Mikroskopie 2: Fluoreszenzmikroskopie - Biofizika
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Mikroskopie 2:
Fluoreszenzmikroskopie
Prof. Dr. Dietmar J. Manstein
Medizinische Hochschule Hannover, Fritz-Hartmann-Centre for Medical Research
Director, Institute for Biophysical Chemistry, MHH
Director, Division of Structural Biochemistry, MHH
Director, Core Unit Laser Microscopy, MHH
Founding Director, Centre for Structural Systems Biology, Hamburg
Medizinische Hochschule Hannover, Carl-Neuberg-Str. 1, 30625 Hannover, Germany
Web: www.mh-hannover.de/bpc.html; www.cssb-hamburg.de/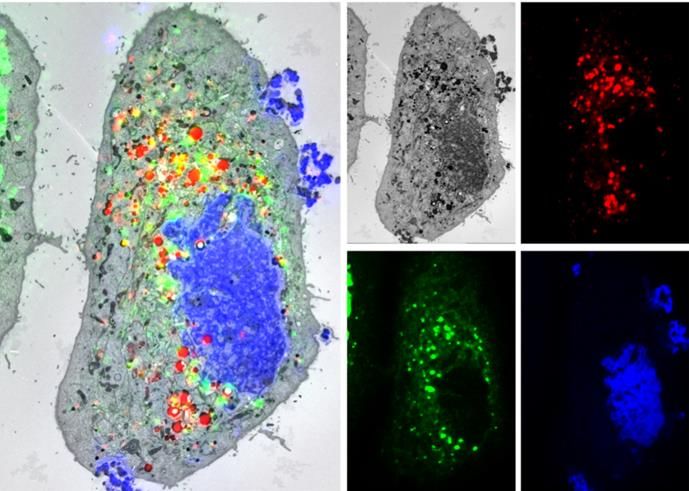
Themen • Fluorophore • Aufbau eines Epifluoreszenzmikroskops • Evaneszentfeldmikroskopie • Konfokalmikroskopie • Multiphotonenmikroskopie • Nanoskopie – Höchstauflösungsmikroskopie • Korrelative Lichtelektronenmikroskopie
Fluoreszenz • Die Fluoreszenz ermöglicht es, Organellen, Zellen und Gewebe mit großer Spezifität, Empfindlichkeit und mit hohem Kontrast zu markieren. • Der molare Extinktionskoeffizient beschreibt das Absorptionspotential des Fluorophors. • Die Fluoreszenzquantenausbeute ist das Verhältnis der Anzahl der emittierten Fluoreszenzphotonen zur Anzahl der absorbierten Photonen. • Die Helligkeit eines Fluorophors ergibt sich aus dem Produkt aus Quantenausbeute und Extinktionskoeffizient. • Die örtliche Umgebung - insbesondere der pH-Wert - kann die Absorptions- und Emissionsspektralprofile von Fluorophoren stark beeinflussen. • Moderne Fluoreszenzmikroskope werden mit einer Epifluoreszenz- beleuchtungseinheit gebaut. Das Objektiv fungiert dabei als Kondensor.
Glossar • Lumineszenz ist die Erzeugung von Licht durch Kaltkörperstrahlung. • Photolumineszenz ist die Absorption von Licht einer Wellenlänge gefolgt von der Emission bei einer längeren Wellenlänge. • Die Größe der dabei beobachteten Stokes-Verschiebung - der Differenz zwischen den Positionen der Bandmaxima des Absorptions- und des Emissionsspektrums - bestimmt, wie leicht das Anregungsspektrum des Fluorophors vom emittierten Signal getrennt werden kann. • Bei der Chemolumineszenz wird Licht durch eine chemische Reaktion erzeugt. • Die Fluoreszenz hört innerhalb von Nanosekunden nach Unterbrechung der externen Photonenquelle oder durch Photobleaching des Fluorophors auf. • Ein Jabłoński Diagramm veranschaulicht die elektronischen Zustände eines Moleküls und die Übergänge zwischen ihnen.
Das Jabłoński Diagramm beschreibt die verschiedenen
Rekombinationsarten, die in fluoreszierenden Medien auftreten
- Die Anregung eines Elektrons erfolgt immer aus dem Grundzustand S0
- Die Rekombination und damit die Emission eines Photons erfolgt immer
aus dem niedrigsten vibronischen Zustand (S1)
- Die Rekombination passiert in einen beliebigen Zustand S0
n
- Übergänge in den Triplettzustand sind möglich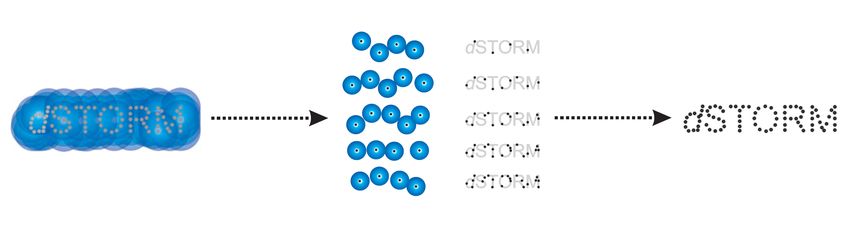
Fluorophore
Gruppen von Fluoreszenzmarkern
Organische Farbstoffe
Fluorescein
Fluoreszierende Proteine
KillerRed protein absorption and emission spectra
Halbleiternanokristalle - Quantendots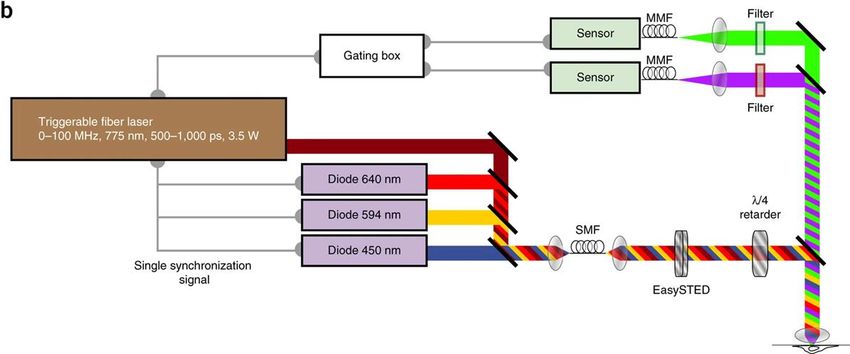
Designprinzipien in der Fluoreszenzmikroskopie
Transmissionsdesign Epifluoreszenzdesign
Detektor
Lichtquelle
Emissionsfilter
Kondensor
Lichtquelle
Anregungsfilter
Objektiv
Probe
Emissionsfilter
Moderne Epifluoreszenzmikroskope enthalten
austauschbare Filterwürfel die jeweils drei
Detektor unterschiedlichen Typen von Filter enthalten:
1: Anregungsfilter
2: Dichroitischer Spiegel
3: Emissionsfilter
Epifluoreszenzmikroskopie
• Moderne Fluoreszenzmikroskope verfügen über eine Epi-
Beleuchtung.
• Das Objektiv fungiert bei der Epi-Beleuchtung gleichzeitig
als Kondensor.
• Die Lichtausbeute im Epi-Fluoreszenzmikroskop ist
proportional zu N.A.4/Mag2.
https://www.laser2000.de/de/filter/20230-simulation-eines-fluoreszenzmikroskopie.html © THORLABS.com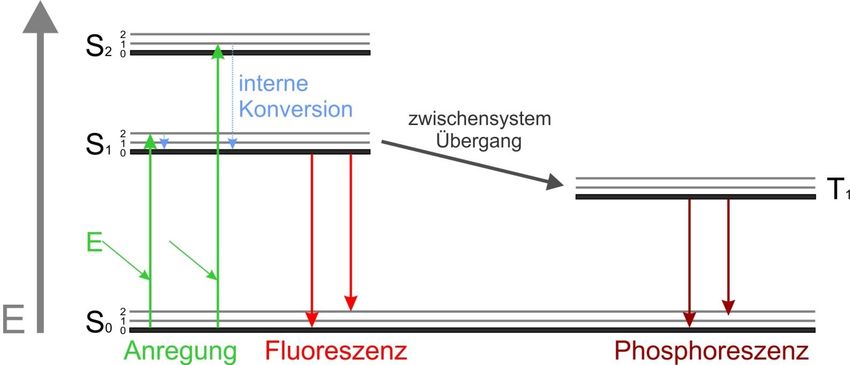
Strahlengang und Probenbeleuchtung in verschiedenen
Arten der Fluoreszenzmikroskopie
Epifluoreszenz Konfokal Evaneszenz - TIRF
Evaneszenz-Wellen-Mikroskopie - TIRF
Toomre D, Manstein DJ. Lighting up the cell surface with evanescent wave microscopy.
Trends Cell Biol 2001; 11(7): 298-303.Evaneszenz-Wellen-Mikroskopie - TIRF
Toomre D, Manstein DJ. (2001)
Fluoreszenzmarkierte Mikrotubuli in Dictyostelium discoideum Zellen mit einem Zytokinesedefekt
Images courtesy by Dr. G. Gerisch, Max-Planck-Institut für Biochemie, MartinsriedLaser-Scanning-Mikroskopie
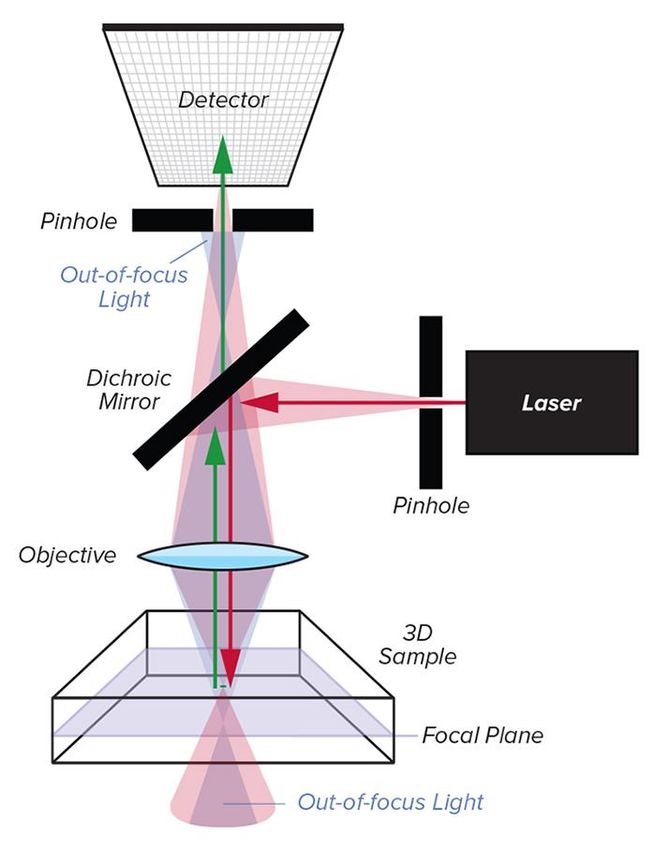
• In der Standard-Epifluoreszenzmikroskopie wird das
gesamte Volumen der Probe beleuchtet.
Fluoreszenzlicht das nicht in der Fokusebene erzeugt
wird reduziert die Auflösung und überdeckt feine
Details, wenn es vom Detektor erfasst wird.
• Eine verbesserte Abbildungsauflösung wird in der
konfokalen Mikroskopie erreicht, indem Fluoreszenz
von außerhalb der Fokusebene durch Hinzufügen
von zwei Lochblenden eliminiert wird.
• Die erste Lochblende wird vor der Anregungsquelle
platziert, um einen beugungsbegrenzten Punkt in
der Probe anzuregen. In den meisten modernen
konfokalen Mikroskopen wird diese
Anregungsblende durch eine Einmoden-
Lichtwellenleiter ersetzt.
• Eine zweite Lochblende vor dem Detektor
verhindert, dass nicht fokussiertes Fluoreszenzlicht
den Detektor erreicht.Laser-Scanning-Mikroskopie
• Zu jedem gegebenen Zeitpunkt sammelt der
Detektor (Photomultiplier-Röhre) nur Licht
aus dem Fokalpunkt.
• Zweidimensionale Schnitte werden durch
Abtasten des Anregungslichts in X und Y mit
einem oder mehreren motorisierten
Schwingspiegeln ("Galvanometer-Spiegel-
Scanner") erfasst.
• Dreidimensionale Bilder werden durch die
Aufnahme und rechnerische Stapelung
aufeinanderfolgender Bildebenen erzeugt.
http://www.microscopist.co.uk/essential-techniques/confocal-microscopy/Mehrfarben-Fluoreszenzmikroskopie
Beleuchtungseinheit Probenträger & Objektiv Detektoreinheit
Typische Effizient von CCDs
Objektive sind auf die Standard-
Deckglasstärke von 0,17 mm korrigiert.
Fokus-Fehler (zufällige Einheiten)Multiphotonenmikroskopie
• Die Multiphotonenmikroskopie ist eine Technik, die in lebendem Gewebe bis zu einer
Dicke von etwa einem Millimeter fluoreszenzmikroskopische Aufnahmen mit hoher
Auflösung ermöglicht.
• Die λ−4-Abhängigkeit der Lichtstreuung bedeutet, dass kürzere Wellenlängen viel
stärker gestreut werden als längere.
• Bilder werden erzeugt, indem eines von zwei unterschiedlichen physikalischen
Phänomenen ausgenutzt wird:
Multiphotonen-Fluoreszenz
Second Harmonic Generation (SHG) oder
Third Harmonic Generation (THG)
Verdopplung oder Verdreifachung der
Schwingungsfrequenz des eingestrahlten
Lichtes.Multiphotonenmikroskopie
• Zwei Photonen, die nur jeweils etwa die Hälfte der zur Anregung des Moleküls
erforderlichen Energie besitzen, können bei ausreichend hoher Photonendichte einen
Fluorophor in einem Quantenereignis anregen.
• Femtosekundenlaser mit Spitzenleistungen von Mega- bis Terrawatt pro cm2 und
Wiederholungsrateb > 80 Mhz werden verwendet, um den erforderlichen hohen Flux
an Anregungsphotonen im Fokusvolumen (1 Femtoliter) zu erreichen.
• Die Wahrscheinlichkeit einer Zwei-Photonen-Anregung ist umgekehrt proportional zur
PulsdauerMultiphotonenmikroskopie
• Da die Absorption von zwei Photonen und die effiziente Anregung auf das winzige
Fokusvolumen beschränkt sind, werden keine Hintergrundsignale erzeugt.
• Dies ermöglicht die Verwendung von sogenannten non-descanned Detektoren ohne
Lochblende wodurch die Effizienz der Lichtdetektion erheblich gesteigert wird.
• Photobleaching erfolgt nur im Fokusvolumen.
Die Anregung und Emission
durch 2 langwellige Photonen
ist auf den Fokus beschränkt.
Die Anregung durch einzelne
kurzwellige Photonen führt zu
Absorption und Emission
entlang des gesamten Strahlen-
ganges.
https://www.univie.ac.at/mikroskopie/3_fluoreszenz/fluoreszenz_mikroskop/5d_multiphoton.htm
Normale Fluoreszenz Multiphoton Fluoreszenz
1 kurzwelliges Anregungsphoton ≥ 2 langwellige Anregungsphotonen
1 langwelliges Emissionsphoton 1 kurzwelliges Emissionsphoton
Anregungsenergie > Emissionsenergie Anregungsenergie > EmissionsenergieNanoskopie (Höchstauflösungsmikroskopie) • Unter den Begriffen „Nanoskopie“ oder „Höchstauflösungs- mikroskopie“ fasst man alle mikroskopischen Verfahren zusammen, die in der Lage sind unterhalb der Beugungsgrenze aufzulösen. • Alle Methoden verwenden dabei Techniken, um Fluorophore durch unterschiedliche Zustände unterscheiden zu können. Dies können beispielsweise der S0- und S1- oder cis- und trans- Zustand sein oder die Unterscheidung erfolgt durch verschiedene Hell- und Dunkelzustände.
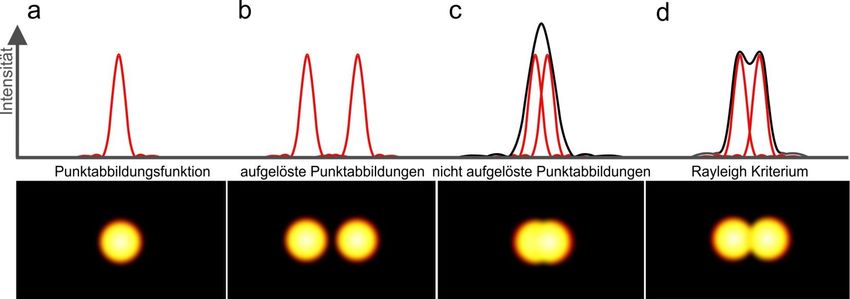
Auflösungsvermögen – Rayleigh Kriterium Rayleigh Kriterium: Zwei beugungsbegrenzte Punkte können noch als zwei einzelne Punkte unterschieden werden, wenn das Intensitätsmaximum des zweiten Punktes mindestens in das erste Intensitätsminimum des ersten Punktes fällt.
Grundlegende Prinzipien der STED-Mikroskop (STED = Stimulated Emission Depletion) Bei der STED-Mikroskopie wird ein Laserstrahl für die Anregung der Fluorochrome in das Präparat fokussiert. Gleichzeitig wird in die Außenbereiche des Fokus ein Ring aus Abregungslicht gelegt, sodass spontanes Fluoreszenzlicht nur aus einem zentralen Bereich abgestrahlt wird, der kleiner ist als der beugungsbegrenzte Anregungsfokus. Der STED-Effekt ist zunächst auf eine Stelle im Präparat begrenzt. Diese Stelle wird daher wie bei anderen Laser-Scanning-Mikroskopen über das Präparat gerastert, um zwei- oder dreidimensionale Bilder zu erzeugen.
Mehrfarben-STED
Fluorophore mit großer Stokes-Shift erlauben die Verwendung eines einzigen STED-
Strahls für die Mehrfarbenanregung. Triggerbare Subnanosekunden-Single Mode-
Faserlaser macht die Pulsvorbereitung überflüssig. Die Detektion mittels Time-Gating
verbessert die Fluoreszenzabreicherung. Durch die Zusammenführung aller Strahlen in
eine SMF-Faser ist das System per Design justiert.
Vicidomini, G., Bianchini, P. & Diaspro, A. STED super-resolved microscopy. Nat Methods 15, 173–182 (2018).Lokalisationsmikroskopie - Prinzip
Detektiertes Positionen der
Signal einzelnen Emitter
Beugungsbegrenztes Bild Detektion der einzelnen Resultierendes
Emitter mit computer- hochaufgelöstes Bild
gestützter Bestimmung des
MaximumsEntscheidungsfindung zur Auswahl von
Nanoskopie-Techniken
Schermelleh L, Ferrand A, Huser T, et al. Super-resolution microscopy demystified. Nat Cell Biol 2019; 21(1): 72-84.Korrelative Lichtelektronenmikroskopie (CLEM)
• CLEM bezieht sich auf computergestützte Kombinationen von Fluoreszenz- und
Elektronenmikroskopie.
• CLEM und cryoCLEM werden verwendet, um biomarkerverstärkte Informationen
auf verschiedenen Längenskalen zu erhalten.
• Die Fluoreszenzmikroskopie liefert Informationen über die Lokalisierung von
Proteinen und Organellen und definiert Regionen von Interesse.
• Die Elektronenmikroskopie liefert hochaufgelöste Informationen.
• In integrierten CLEM-Systemen erfolgt die Aufnahme der Probe simultan mit
einem Elektronenstrahl und über einen optischen Lichtweg.
Schematische Darstellung der Realisierungen für integrierte Korrelative Licht- und Elektronenmikroskopie menschlicher
Fluoreszenzmikroskopie innerhalb von ((a), (b)) SEMs ((c), (d)) Hepatomzellen, die ein GFP-markiertes virales Protein (grün)
TEMs. Der Elektronenstrahl ist grün, der Lichtstrahl blau und ein EGFP-markiertes zelluläres Markerprotein produzieren;
dargestellt. Lipidtröpfchen (rot); Kern (blau).
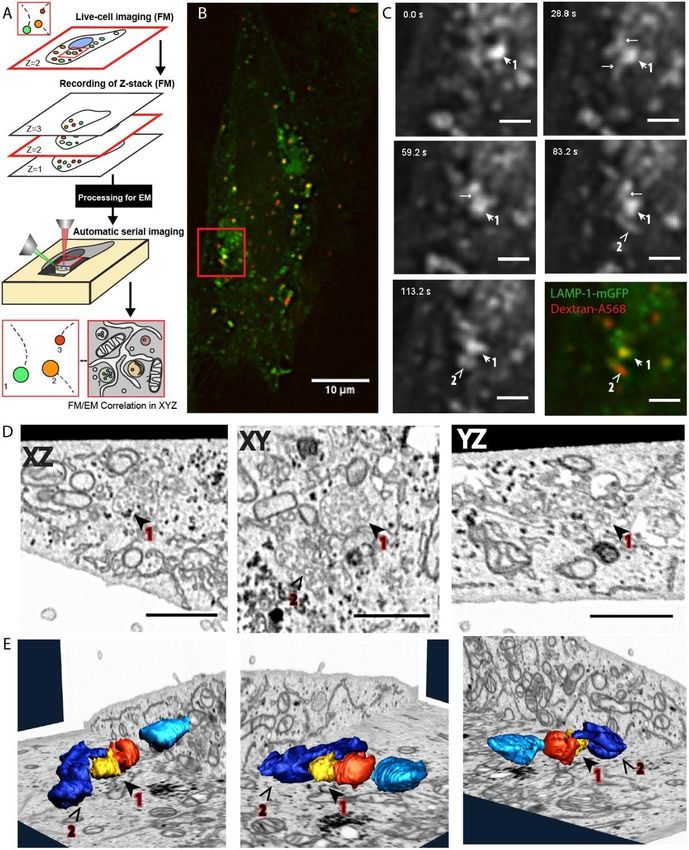
Toshio Ando et al. J. Phys. D: Appl. Phys. 51 (2018) 443001 (42pp) © SFB 1129 Mirko Cortese and Volker LohmannVolumen-CLEM bietet eine direkte Verbindung zwischen der Dynamik lebender Zellen und der 3D-Ultrastruktur auf der Ebene einzelner Organellen (A) Schematische Darstellung des Volumen- CLEM-Workflows. (B) Fluoreszenzbild von Zellen, die GFP- markiertes Lysosom-assoziiertes Membranglykoprotein 1 produzieren, nach Inkubation mit dem Endocytosemarker Dextran-Alexa568. (C) Lebendzellfluoreszenzmikroskopie gefolgt von einer in-situ-Fixierung. (D) Zeigt Schnitte des rekonstruierten Focused Ion Beam SEM (FIB-SEM) Datensatzes entlang der drei Betrachtungsachsen (XZ/XY/YZ), der die ROI der lebenden Zelle enthält ((B), (C) rotes Quadrat). Die beiden Spots 1 und 2 werden als späte Endosomen klassifiziert. (E) FIB-SEM-Segmentierung und 3D- Rekonstruktionen der Spots 1 und 2; die in der Lebendzell-Fluoreszenzmikroskopie abgebildeten Organellen (1,2) wurden segmentiert und mit den Referenz- Fluoreszenz Mikroskopie Daten korreliert.
Danke für Ihre Aufmerksamkeit!
Sie können auch lesen

















































