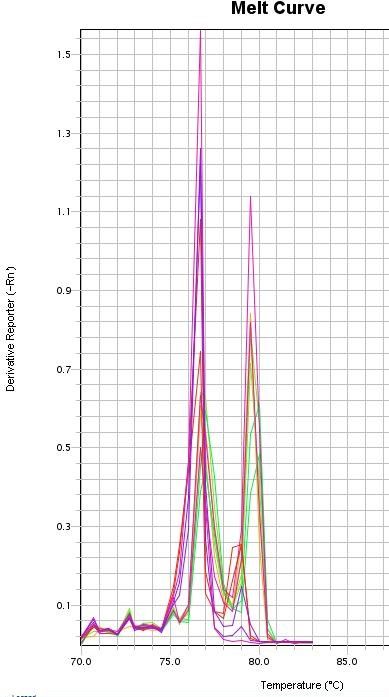
Die zwei oben genannten rRNAs, 16S und 12S rRNA, bilden, zusammen mit Proteinen,
das Ribosom, welches den Ort der Proteinbiosynthese darstellt. Die 16S Region ist circa
1500 bp lang (Yang B. et al. 2016;Keller P. M. et al. 2010). Da diese kodierenden
Regionen bei jedem Individuum identische Sequenzen aufweisen und die 16S-Region
für Mutationskrankheiten nur wenig anfällig ist (Elson J. L. et al. 2015), wurde in
diesem Versuch die 16S- Region als Referenzregion gewählt.
1.2. MTDNA SEQUENZIERUNG UND DEREN NUTZUNG IN DER
FORENSISCHEN SPURENANALYSE
Mitochondriale DNA ist ein wichtiger Teil der forensischen Spurenanalyse. Wenn
Tatortspuren gesichert werden, handelt es sich häufig um sehr geringe
Zellantragungen mit zum Teil degradierter DNA. Bei so geringen DNA-Mengen von
maximal 100pg spricht man geläufig von „Low Template DNA" (Buckleton J. 2009).
Die Sequenzierung von low-template DNA ist, mit der Identifizierung bedeutender
Personen, in den letzten Jahren in der Gesellschaft bekannt geworden. 2007 die
Identifizierung der russischem Zaren-Familie (Clark D. P., Pazdernik N. J. 2009) und
2014 die von mexikanischen Studenten, welche von einem mexikanischen
Drogenkartell ermordet und verbrannt wurden (Eades L. 2015). Bei beiden Bespielen
handelte es sich um stark degradierte Proben, wie verbrannter Knochen. Die Analyse
genomischer DNA ist dadurch besonders beeinträchtigt. Die verhältnismäßig große
Länge der typisierten Kern-DNA Sequenzen und die Tatsache, dass pro Zelle nur eine
Kopie der genomischen DNA vorliegt, können eine konventionelle Analyse mittels STR-
Profil massiv erschweren. Die Masse eines nukleären Genoms einer Zelle liegt bei circa
6pg. Mit einer Probe, die diese 6pg oder weniger DNA-Menge enthält, ist man an der
Detektionsgrenze für humane diploide Zellen angelangt und die Erlangung eines
vollständigen DNA-Profils ist nicht mehr zu erwarten. Im Gegensatz dazu gibt es pro
Zelle 102 bis 104 Kopien mitochondrialen Genoms (Rooney J. P. et al. 2015;Bogenhagen
D., Clayton D. A. 1974, 1974), je nach Metabolismus der Zelle. Des Weiteren ist die
mitochondriale DNA weitaus kürzer und durch ihre Ringstruktur stabiler (Budowle B.
et al. 2005) gegenüber Umwelteinflüssen und Degradation. So kann es sein, dass eine
4
Analyse der gleichen Probe von mitochondrialer DNA zu einem validen Ergebnis führt,
jedoch nicht auf Basis genomischer STRs.
Bei der Analyse und Interpretation von solch geringen und degradierten DNA-Mengen
mittels mitochondrialer Marker ist es besonders wichtig die Art und Weise der
Spurenantragung zu bedenken. Sekundär- oder Tertiärtransfer (Lowe A. et al. 2002),
das heißt eine Zellübertragung von einer Person auf eine Weitere, oder auf ein Objekt
oder gar wiederum auf eine weitere Person oder ein weiteres Objekt, ist häufig und
schwer detektierbar und kann möglicherweise zu Schwierigkeiten bei der
Ergebnisinterpretation führen. Dieser Sekundär- oder Tertiärtransfer kann natürlich
auch von bekannten Einflussfaktoren herrühren, daher ist Kontamination ein weiterer
Punkt, den es zu beachten gilt, wenn man eine sehr geringe Spur interpretiert. Eine
Kontamination ist sowohl während der Spurensicherung als auch im Labor möglich.
Daher sind strenge Arbeitsschutzregeln wie regelmäßiger Handschuhwechsel, das
Tragen eines Mundschutzes und getrennte Räume von prä- und post-PCR Schritten
unerlässlich. Nichtsdestotrotz kann eine Kontamination nie ganz ausgeschlossen
werden.
Wie bereits erwähnt, besitzt die mtDNA kaum Introns sondern fast nur kodierende
DNA. Will man eine Analyse mit mtDNA-Proben durchführen, bietet sich die D-Loop
Region und darin insbesondere die Hypervariablen Regionen an. Da diese
Sequenzbereiche nicht kodierend sind, können sie eine natürliche Varianz zwischen
Personen unterschiedlicher maternaler Abstammung zeigen. Hierbei ist jedoch zu
beachten, dass innerhalb einer Familie mit gleicher mütterlicher Abstammung keine
maßgebenden Unterschiede zu erwarten sind. Die Analyse ist also weniger
diskriminativ und dient eher dem Ausschluss von berechtigten Personen. Was für die
Forensik eher eine negative Auswirkung hat, wird in der Biogeographie dafür genutzt,
anhand von Haplogruppen Bevölkerungsgruppen verschiedenen Ursprungsorten
zuzuordnen. Menschen mit der gleichen Mutation von Nukleotidsequenzen an
bestimmten Orten des Genoms haben somit einen bestimmten Haplotyp und gehören
gemeinsam einer Haplogruppe an. Anhand von Phylotrees (van Oven M. 2015) oder
ähnlichen Programmen, kann man die Haplogruppen klassifizieren und geographisch
ihrer Abstammung einordnen. Um einen wissenschaftlichen Vergleich herstellen zu
können, wurde 1981 an der Cambridge Universität das Genom einer aus Europa
5stammenden Frau vollständig sequenziert und als Cambridge Referenzsequenz (CRS)
benannt. Heute gilt diese in vielen Ländern als Vergleichsreferenz.
Mitochondriale DNA ist 5-10 mal anfälliger für Mutationen (Giles R. E. et al. 1980) als
nukleäre DNA. Dies wird durch verschiedene Faktoren erklärt. In Mitochondrien gibt
es keine schützenden Histone, die Reparaturmechanismen sind weniger effizient als
im Zellkern und es besteht eine höhere Konzentration an reaktiven Sauerstoffspezies,
die als Nebenprodukt der Zellatmung in den Mitochondrien entstehen.
Im Gegensatz zu der Detektion von Längenunterschieden, bei der STR-Analyse der
Kern-DNA, werden bei der mitochondrialen DNA-Analyse Mutationen einzelner Basen
untersucht. Punktmutationen, welche eine Purinbase in die jeweils andere oder eine
Pyrimidinbase in die jeweils andere verwandelt, nennt man Transition (Clauss W. ,
Clauss C. 2009). Dabei kann ein Wechsel von A zu G und umgekehrt und von T zu C und
umgekehrt stattfinden. Bei einer Transversion wird eine Purinbase in eine
Pyrimidinbase oder umgekehrt verwandelt (zB. Springer Klinisches Wörterbuch,
2007, ISBN 978-3-540-34602-9). Aber auch Längenheteroplasmien kommen vor. In
der mitochondrialen Kontrollregion ab Basenpaar 303 – 309 kommt es gehäuft zu C-
Strech Längenheteroplasmien. Wie in Lutz-Bonengel S. 2001 beschrieben, werden
hierfür „Insertionen (…) von Cytosin-Resten an der Position 309.x und ein „slippage“
der DNA-Polymerase während der Replikation“ verantwortlich gemacht. Dies ist
beispielhaft Abbildung 21 im Anhang dargestellt.
Diese Mutationen können sich stabil über viele Generationen weitervererben und
verbreiten. Wenn man bedenkt, dass alle heute lebenden Menschen von ein paar
wenigen weiblichen Vorfahren abstammen, kann man sich gut vorstellen, dass diese
ehemaligen Punktmutationen zu einem Marker für genetische Abstammung werden,
die sogenannten Single Nucleotide Polymorphismus (SNPs). Dabei sind circa 2/3 aller
Polymorphismen C zu T Transformationen, da dieser Austausch durch Methylierung
und anschließende Desaminierung verhältnismäßig einfach von statten geht. Eine
andere Art von Punktmutationen sind Insertionen beziehungsweise Deletionen.
Hierbei werden die Basen nicht verändert, sondern es bilden sich neue zusätzliche
Basen, was man Insertionen nennt oder Basen verschwinden, was zu einer Deletion
führt.
61.3. BEDEUTUNG DER MTDNA QUANTIFIZIERUNG
Das Prinzip der Real-Time PCR beruht auf der herkömmlichen Polymerase-Ketten-
Reaktion mit zusätzlicher Quantifizierung der DNA einer Probe in Echtzeit. In dieser
Arbeit wurde dazu die ABI-Prism-7500-Real-Time PCR genutzt. Das Prinzip dahinter
verläuft nach den Phasen „Erregung, Emission und Kollektion“ (Applied biosystems
28.08.2019a). Über einen optischen Heizdeckel wird sowohl die Temperatur, um die
Zyklen der PCR zu temperieren, geregelt, als auch die Energie aus einem Argon-Laser
gelenkt (Seifert J. u. Ekkernkamp A. 2006). Nach jedem PCR Zyklus wird ein Farbstoff,
beispielsweise SYBR-Green, in den Reaktionstubes angeregt. Das gewählte Kit SYBR
Green Power Up beinhaltet eine DUAL-Lock-DNA Polymerase, die bei niedrigen
Temperaturen noch keine Funktion ausübt und so die eigene Aktivität kontrolliert, den
interkalierende Fluoreszenzfarbsoff SYBR Green (Abbildung 19) und ein
Referenzfarbstoff ROX Dye, durch den die Fluoreszenz zwischen den einzelnen
Reaktionen für das Messinstrument vergleichbar gemacht wird. Weitere Reagenzien
sind MgCl2, dNTPs und Stabilisatoren. SYBR Green hat eine maximale Emission von
grünem Licht bei 520 nm (Sigma-Aldrich 2019)(Abbildung 20)Diese Fluoreszenz kann
dann von dem ABI-Prism®-7500-Sequence-Detection-System erkannt und in ein
elektrisches Signal umgewandelt werden. Die resultierende emittierte Fluoreszenz
wird über ein optisches System erkannt und gemessen (Abbildung 18). Je mehr DNA
amplifiziert wird, desto mehr Fluoreszenz wird emittiert.
Die Quantifizierung, also Messung der DNA-Menge einer „Spur“ hat vor allem vor
Gericht hinsichtlich der Interpretation von Wahrscheinlichkeiten verschiedener
Hypothesen für Tathergänge Bedeutung. Hierbei beruht der Großteil der Studienlage
bisher auf Versuchen mit Kern-DNA (Reuss E. 2008). DNA-Spuren am Tatort können
zunächst einmal in Tat-relevante Spuren durch „aktiven Transfer“ und Spuren die nicht
direkt mit dem Tathergang in Verbindung gebracht werden können, „passiver
Transfer“ (Gill P. 2014;Fonneløp A. E. et al. 2015) unterteilt werden. Ein passiver
Transfer wird beispielsweise im Rahmen von Unbeteiligten vor der Tat am Tatort
angetragen oder kann von Ermittlern nach der Tat durch Ermittlungsarbeit entstanden
sein. Es ist also eine Kontamination hinterher oder eine bereits bestehende
„Verunreinigung“ eines Objekts vor der Tat. Eine DNA-Spur, welche passiv angebracht
7wurde, hat also nichts mit dem Tathergang zu tun. Ein aktiver Transfer wird von der
Person, welche die eigentliche Tat durchführt, angebracht. Dies kann auf
unterschiedliche Art, beispielsweise über Aerosol oder direkt beim Berühren von
Objekten passieren (Fonneløp A. E. et al. 2015). Wenn man nun von einer aktiven Spur
ausgeht, gilt es diese im Bezug auf mögliche Arten der Spurenantragung zu vergleichen.
Hierfür wird unter den Möglichkeiten an Tathypothesen „jene Hypothese (bestimmt),
die unter Berücksichtigung der Art des Beweismittels und der Umstande des Falles
wirklich angewendet werden können“ (Fonneløp A. E. et al. 2015). Die gewählte
Hypothese des Tathergangs und somit der Spurenantragung kann dann mit der Art und
Menge der Spur verglichen werden. Die Spurenantragung hat viele verschiedene
Einflussfaktoren. Eine gelungene Zusammenfassung bietet „Die Analyse von
Hautkontaktspuren“ von (Pfeifer C. et al. 2016). Dabei wird ersichtlich, dass zusätzlich
zur Ursprungsebene (Cook R. et al. 1998), also der ursprüngliche Spurenverursacher,
auch die Aktivitätsebene, wie beispielsweise Sekundärantragungen, von Bedeutung
sein können. So kann zum Beispiel eine durch Primärantragung angebrachte DNA-Spur
auf einer Plastik-Fläche relativ leicht durch Reibung auf eine raue Sekundärfläche
übertragen werden (Pfeifer C. et al. 2016). Ein solches Szenario ist bereits mit Kern-
DNA Mengen der Primärspur von 2ng möglich und kann bei optimalem Transfer (glatte
Oberfläche auf raue Oberfläche, Reibung als Kontaktart und einer zeitlich noch frischen
Spur) eine Spur mit 1ng ergeben. Wenn der DNA-Gehalt einer Sekundär-Spur also sehr
hoch ist, können bestimmte Hypothesen der Antragung von einer Primärspur mit
niedrigerem DNA-Gehalt aufgrund der geringen Wahrscheinlichkeit bereits
ausgeschlossen werden. Andersherum kann eine Spur mit geringem DNA-Gehalt
ausgeschlossen werden, wenn die Antragungsmöglichkeiten sehr vielseitig sind und
somit keinen tatkräftigen Beweis darstellen, da beispielsweise niedrigste DNA-Spuren
mittels Tertiärtransfer sehr große Schwankungen im Bezug auf
Antragungswahrscheinlichkeiten aufweisen. Es gilt immer eine Spur nicht nur ihrem
Besitzer zuzuordnen, sondern auch die Aussagekraft dieser Spur hinsichtlich einer
Tathypothese zu vergleichen.
8Zielsetzung der Arbeit:
Die Quantifizierung von mtDNA hat, aus forensischer Sicht, verschiedene Vorteile. Ein
Vorteil ist die oben genannte Rekonstruktion der Tathypothese. Ein weiterer Vorteil
soll in dieser Arbeit verifiziert werden, also ob es möglich ist, ausgehend von der
Quantifizierung die Qualität der Sequenzierung vorherzusagen. Ebenso stellt sich die
Frage, wie niedrig die Qualität der Sequenzierung sein kann um diese Ergebnisse noch
zu nutzen und wie viel DNA dafür mindestens gebraucht wird. Es wird nach einem Cut-
off der einzusetzenden DNA-Menge gesucht, die es für eine Sequenzierung nach Sanger
bedarf. Diese Erkenntnis ist nicht nur wichtig um spurenschonend mit quantitativ
niedrigen Spuren umzugehen, sondern könnte auch zu einem ökonomischeren
Arbeiten in der Forensik führen, sodass man vor der Sequenzierung einer Probe eine
Quantifizierung voranstellt und somit deren Sequenziererfolg vorhersagen kann. In
dieser Arbeit wird der Sequenziererfolg in % angegeben, also wie viel % der Länge des
gewünschten DNA-Abschnittes ergeben eine Nukleotid-Abfolge. Ziel dieser Arbeit war
also die Entwicklung eines mtDNA-Quantifizierungs-Assays bei gleichzeitiger
Bestimmung des Degradationsstatus.
92. MATERIAL UND METHODEN
2.1. MATERIAL
i. DNA Extraktion: Chelex 5% (BioRad), Proteinase K Solution
(Promega, Mannheim)
ii. PCR-Reaktion: MgCl2 Solution 25mM, BSA 10mg/ml, 10x PCR Puffer
2, 10µM dNTP, Ampli Taq Gold –Polymerase 250 Units (alle Thermo
Fisher Scientific, Dreieich)
iii. Amplifikationskontrolle mit Agarose-Gel: Hyperladder 1 (200-
10000 bp) inklusive 5x loading buffer (bioline, Luckenwalde),
Agarose High Resolution (Carl Roth, Karlsruhe) , Ethidiumbromid
0,5mg/ml , TAE-Puffer (96,8g Tris, 22,8ml Essigsäure, 7,4g EDTA, pH
8,0)
iv. Aufreinigung PCR-Produkt: EXOI Exonuclease I 20U/µl, FastAP
Thermosensitive Alkaline Phosphatase 1U/µl (Fermentas GmbH, St.
Leon-Roth)
v. BigDye Sequenzierung nach Sanger: Big-DyeTM Terminator v1.1
Cycle Sequencing Kit (Thermo Fisher Scientific)
vi. Aufreinigung DyeEx: DyeEx 2.0 Spin Kit (Qiagen, Hilden)
vii. Kapillarelektophorese: Hi-Di Formamid, POP7, 36-cm-Kapillare
(alle Thermo Fisher Scientific)
viii. Gel-Extraktion: QIAEX II Gel Extraction Kit (Qiagen)
ix. Photometrische DNA-Messung: QuantiFlour dsDNA System
(Promega)
x. 16S-Amplifizierung mittels Vectortransformation in E. coli-Zellen:
Vector (), kompetente Zellen E. coli (), Nährmedium/Platten (),
Flüssig-Nährmedium ()
xi. Klonierung: Lysogeny Broth-Medium, Sfi1- Enzym, pAC1- Vector
(Promega)
xii. DNA-Aufreinigung aus den Zellen: GenElute Plasmid Miniprep Kit
(Sigma Aldrich, Taufkirchen)
xiii. qPCR: SYBR GreenER qPCR SuperMix Universal (Thermo Fisher
Scientific)
102. Primer (biomers.net, Ulm)
Detaillierte Liste im Anhang
2.2. GERÄTE
1. Thermomixer comfort 1,5ml (Eppendorf)
2. Peqlab perfect Spin 24 Plus- Zentrifuge (PEQLAB)
3. Labnet International Inc. Vortex Mixer (Labnet)
4. Sarstedt Handzentrifuge (Sarstedt)
5. Alpha Mager EP + PC-Programm Alpha ease FC
6. Electrophoresis Power Supply EPS 301 (Amersham pharmacia
biotch)
7. Quantus Flourometer (Promega)
8. Standard-Thermocycler Biometra (Analytik Jena, Jena)
9. Geräte für die Real-Time-PCR: ABI-Prism-7500-Real-Time PCR
(Thermo Fisher Scientific)
10. 3130 Genetic Analyser -Sequenzierung (Thermo Fisher Sientific)
2.3. PROBEN
- Mundschleimhautproben wurden von 20 freiwilligen Probanden mittels DNA-
freier Watteabriebtupfer abgenommen
- Haarproben mit telogener Wurzel, welche natürlicherweise ausgefallen waren
- Haarschäfte
- Einzelhautschuppen, geprickt
112.4. VERSUCHSAUFBAU
Da mitochondriale DNA bei einem Tathergang dann zu Rate gezogen wird, wenn es sich
bei einer Spur um wahrscheinlich zu geringe Mengen genomischer DNA handelt, wurde
hier Probenmaterial von den Probanden entnommen welches voraussichtlich
unterschiedliche mtDNA-Mengen beinhaltet. Es wurden telogene Haare gesammelt
und deren Wurzeln und Schäfte untersucht, sowie Einzelhautschuppen gepickt um
Proben mit geringem DNA-Material zu erhalten. Zur Referenz wurden
Mundschleimhautproben der betreffenden Personen aus deren Wangeninnenseiten
zum Abgleich mit den untersuchten Spuren entnommen.
Für diesen Versuch wurde die 16S-Region als Referenzregion gewählt. Dieser
Abschnitt wurde mittels der Primer L2025 und H3108 amplifiziert. Auf der 16S-Region
wiederum wurden ein kurzes Segment gewählt, mit 167bp (Short-Fragment) und ein
langes mit 492bp (Complete-Fragment) Länge, die für die spätere Quantifizierung, als
Short- und Long-Segment wichtig werden. Aufgrund von Schwierigkeiten in der
Versuchsdurchführung wurde während der PCR das Short- und Complete-Fragment
genutzt und bei der qPCR das Short- und Long-Fragment. Die Primer auf der 16S
Region wurden nach (Fregel R. et al. 2011) ausgesucht und zu einer multiplex qPCR
optimiert, sodass sowohl das Short als auch das Long-Fragment, zu ähnlichen Teilen
amplifiziert wurden.
L2025 1083pb H3108
S 167- SR
F Short
SF 492-complete LR
LF 314-LONG LR
ABBILDUNG 3) 16S -REGION MIT DEM KURZEM, LANGEM UND COMPLETE-FRAGMENT SOWIE DEREN
PRIMERN UND LÄNGE ALS BASENPAAREN VEREINFACHT DARGESTELLT, QUELLE: EIGENE DARSTELLUNG
DIE 16S-REGION MITTELS DER PRIMER L2025 UND H3108 AMPLIFIZIERT
UND DIE DARAUF ENTHALTENEN FRAGMENTE, DAS COMPLETE-SEGMENT ÜBERSCHNEIDET DAS SHORT-
UND LONG-SEGMENT UND WIRD AUS DEREN PRIMERN AMPLIFIZIERT
12Darüber hinaus wurde die komplette 16S-Region mit den Primern L2025 und H3108
aus (Maca-Meyer N. et al. 2001)amplifiziert. Daraufhin wurde die 16S-Region mit den
Primern L2015_Sfi1_neu und H3108_Sfi1_neu, die jeweils um die Randsequenzen der
Restriktionsstelle des Endonukleaseenzyms SfiI erweitert wurden, aus einer MSH-
Probe amplifiziert und anschließend in den Vektor pAC1 ligiert. Durch eine Klonierung
in E. coli Zellen wurde die 16S-Region stark vervielfacht. Die nach der Plasmid-
Aufreinigung hochkonzentrierte Lösung aus 16S-Fragment in Plasmiden wurde nun in
bestimmten Verdünnungen als Standardreihe bei der qPCR verwendet. Diese Real-
Time-PCR quantifiziert mitochondriale DNA mittels der Relation zur vorgegebenen
Standardreihe. Die zwei unterschiedlich großen Fragmente in der 16S-Region lassen
zudem eine Einschätzung des Degradationsgrades einer Probe zu. Die D-Loop-
Regionen der unterschiedlich konzentrierten DNA-Extrakte aus MSH und Haar-Proben
wurden dann in einer Sequenzierreaktion nach Sanger analysiert und zur Kontrolle mit
der rCRS abgeglichen.
2.5. DNA-EXTRAKTION
Die Mundschleimhautproben wurden durch einen Abrieb beider Wangeninnenseiten
mit einem DNA-freien Wattetupfer entnommen. Die Haarwurzelproben waren
telogene Haare, also solche, die von allein ausgefallen waren. Die Haarschäfte wurden
vorher leicht mit 10%iger Ethanollösung abgerieben. Für die DNA-Proben aus
telogenen Haarwurzeln wurden circa 1,5 bis 2 cm lange Stücke eines Haares ab der
Wurzel abgeschnitten. Von den Haarschaftproben wurden circa 5 cm Haarschaft in
einem Tube aufgefangen. Hautschuppen wurden durch Abklebung mit Neschenfolien
von getragener Kleidung gewonnen und für die Analyse mit einem Skalpell aus den
Folien ausgeschnitten. Die Extraktion verlief wie in 2.b.i. beschrieben. Die extrahierte
DNA der Proben wurde zwischen den Versuchen bei -20°C im Gefrierschrank
aufbewahrt. Die verdünnten MSH-Proben wurden zu Beginn der Versuchsreihe
hergestellt und waren somit bis zu 10 Monate alt. Der zweite Teil, die Haarproben,
wurde erst später hergestellt und war somit nur circa bis zu 8 Monate alt.
Nun wurde die DNA aus ihren, zum Teil, noch intakten Zellen herausgelöst und
aufgereinigt. Die hier verwendete Extraktion war die sogenannte Kochlyse. Dazu
wurden eine 5% Chelexlösung und Proteinase K verwendet. Bei den höher
13konzentrierten MSH-Proben wurde pro Tupfer 195µl 5% Chelex mit 5µl Proteinase K
und bei den niedrigen Haarproben pro Haar 50 µl Chelex mit 10 µl Proteinase K
verwendet. Durch mechanische Schüttel-Bewegung werden die Zellen bei einer
optimalen Inkubationstemperatur für Proteinase K von 56°C aufgebrochen. Dies
braucht für die MSH-Proben circa eine Stunde und für die Haarproben circa 16
Stunden.
Nach der Extraktion wird die Proteinase K durch 9-minütiges Kochen bei 92°C
denaturiert und somit deaktiviert. Das Chelex wird bei 10.000rpm abzentrifugiert.
Übrig blieb im Überstand aufgereinigte DNA, welche eingefroren gelagert werden
kann.
2.6. PCR-AMPLIFIKATION UND AGAROSE KONTROLLE
Es wurden verschiedene PCR-Reaktionen durchgeführt:
1. 16S Region und darauf liegendes Short- und Complete-Fragment, Kontrolle der
Klonierung
2. Short und Complete-Fragment zur Optimierung der PCR zur späteren
Verwendung in der qPCR
3. Midi-Ansatz zur Sequenzierung der qPCR-Proben
1) 16S Region
Die 16S- Region, der gewählte Abschnitt auf der mtDNA, wurde nach Fregel et al.(
2011) ausgewählt. Dies ist eine Gensequenz, die für die 30S Untereinheit des
mitochondrialen Ribosoms kodiert. Die komplette 16S-Region wird mit den Primern
H3108 (TCGTACAGGGAGGAATTTGAA) und L2025 (GCCTGGTGATAGCTGGTTGTCC)
amplifiziert. Innerhalb dieser Sequenz befinden sich ein Short- und ein Long-Segment.
Durch die Primer mtDNA_SF und mtDNA_SR wird das Short-Segment mit 167bp und
durch die Primer mtDNA_SF und mtDNA_LR das Long-Segment mit 314bp gebildet.
Diese sind ebenfalls ausgewählt aus (Fregel, R., et al 2011). Der Vorteil dieser
Fragmente ist ihre unterschiedliche Länge. Das Verhältnis dieser Fragmente kann zu
einer Einschätzung des Degradationszustandes der DNA verhelfen. Ist eine
Amplifizierung des Long-Fragments nicht möglich während das kurze Fragment
14optimal amplifiziert wird, so kann dies ein Hinweis darauf sein, dass die DNA-Probe
bereits degradiert ist oder die Probe insgesamt zu wenig DNA-Menge enthält.
Der Unterschied des genutzten Programmes zur PCR-Amplifikation zum publizierten
PCR-Programm von (Fregel, R., et al 2011) liegt in der Zeit und Temperatur für den
Annealing-Schritt. Im Original wird dies bei 62°C für eine Minute durchgeführt. In
Testreihen zeigte sich jedoch, dass diese Primer besser bei niedrigeren Temperaturen
in 2 Schritten funktionieren.
Der PCR-Ansatz belief sich auf 20µl pro Probe. Eingesetzt wurde 11 µl H2O, 1,4 µl MgCl2,
0,6 µl BSA, 2 µl Puffer, 1 µl dNTPs, 0,6 µl Taq Polymerase und insgesamt 1 µl 10 µM
Primer aus einer 1:1 Mischung aus L2025 und H3108. Zur den 18 µl Mastermix wurden
2µl DNA-Extrakt aus einer MSH-Probe hinzugefügt. Das PCR-Programm beginnt bei
96°C für 8 Minuten. Dabei trennt sich der Doppelstrang zu Einzelsträngen und die
Polymerase wird aktiviert. Darauf folgt 1 Minute lang 93°C. Nun folgen 50 Sekunden
bei 55°C und 20 Sekunden bei 58°C. Hierbei binden die Primer an die passende Stelle
auf der DNA. Die Elongation, also Verlängerung des neu amplifizierten Stranges durch
die Taq-Polymerase, findet bei 72°C für 30 Sekunden statt. Nach 30 Zyklen schließt die
PCR mit einer finalen Extension für 45 sec bei 72°C ab.
Zur Kontrolle, dass die ausgesuchten Primer die auf 16S liegenden Short und Long
Fragmente amplifizieren, wurde zuerst eine PCR der 16S-Region und anschließend mit
den daraus resultierenden PCR-Produkten eine PCR mit den Primern für Short und
Long durchgeführt. Hierbei zeigte sich, dass sich alle Produkte auf 16S befinden.
16S+Adapter
Die PCR zur Amplifizierung der 16S Region mit angehängten Adaptern zur Ligation in
die Schnittstelle des Restriktionsenzyms SfiI wurde ebenfalls auf 20 µl mit dem
gleichen Mastermix und der gleichen Menge MSH-Probe durchgeführt. Anstatt der
vorher benannten Primer L2025 und H3108 wurden nun L2520_Sfi1_neu und
H3108_Sfi1_neu ebenfalls in einer 1:1 Mischung mit insgesamt 1 µl 10 µM Primer auf
20 µl Ansatz pro Probe verwendet. Aufgrund der längeren Primer wurde ein
TouchDown-Programm anstelle des normalen PCR-Programms angewandt. Dabei
begann das PCR Programm ebenfalls bei 96°C für 8 Minuten, dann 93°C für eine Minute
und dann jeweils 15 Sekunden in 2°C-Schritten eine Erniedrigung der Temperatur von
61°C auf 51°C und am Ende 72°C für 45 Sekunden. Die PCR betrug 22 Zyklen.
152) Short- und Complete-Fragment
Das PCR-Programm und die Reagenzien wurden nach dem gleichen Prinzip wie in der
Amplifizierung der 16S-Region genutzt. Die Primer der Real-Time PCR aus Fregel, R.,
et al 2011 wurden bei der Standard-Amplifizierung leicht verändert genutzt, da sich
das Long-Fragment aus LF und LR schlecht amplifizieren ließ. Deshalb wurde der LF-
Primer weggelassen und somit ein Complete-Fragment mit 492 bp aus dem SF- und
LR-Primer amplifiziert. Das Short-Fragment aus SF und SR mit 167 bp wurde belassen.
Die Konzentrationen der drei Primer wurde vom genannten Paper von 8 µM Short-
Primer und 16 µM Long-Primer auf 10 µM angepasst. Zusätzlich wurde in der PCR ein
Verhältnis von 2,3:1 der Short-Primer zu dem Long-Primer benötigt, um eine ähnlich
starke Ausbeute beider Fragmente zu erhalten. Pro Reaktion wurde wie bei der 16S
Region 1 µl Primer eingesetzt, für das Short und Long-Segment also 0,5 µl SF, 0,35 µl
SR und 0,15 µl LR. Das PCR-Programm war das gleiche wie zur 16S-Amplifizierung. Es
beginnt bei 96°C für 8 Minuten, nun 1 Minute lang 93°C. Nun folgen 50 Sekunden bei
55°C und 20 Sekunden bei 58°C. Darauffolgend 72°C für 30 Sekunden. Nach 30 Zyklen
schließt die PCR mit einer finalen Extension für 45 sec bei 72°C ab.
3) PCR für Midi Ansatz
Die eigentliche Sequenzierung zur Identifizierung von SNPs findet auf nicht-
kodierenden Segmenten statt. Hierzu wurde der D-Loop auf dem Mitochondrium
gewählt. Der komplette D-Loop, auch Kontrollregion genannt, ist 1180 Basenpaare
lang. Er beginnt ab Position 16024 und geht über den Origin of Replication bis Position
576. Um diesen großen Bereich abzudecken wurde die Midi-Methode nach (Berger u.
Parson 2009) gewählt. Hierbei wird das circa 1200 bp lange Stück in 5 überlappenden
Fragmenten amplifiziert. Die Fragmente haben eine Länge zwischen 280 bp bis 444 bp.
Dadurch ist es möglich, auch degradierte Proben umfassend zu sequenzieren.
Der Midi-Ansatz (Berger u. Parson 2009) wird in 2 Reaktions-Ansätze geteilt. In Ansatz
I werden 3 „Hauptfragmente“, hier benannt als I1, I2, I3, gebildet. Im Ansatz II zwei
überlappende Fragmente, II1 und II2. Anders als im Paper beschrieben wurde der
Ansatz I in 3 Singleplex-Reaktionen durchgeführt, da eine Multiplex-Reaktion keine
zufriedenstellenden Ergebnisse brachte. Hierbei wurde das 444 bp lange Fragment I1
mittels der Primer F15989 und R16433, das 250 bp lange Fragment I2 mittels den
Primern F16450 und R130 und das 280 bp lange Fragment I3 durch die Primer F317
16und R599 amplifiziert. Im Multiplex-Ansatz II reagieren alle 4 Primer zusammen als
Multiplex-PCR. Hierbei bildeten F16197 und R16509 ein 312 bp langes Fragment und
F109 und R460 ein 351 bp langes Fragment.
Da die Primer der Rückreaktionen besonders niedrige Annealing-Temperaturen (zum
Teil bis zu 13 °C Unterschied des Forward zum Reverse-Primer) haben, wurde die
Annealing-Temperatur von 57°C auf 45°C herabgesetzt.
Die 3 Ansätze von Midi I, Midi I1, I2, I3, wurden jeweils auf 15 µl pipettiert. Davon
waren 5 µl DNA-Extrakt, 1,4 µl Primer (jeweils die forward und reverse Primer für ein
Fragment des Ansatz 1 in 1:1 Mischung), 1,1 µl MgCl2, 1,5 µl Puffer, 0,5 µl BSA, 0,8 µl
dNTPs, 4,1 µl DNA-freies Wasser und 0,6 µl Taq-Polymerase. Die Ansätze von II wurden
auf 30 µl pipettiert, da aus ihnen insgesamt 4 Sequenzierreaktionen folgen sollten. Der
Ansatz II enthielt 10 µl DNA-Extrakt, 2,1 µl MgCl2, 3 µl Puffer, 0,9 µl BSA, 1,5 µl dNTPs,
10,5 µl DNA-freies Wasser und 1,2 µl Taq-Polymerase. Dazu kamen 1,8 µl Primermix
hinzu aus 0,3 µl F16197, 0,3 µl R16509, 0,45 µl F109 und 0,45 µl R460. Alle Primer
hatten eine Konzentration von 200 µM. Das PCR-Programm begann bei 5 Minuten 95°C,
hatte dann die Schritte Denaturierung, Annealing und Elongation in 36 Zyklen bei 20
sec 95°C, 30 sec 45°C und 30 sec 72°C.
Agarose-Gelelektrophorese-Kontrolle:
Zur Amplifikationskontrolle der PCR wurde ein Agarose-Gel benutzt. Hierbei wird ein
1%iges Agarosegel aus 1x TAE-Puffer und Agarose in Kammern gegossen. In deren
Slots wurden, zum einen mit Loading Buffer versetzte PCR-Produkte, und zur
Längendifferenzierung ein Längenstandard separat pipettiert. Der Loading Buffer war
zur besseren Visualisierung beim Einpipettieren mit Bromphenolblau angefärbt und
stabilisiert die DNA während der elektrophoretischen Auftrennung. Um die Fragmente
einer bestimmten Länge zuordnen zu können wurde gleichzeitig zu den DNA–Proben
ein Längenstandard aufgetragen. Da die Proben und die Ladder unter gleichen
Bedingungen liefen, konnten die Fragmente der DNA-Proben somit einer Länge
zugeordnet werden. Zur Kontrolle der 16S PCR wurde 5µl HyperLadder 1 Bioline
benutzt, welche PCR-Produkte zwischen 200 bp und 10000 bp anzeigte. Für das Short-
und Long-Segment wurde 5µl HyperLadder 25bp benutzt, welche PCR-Produkte
zwischen 25 bp und 500 bp anzeigt. In beiden Fällen wurde 1µl 5x sample Loading
Buffer mit 3µl DNA-Extrakt verwendet. Die Proben, die auf dem Gradienten-Cycler
17amplifiziert wurden, also die 16S-Region, welche zur Klonierung und dann als
Standardkurve für die qPCRs genommen werden sollte, wurden mit 18µl DNA-Extrakt
und 6µl Puffer in die Gel-Kammern eingegeben, um einen möglichst hohen Ertrag zu
gewinnen.
Das beladene Gel lief bei 110V und 200mA für circa 45 Minuten in einer
Elektrophoresekammer. Zur Visualisierung unter UV-Licht wurde die DNA durch
Zugabe von Ethidiumbromid zu Agarosegel und Laufpuffer in der
Elektrophoresekammer angefärbt. Zur Aufnahme eines Bildes wurde das System
AlphaEase FC verwendet.
2.7. AUFREINIGUNG PCR-AMPLIFIKAT
Die Aufreinigung der PCR-Produkte zur Sequenzierung als auch zur Klonierung wurde
nach der Amplifizierung enzymatisch durchgeführt. Überschüssige Primer und
einzelne Nukleotide wurden hierbei durch enzymatischen Verdau entfernt. Hierzu
wurde pro 1 µl Probe, 0,26 µl FastAP und 0,14 µl EXOI 10 µM hinzu pipettiert und bei
37°C 30 Minuten verdaut. Um die hinzugegebenen Enzyme danach wieder zu
deaktivieren, wurde die Lösung bei 95°C für 5 Minuten erhitzt.
2.8. KLONIERUNG
Für eine Real-Time PCR benutzt man eine Standardkurve, womit die eingegebenen
Proben verglichen werden. Diese Standardkurve besteht aus Vergleichsproben aus
einer Verdünnungsreihe von bekannter Konzentration. Um eine stabile und
reproduzierbare DNA-Amplifikation zu gewährleisten, wurde in diesem Fall eine
Klonierung der 16S Region in E. coli-Zellen vorgenommen. Dabei wird eine PCR der 16S
Region durchgeführt, wobei die Primer an den 5‘- Enden die Sequenz von Schnittstellen
enthalten, welche später auch im Vektor von einem bestimmten Enzym, diesem Fall
Sfi1, geschnitten werden können. Dies ermöglicht die Ligation beider Stücke mit Hilfe
der produzierten „sticky ends“, der überstehende Enden, welche durch das Schneiden
mit dem Enzym Sfi1 entstanden sind, die sich im folgenden Arbeitsschritt gut
verbinden lassen. In diesem Fall wurde der Primer H3108_Sfi1_neu und
L2025_Sfi1_neu gewählt. Mit diesen modifizierten Primern wurde eine PCR
durchgeführt. Diese funktionierte mit einem Gradienten-Cycler mittels Touchdown-
Programm wie in 2.6. beschrieben. Das amplifizierte 16S Stück mit SfiI-Adaptern
18konnte im Agarose-Gel verifiziert und für die weitere Analyse ausgeschnitten werden.
Um einen möglichst konzentrierten Ertrag zu bekommen, wurden circa 18 μl PCR-
Produkt und 6 μl Loadingbuffer in die Taschen gefüllt. Zwei der stärksten Banden
wurden ausgeschnitten und aufgereinigt. Die Aufreinigung wurde mit dem QIAEX
Purifucation Kit durchgeführt. Hierbei wird das Agarose-Gel-Stück mit dem PCR-
Amplifikat in QX1-Puffer in einem Verhältnis von 3x Puffer zu 1x Gel aufgelöst. Nun
wird 10μl gevortextes QIAEX2 hinzugefügt und unter Schütteln bei 50°C für 10
Minuten bei saurem pH inkubiert. Dabei wird die DNA an QIAEX2 Partikel gebunden.
Die Mischung wurde 30 Sekunden zentrifugiert und der Überstand abpipettiert. Das
übergebliebene Pellet wurde mit QX1 Puffer gewaschen, zentrifugiert und der
Überstand abpipettiert. Dieser Waschschritt entfernt die Agarose-Bestandteile.
Darauffolgend wurde das Pellet zweimal mit PE-Buffer gewaschen, zentrifugiert und
der Überstand abpipettiert. Hierbei werden Salzkontaminationen entfernt. Das Pellet
mit der DNA wurde für 15 Minuten luftgetrocknet und danach mit DNA freiem Wasser
für 5 Minuten gelöst. Nach erneutem Zentrifugieren erhält man gereinigte DNA im
Überstand.
Sowohl das PCR-Produkt als auch der gewählte Vektor wurden dann getrennt
voneinander mit dem Restriktionsenzym verdaut und die bereits beschriebenen
„sticky ends“ bildeten sich an deren Ende. Als Restriktionsenzym wurde Sfi_1 gewählt,
welches auf dem Vector pAc1 schneidet. Es wurden jeweils 1 µl Sfi_1, 5µl Buffer Cut
smart und 14 µl H2O mit 1) 30 µl PCR Produkt und
2) 14 µl pAc1
für 4 Stunden bei 50°C verdaut. Am Ende dieses Schrittes erhält man ein PCR-Produkt,
in diesem Fall die 16S Region mit den Enden die von Sfi_1 erkannt und geschnitten
wurden, sowie den gewählten Vektor, welcher ebenfalls an der gewünschten Stelle
geschnitten wurde.
Daraufhin wurden beide Ansätze mittels einer Säulenaufreinigung der GenElute
Miniprep Binding Column mit 15 µl Elution Buffer aufgereinigt. Hierdurch wurde die
DNA wieder von anderen Inhaltsstoffen getrennt.
Die Ligation der oben beschriebenen „sticky ends“, also der passenden Enden des PCR-
Produkts mit dem Vector, erfolgte mit 6 µl PCR-Produkt, 2 µl Vector, 1 µl
Ligationspuffer und 1µl Ligase bei 14°C für 2 Stunden. Durch die Ligation erhält man
19das modifizierte Plasmid, also einen Vektor mit der eingebauten gewünschten Sequenz,
dem PCR-Produkt.
Das entstandene Plasmid-Produkt konnte nun durch Transformation in E. coli Zellen
überführt werden. Dabei wurden 5 µl Plasmid-Produkt mit 100 µl E. coli Zellen zuerst
leicht vermischt und 10 Minuten auf Eis inkubiert und dann bei 42°C für 90 Sekunden
aufgeheizt. Nun wurden 700 µl Lysogeny Broth-Medium ohne Antibiotika zu dem Mix
hinzugefügt und die Zellen für eine Stunde bei 37°C und 1000rpm im Thermoschüttler
geschüttelt. Daraufhin wird der Mix durch Zentrifugieren und durch Abpipettieren des
Überstandes höher konzentriert und auf LB-Agarplatten (Bertani G. 1951) mit
Antibiotikum ausgestrichen. Die Platte wird über Nacht bei 37°C inkubiert. Am
nächsten Tag wurden 25 Kolonien gepickt und zuerst in einen PCR-Mix mit den bereits
vorher genutzten Primern H3108_Sfi1_neu und L2025_Sfi1_neu gemischt und dann auf
einer weiteren Agarplatte ausgestrichen. Am folgenden Tag konnte man durch die PCR
sehen, welche Kolonien die ausgewählte DNA-Sequenz aufgenommen hatten. Diese
Zellen wurden wiederum in Flüssignährmedium (LB) angesetzt und bei 37°C über
Nacht inkubiert. Nun konnte die eigentliche Aufreinigung des Plasmids mittels Mini-
Prep erfolgen. Die Zellen wurden dabei in Resuspensions-Puffer gelöst und mit Lyse-
Puffer lysiert. Diese Lösung wurde wiederum neutralisiert und mittels
Säulenaufreinigung die DNA herausgetrennt.
2.9. REAL TIME PCR
Zu beachten ist zu zunächst, dass in der Real-Time-PCR nicht wie in der PCR das Short-
und Complete-Fragment amplifiziert wurden, sondern das Short- und Long-Fragment.
Allgemein: Es wurden insgesamt 131 Proben mittels Echt-Zeit PCR auf ihre mtDNA-
Quantität untersucht. Bei der Erstellung des Real-Time-Quantifizierung Designs
wurden die MIQE Guidelines von (Bustin 2013) im Versuchsaufbau berücksichtigt.
Das Prinzip einer Real-Time PCR beruht auf dem gewöhnlichen Ablauf einer PCR
(Denaturierung, Annealing, Extension). Während der Extension interkaliert der
Farbstoff SYBR Green in die doppelstränigen DNA Amplifikate und gibt ein
fluoreszierendes Signal ab. Die PCR Reaktion wiederholt sich 40 Zyklen, sodass sich die
Menge der Proben-DNA und somit auch die Intensität des Farbsignals, bei jedem
Zyklus der exponentiellen Phase, vervielfacht. Um die gemessenen Proben einem DNA-
20Masse-Wert zuordnen zu können, wird eine Standardkurve angelegt. Hierbei werden
6 Proben mit bekannter DNA-Konzentration zu einer Verdünnungsreihe hergestellt
und bei jedem Lauf ebenfalls gemessen. Unbekannte Proben werden dann mit der
jeweils laufeigenen generierten Standardkurve verglichen und hierdurch die DNA-
Menge der Probe extrapoliert bestimmt. Dabei ist es wichtig, dass die Stammlösung für
den Standard aus aufgereinigter DNA besteht und eine geeignete Referenzsequenz
enthalten ist. Da man den gleichen Stock für die Standardkurven verschiedener
Analyseläufe verwendet, sind die Ergebnisse vergleichbar. Hierfür ist eine stabile
Lagerungsmöglichkeit notwendig wie beispielsweise in Plasmid-DNA eingebettet und
tiefgefroren. In dem benutzen qPCR-Kit ist eine ROX Referenz (Jordan, L., Kurtz, R.
2010) bereits enthalten. Dieser Referenzfarbstoff normalisiert das Fluoreszenzsignal.
Die Lichtquelle des Messinstrumentes hat einen unterschiedlichen
Einstrahlungswinkel je nach Tube. Dadurch kommen verschiedene Lichtwege und
Intensitäten beim Detektor an. Diese Variationen in der Messung werden durch die
ROX Referenzfluoreszenz ausgeglichen, da die Unterschiede in der ROX Fluoreszenz
mitbestimmt und von der Messung der SYBR Green Fluoreszenz abgezogen. Somit
können Messungenauigkeiten korrigiert werden. Der SYBR Green Farbstoff
interkaliert unspezifisch mit jeder Art doppelsträngiger DNA, weswegen anschließend
eine Schmelzkurvenanalyse (Ririe et al. 1997) durchgeführt wurde. Dabei erhöht sich
die Temperatur schrittweise (meist 0,5°C-Schritte) und die DNA trennt sich abhängig
von ihrer Sequenzlänge voneinander in einzelsträngige Fragmente. Anhand der
Temperaturkurve und der Temperaturspitze einer Kurve können dann Rückschlüsse
auf die Länge der amplifizierten DNA-Fragmente gezogen werden. Somit kann
kontrolliert werden, ob das gewünschte Fragment und in diesem Fall auch, welches der
beiden Fragmente stärker amplifiziert wurde.
Dieser Assay wurde im halben Ansatz durchgeführt und gegen den vollen Ansatz
validiert. Der gewählte SYBR Green Mastermix ist für beide Ansatzmengen vom
Hersteller optimiert.
Die Real-Time PCR wurde für eine Konzentrationsspanne von 10 pg/µl – 0,0001 pg/µl
optimiert. Dies ist die niedrigste Konzentrationsspanne, welche noch verlässliche
Werte in wiederholten Messungen ergaben und realistisch bei degradierten
Tatortspuren, z.B. ausgefallene oder ausgerissene Haare oder Hautschuppen
gewonnen werden können. Es wurden insgesamt 25 qPCR Läufe durchgeführt wovon
2117 eine Effizienz von mindestens 80% aufwiesen. Davon wurden wiederum 15 in diese
Versuchsreihe eingeschlossen, da zwei Läufe Kontaminationen in der Negativ-
Kontrolle aufwiesen. Die Werte der Proben lagen zwischen 0,001 ng/µg und 0,000 000
009 ng/µl (9E-09ng/µl), wobei Konzentrationen kleiner 0,000 09ng/µl unterhalb des
niedrigsten Punktes der Standardkurve S5 liegen und somit nur extrapoliert sind. In
die Bewertung der Probenergebnisse wurden also nur Proben von Läufen mit
einbezogen, deren Negativ-Kontrollen leer waren und deren Standardkurve eine
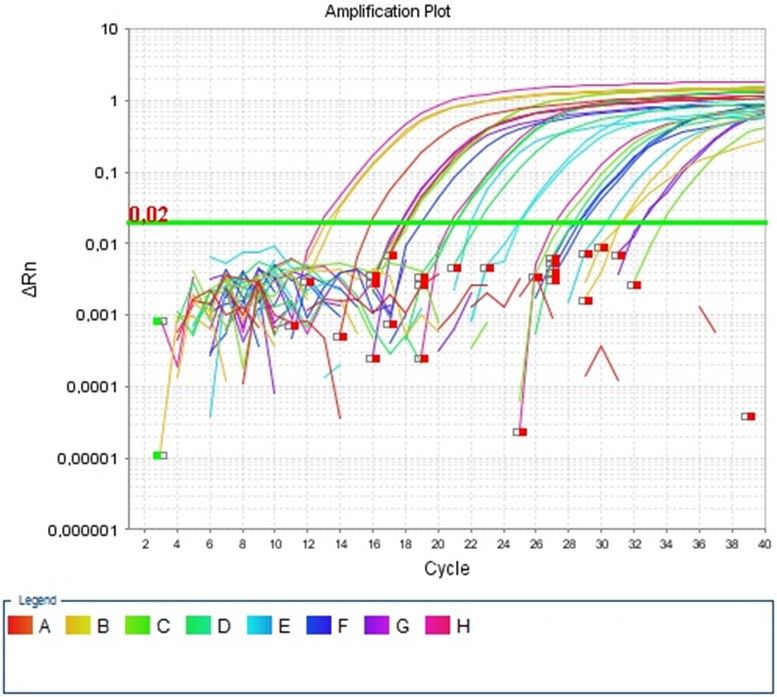
Effizienz oberhalb des Threshold von 0,02ΔRn aufwies.
Standardkurve: Da man, wie bereits erwähnt, nicht von der Fluoreszenzstärke direkt
auf eine DNA-Konzentration schließen kann, bedarf es Referenzwerte. Diese müssen
vorher bekannt sein und in Verdünnungsreihen in mindestens 5 verschiedenen
Konzentrationen mitanalysiert werden. Da es sich bei den DNA-Sequenzen um
mitochondriale DNA handelt, ist es wichtig, dass die Standardreihe eine konservative,
also mutationsunanfällige mtDNA-Sequenz enthält, auf dem die zu amplifizierenden
Stücke erhalten sind. Dies ist hier durch die Klonierung der 16S Region in ein Plasmid
als Ausgangsmaterial für die Standardkurve gelungen. Die komplette
Plasmidamplifizierung ist im Absatz 2.b.v. beschrieben. Daraus erhielt man circa 300μl
aufgereinigtes pAc1-Plasmid, welches zwischen den Sfi1-Schnittstellen (2525) die
Sequenz der 16-S-Region enthält. Die Konzentration dieser DNA in Lösung wurde
erstmals mit dem Nanodrop gemessen. Für diesen Assay wurden Konzentrationen
gewählt, welche in der forensischen Spurenanalyse bei schwierigen und degradierten
Proben häufig vorkommen.
Zur Herstellung der Standardreihe wurde die Stammlösung der Klonierung mit
31ng/µl 16S-Region in 0,01ng/µl konzentrierte Stocklösungen aliquotiert. Diese
dienten dann als S1 Verdünnung, woraus dann jedes Mal eine neue Verdünnungsreihe
aus 5 µl S1 Lösung mit 45 µl DNA-freiem Wasser hergestellt wurde. Dies ergab die S2
Lösung, welche dann wiederum zehnfach verdünnt zur S3 Lösung wurde.
S1 - 10pg
S2 - 1pg
S3 - 0,1pg
22Sie können auch lesen

















































