Molekülspektroskopisches Praktikum - SS 2020 Infrarot- und UV-Spektroskopie Flüssig-NMR-Spektroskopie Massenspektrometrie 1 6 20 24
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Molekülspektroskopisches Praktikum SS 2020 Infrarot- und UV-Spektroskopie 1–6 Flüssig-NMR-Spektroskopie 7 – 19 Massenspektrometrie 20 – 24
Infrarot-Spektroskopie 1. Vorbereitung Voraussetzung für die Durchführung des Praktikums ist die Grundlagenkenntnis von UV/Vis - und IR-Spektroskopie sowie von den Prinzipien der angeführten Geräte und Methoden. Insbesondere wird auf die folgenden Punkte Wert gelegt: elektromagnetische Wellen und ihre Messgrößen Wechselwirkung elektromagnetischer Wellen mit Materie praktikumsrelevante physikalische Modelle (z.B. harmonischer Oszillator, starrer Rotator) Herleitung und Besonderheiten des Lambert-Beerschen Gesetzes Aufbau und Funktionsweise eines optischen Spektrometers (dispersive Spektrometer und Fourier-Transform-Spektrometer mit Michelson-Interferometer) praktische Durchführung einer IR-Messung (Probenvorbereitung, Küvettenmaterial, Lösungsmittel, Messbedingungen) ATR-Messprinzip (Abgeschwächte Totalreflexion) Auswertung von Schwingungs- und Rotationsschwingungs-Spektren Literaturempfehlung: [1] C.N. Banwell, E. M. McCash "Molekülspektroskopie: Ein Grundkurs" R. Oldenbourg Verlag, 1999 [2] M. Hesse, H. Meier, B. Zeeh, "Spektroskopische Methoden in der Organischen Chemie", 8. überarbeitete Aufl., Thieme, Stuttgart, 2011 [3] W. Gottwald, G. Wachter "IR-Spektroskopie für Anwender" Wiley-VCH Weinheim, 1997 u.a. Bitte bringen Sie zum Praktikum neben Schreibmaterial auch Schutzkleidung (Kittel, lange Hose, geschlossenes Schuhwerk) und Schutzbrille mit. Praktikumsort ist das Technikum/Analytikum Linnéstr. 3, Raum 606. Benutzen Sie bitte das hintere Treppenhaus am Hofeingang. 1
2. Aufgabenstellungen für das Praktikum 2.1. UV/Vis-Spektren von der zu identifizierenden und von der gemeinsamen Substanz Nehmen Sie von der zu identifizierenden Substanz ihrer Gruppe ein UV/Vis-Spektrum im Bereich 200 bis ca. 700 nm auf. Stellen Sie dazu eine geeignete Verdünnung der Probe her. Als Lösungs- mittel steht abs. Ethanol (in UV/Vis-reiner Qualität) bereit. Das UV/Vis-Spektrum der gemeinsamen Substanz für alle Gruppen bekommen Sie als pdf-Datei ausgehändigt. Berechnen Sie daran für die angegebenen Wellenlängen jeweils den molaren Extinktionskoeffizienten (in der Einheit L/(mol*cm). Alle benötigen Angaben finden Sie auf dem Spektrum. Beide Spektren sind bezüglich des Gehalts an Strukturinformationen zu diskutieren. 2.2. IR-Spektren von der zu identifizierenden und von der gemeinsamen Substanz Nehmen Sie das IR-Spektrum von ihrer zu identifizierenden Substanz im Bereich von 400 bis 4000 cm-1 auf. Verwenden Sie die Dünnfilmtechnik (Flüssigkeitsfilm zwischen zwei KBr-Platten) für flüssige Proben. Von Feststoffen ist ein KBr-Pressling herzustellen. Das IR-Spektrum der gemeinsamen Substanz für alle Gruppen bekommen Sie als pdf-Datei aus- gehändigt. Klassifizieren Sie die Substanzen mit Hilfe von IR-Absorptionstabellen, z. B. in [2]. Diskutieren Sie die gefundenen Absorptionsbanden beider Spektren und geben Sie einen Strukturvorschlag für die zu identifizierende Substanz ihrer Gruppe an. 2.3. Wirkung von Massenänderung auf die Lage von Schwingungsbanden Untersuchen Sie den Einfluss der Masse auf die C-H-Valenzschwingung am Beispiel von Aceton. Vergleichen Sie dazu die Lagen der Schwingungsbanden der C-H- bzw. der C-D- und der C=O-Valenzschwingung von „normalem“ Aceton (Aceton-D0, CH3-CO-CH3) und Deuterium- substituierten Aceton (Aceton-D6, CD3-CO-CD3). Berechnen Sie die Verhältnisse der Wellenzahlen der C-H- und der C-D-Valenzschwingung nach dem Modell des harmonischen Oszillators (unter Annahme von gleichen Kraftkon- stanten). Vergleichen Sie Ihr Ergebnis mit dem Verhältnis der gemessenen Wellenzahlen. Ist die An- wendung des einfachen Modells sinnvoll? 2
2.4. IR-Spektren von amorphen Feststoffen – ATR-Technik Messen Sie die IR-Spektren zweier Polymere. Die Proben liegen als feste Folien/Streifen vor, bei denen übliche Transmissionsmessungen nicht möglich sind. Die IR-Spektren sind mit der ATR- Technik aufzunehmen. Identifizieren Sie anhand des IR-Spektrums charakteristische Banden und nehmen Sie eine möglichst genaue Bestimmung der vorliegenden Kunststoffe vor (siehe auch Flußdiagramm im Anhang). 2.5. Rotationsschwingungsspektren von Gasen Es soll das Rotationsschwingungsspektrum von Kohlenmonoxid ausgewertet werden. Befüllen Sie eine Gasküvette mit dem Rauch einer Filterzigarette. Dazu stehen Ansatzstücke bereit, mit denen die brennende Zigarette und eine Saugvorrichtung an den Küvettenstutzen angesetzt werden können. Das Gasspektrum muss mit hoher Auflösung (1 cm-1) auf- genommen werden. Die Valenzschwingungsbande von CO ist aus dem Spektrum zu ermitteln. Bestimmen Sie am Spektrum die Rotationskonstante B. Berechnen Sie die Bindungslänge der CO-Bindung anhand des Modells des starren Rotators und die Kraftkonstante anhand des Modells des harmonischen Oszillators. Die Bindungs- länge läßt sich aus dem Trägheitsmoment bestimmen, wenn man ein einfaches Hantelmodell annimmt. Das Trägheitsmoment I des zweiatomigen Moleküls ist dann I µ r2 mit r Abstand zwischen beiden (Punkt-)Massen (d. h. die gesuchte Bindungslänge) µ reduzierte Masse Das Trägheitsmoment I selbst kann man aus der gemessenen Rotationskonstante B ermitteln mit: h B 8 c I 2 mit: h PLANCKsches Wirkungsquantum c Lichtgeschwindigkeit Hinweis: Die Rechnung wird wesentlich erleichtert, wenn man konsequent SI-Einheiten ver- wendet und alle Zahlen in Zehnerpotenz-Schreibweise einsetzt. Protokollieren Sie Ihren Rechen- weg so, daß er nachvollziehbar ist. 3
Berechnen Sie die Kraftkonstante k nach folgender Gleichung: mit: ῦ Schwingungsübergang im CO µ reduzierte Masse Beurteilen Sie die Bindungsstärke von Kohlenmonoxid im Vergleich zu anderen linearen Molekülen anhand der berechneten Kraftkonstante k. Im Folgenden sind beispielhaft einige Moleküle und ihre Kraftkonstanten zum Vergleich angeführt: HCl 481 Nm-1 SO2 1.001 Nm-1 Weitere relevante Moleküle können O2 1.141 Nm-1 aus eigenständiger Literaturrecherche N2 2.242 Nm-1 dem Vergleich zugeführt werden. Bedenken Sie auch die elektronische Struktur der betrachteten Moleküle. Welche Rolle spielen induktive und mesomere Effekte? Wie ist hier Kohlenmonoxid einzuordnen? Zeigen sie mögliche Grenzstrukturen. 2.6. Quantitative IR-Spektroskopie Ein kommerzieller Farbverdünner besteht aus n-Hexan als Hauptkomponente. Als eine der Nebenkomponenten im einstelligen Vol.-%-Bereich ist auch Aceton enthalten. Bestimmen Sie den Volumenanteil einer Lösung von Aceton in n-Hexan. Stellen Sie dazu sieben Kalibrierlösungen mit Volumenanteilen von 1,0 – 4,0% (v/v) Aceton (in 0,5%-Schritten) in n-Hexan her, indem Sie das Aceton mit einer Bürette genau abmessen und dann in einem 10-ml-Messkolben mit n-Hexan auffüllen. Nehmen Sie die IR-Spektren mit Standardauflösung (4 cm-1) im Transmissions-Modus im Bereich 2000 – 1000 cm-1 auf. Das Leer-Spektrum (Background) muss gegen Luft (d.h. ganz ohne Küvette) aufgenommen werden, weil einerseits eine Aufnahme mit Lösungsmittel eine sehr starke Absorption und damit ein schwaches Signal ergibt, was Probleme bei der Quo- tientenbildung (I / I0) verursacht, und sich andererseits die Aufnahme einer Leerküvette wegen Interferenzbildung verbietet (die Schichtdicke ist hier in der Größenordnung der Wellenlänge des Lichtes). Damit steht allerdings für die quantitative Auswertung die benötig- te I0-Intensität nicht zur Verfügung. Die gut erkennbaren Keton-typischen Aceton-Banden liegen bei 1719 cm-1 und 1213 cm-1, die beide einzeln auszuwerten sind. Um die Intensität I0 zu korrigieren, legt man als Grund- 4

linie eine Gerade durch den linken und rechten Fußpunkt der jeweiligen Bande. Als Mess- werte werden die Intensität I an der Spitze der Bande und die Intensität I0 am Schnittpunkt des Lots mit der Grundlinie entnommen. Tragen Sie für die sieben Lösungen die Extinktionen beider Banden in eine Tabelle ein und berechnen Sie je eine Ausgleichsgerade (lineare Regression). Aus dieser bestimmen Sie nach Messung des Farbverdünners dessen Acetongehalt. Schätzen Sie die Fehlergröße Ihrer Messung (Grösstfehler-Rechnung) und geben Sie das Messergebnis mit der richtigen Einheit und unter Beachtung der abgeschätzten Genauigkeit an. Diskutieren Sie mögliche Ursachen des Fehlers und die Auswirkung verschiedener Fehlerquellen auf die Richtigkeit der Kalibrierung. Inwiefern lässt sich anhand der abschätzbaren Fehler eine Aussage über die Richtigkeit des Ergebnisses treffen? 3. Anmerkungen zur Protokollführung Das Protokoll sollte durchgehend lesbar und übersichtlich formatiert sein. Bei Verwendung von Blocksatz ist die Aktivierung der Silbentrennung obligatorisch, um unnötige Leerzeichen zu vermeiden. Alle Betrachtungen sollten möglichst in Text ausformuliert werden. Resultate müssen in einem erkennbaren Abschlusssatz zu finden sein. Protokolle sollten eine Rekonstruktion der Experimente durch dritte Personen ermöglichen. Prüfen Sie bitte die Erfüllung dieser Anforderung. Aufgabenstellung und Durchführung sollten in Kurzform zu Beginn des jeweiligen Teilver- suchs wiedergegeben werden. Grundlegende theoretische Einführungen können vorausgesetzt werden und müssen hier nicht wiedergegeben werden. Die Einleitung sollte lediglich kurze physikalische und technische Grundlagen bereitstellen, die für Verständnis und Durchführung der jeweiligen Versuche notwendig sind. Zu allen abgespeicherten pdf-Spektren wird eine Spektrendiskussion entsprechend der Aufgabenstellung und im Rahmen der jeweiligen Interpretierbarkeit erwartet. Spekulationen sollten ggf. durch zusätzliche Informationen gestützt werden. Auf eine sorgfältige Kenntlichmachung von benutzten Quellen wird Wert gelegt. 5
Flußdiagramm zur Polymeren-Bestimmung 6
Einführung in die NMR-Spektroskopie 1 Übersicht Diese Versuche behandeln die Grundlagen der Flüssig-NMR-Spektroskopie sowie praktische Aspekte bei der Aufnahme von ein- und zweidimensionalen 1H- und 13C-Spektren. Vor der Durchführung des Versuches findet ein Antestat statt, in dem folgende Schwerpunkte besprochen werden: Flüssig-NMR: Grundgleichung NMR, Zeeman-Aufspaltung/Energie-Niveaus, Magnetisierungs-Vektor- Modell, Messprinzip Aufnahme und Verarbeitung des FID Aufbau eines FT (Fourier-Transform)-NMR-Spektrometers, Durchführung der Messung (Probenvorbereitung, Vorbereitung und Durchführung der Messung) Spektrale Parameter eindimensionaler NMR-Spektren: chemische Verschiebung, skalare Kopplung Relaxationsmechanismen Experimente: Wie werden 90°-Pulse bestimmt? Wie funktioniert das Inversion-Recovery- Experiment? 2D-NMR: allgemeiner Ablauf, Informationsgehalt von grundlegenden 2D-Spektren (COSY, NOESY, HSQC/HMQC, HMBC), Auswertung Informieren Sie sich im Vorfeld bitte auch anhand der entsprechenden Literatur über die theoretischen Sachverhalte. Hierbei wird insbesondere [1] und [2] empfohlen, falls die Antestat- Schwerpunkte zum Zeitpunkt Ihres Praktikumstermins noch nicht oder nur teilweise in der Vorlesung besprochen wurden. Es wird erwartet, dass Sie unabhängig vom Stand der Vorlesung in der Lage sind, Fragen zu allen Schwerpunkten sowie die unter 4.7 (Flüssig-NMR) gestellten Fragen zu beantworten. Es werden alle Spektren digital zur Verfügung gestellt. Die Dateien finden Sie auf dem Spektrenserver des Instituts für Analytische Chemie; für den Download ist ein FTP-Programm erforderlich (Adresse des Spektrenservers: spekserv.chemie.uni-leipzig.de, die Zugangsdaten werden Ihnen während des Praktikums mitgeteilt). Die zur Prozessierung und Darstellung der Spektren benötigte Software MNOVA kann unter folgendem Link heruntergeladen werden: http://research.uni-leipzig.de/nmr/MNOVA. Zur Aktivierung wird eine Lizenzdatei benötigt, die per Uni-Mail bei Dr. Findeisen angefordert werden kann. Dieses Programm ist ebenfalls an einem der Rechner im PC-Pool installiert. 7
Literatur: [1] M. Hesse, H. Meier, B. Zeeh, Spektroskopische Methoden in der organischen Chemie, 8. Auflage, Georg Thieme Verlag, Stuttgart, 2012. [2] H. Günther, NMR-Spektroskopie, 3. Auflage, Georg Thieme Verlag, Stuttgart, 1992. [3] H. Friebolin, Ein- und zweidimensionale NMR-Spektroskopie - Eine Einführung, 5. Auflage, Wiley-VCH, Weinheim, 2013. [4] M. Findeisen, S. Berger, 50 and More Essential NMR Experiments, 1. Auflage, Wiley-VCH, Weinheim, 2014. [5] M. Duer, Introduction to Solid-State NMR Spectroscopy, Blackwell Publishing Ltd, Oxford, 2004. 2 Theoretische Grundlagen 2.1 Zeeman-Effekt Kernresonanzspektroskopie (Nuclear Magnetic Resonance) beruht auf der energetischen Aufspaltung der sonst entarteten Kernspinniveaus in einem Magnetfeld. Dieser Effekt wird als Zeeman-Effekt bezeichnet und tritt für alle Kerne auf, die keine gerade Anzahl an Protonen und Neutronen besitzen. Beispiele hierfür sind 1H, 13C, 19F, 29Si oder 31P. Die aufgespalteten Energieniveaus werden bei Kernen mit der Kernspinquantenzahl I = 1/2 als und bezeichnet und ihre Besetzung erfolgt gemäß der Boltzmann-Verteilung: ∆ (1) mit (2) ∆ ℎ ℎ 2 Dabei ist die absolute Temperatur, die Boltzmann-Konstante, das angelegte Magnetfeld, das gyromagnetische Verhältnis und ℎ das Plancksche Wirkungsquantum. 2.2 Resonanzfrequenz Die Energiedifferenz der beiden Besetzungszustände und ist laut Gleichung (2) linear vom Magnetfeld und dem gyromagnetischen Verhältnis des Kerns abhängig. Die daraus resultierende Frequenz wird als Larmorfrequenz bezeichnet. Diese lässt sich veranschaulicht als diejenige Frequenz beschreiben, mit der ein Kernspin um die Richtung des Magnetfelds (z- Richtung) präzessiert. Die Larmorfrequenz kann entsprechend in Hz oder als Kreisfrequenz in rad ∙ s-1 dargestellt werden. rad ∙ s -1 Hz ∙ 2 ∙ (3) 8
Da das angelegte externe Magnetfeld durch die Elektronen am Atomkern abgeschirmt wird, ergibt sich für jeden unterscheidbaren Atomkern ein spezifisches effektives Magnetfeld , was die Resonanzfrequenz eines jeden Kernes ändert und somit zu unterschiedlichen Signalen im Spektrum führt. Diese spezifischen Frequenzen werden gegen die Referenzfrequenz eines Standards, für 1H und 13C meist Tetramethylsilan (TMS), referenziert und ergeben die chemische Verschiebung mit (4) ppm ∙ 10 . 2.3 Das Vektormodell Zur Veranschaulichung der Kernspins kann das Vektormodell herangezogen werden. Es gilt allerdings zu beachten, dass es sich hierbei um eine Näherung handelt, die das Verständnis der NMR vereinfacht, aber nicht alle beobachteten Wechselwirkungen beschreibt. Weiterführende Literatur hierzu finden Sie unter anderem hier: [1], [2]. Das Modell vereinfacht die Gesamtheit aller gleichartigen Kernspins (zum Beispiel alle Protonen) in einer Probe als makroskopischen Vektor. Dieser ist zunächst entlang des äußeren -Feldes ausgerichtet und präzessiert mit der Larmorfrequenz um die z-Achse. Spin-1/2-Kerne können in diesem Modell prinzipiell zwei Orientierungen einnehmen – parallel und antiparallel zur z-Richtung, wobei die positive z-Richtung dominiert. Der Magnetisierungsvektor Mz des Vektormodells ist also im thermischen Gleichgewicht in positiver z-Richtung orientiert. Abb. 1: Ausrichtung des Magnetisierungsvektors im thermischen Gleichgewicht entlang der z-Richtung im starken äußeren Magnetfeld. 9
3 Experimentelle Anforderungen 3.1 Der Aufbau eines FT-NMR-Spektrometers Ein NMR-Spektrometer ist aus den folgenden, wesentlichen Komponenten aufgebaut: Supraleitender Magnet Probenkopf (beinhaltet u.a. die Sender- und Empfängerspulen für 1H- und Heterokerne, wie 13C) HF (high-frequency)-Sender mit Synthesizer HF-Empfänger & Verstärker Steuer-, Bedien- und Ausgabeeinheiten Abb. 2: Schematische Darstellung des Aufbaus eines NMR-Spektrometers (links) und eines Flüssig-NMR-Probenkopfs. 3.2 Eindimensionale NMR-Experimente Ein einfaches eindimensionales Experiment in der NMR-Spektroskopie soll hier beispielhaft beschrieben werden. Ausgangspunkt ist das Vektormodell mit der Gleichgewichtsmagnetisierung entlang des äußeren Magnetfeldes in positiver z-Richtung. Um den Beitrag der Präzession vernachlässigen zu können, bewegt sich der Beobachter ebenfalls mit derselben Frequenz. Man spricht vom rotierenden Koordinatensystem. Für ein NMR-Experiment werden alle Kernspins derselben Sorte (zum Beispiel alle Protonen) mit einem Radiofrequenzpuls (rf-Puls) angeregt. Das geschieht mittels eines senkrecht zum -Feld angelegten schwächeren Magnetfeldes . Dieses zusätzliche Feld wird für eine definierte Dauer tp eingestrahlt, die typischerweise im Bereich von einigen Mikrosekunden liegt. Die 10
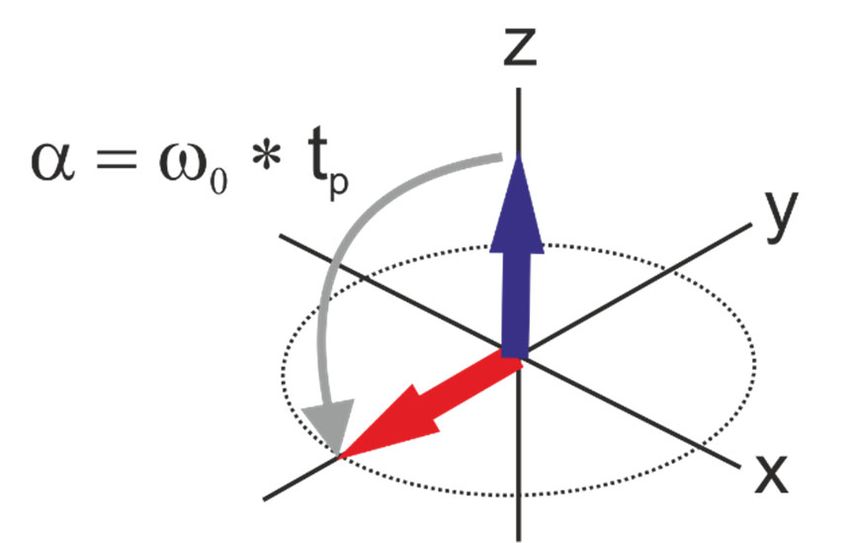
Wechselwirkung der Kernspins mit den beiden Magnetfeldern und kann als Rotation des Magnetisierungsvektors um die Achse der Richtung des zusätzlichen Feldes verstanden werden. Konkret bedeutet das, dass ein Puls aus x-Richtung zur Drehung des Magnetisierungsvektors um die x-Achse von der z-Richtung in Richtung der xy-Ebene erfolgt. Der dabei überstrichene Winkel α berechnet sich aus der Larmorfrequenz der untersuchten Kernsorte und der Pulslänge tp. Abb. 3: Ein Puls aus x-Richtung führt zur Rotation des Magnetisierungsvektors um die x-Achse in der yz-Ebene um den Winkel α. Für die meisten NMR-Experimente sind der 90°- und der 180°-Puls von besonderer Bedeutung. Nach Abschalten des -Feldes kehrt das Spinsystem wieder in den Gleichgewichtszustand zurück. Dieser Vorgang heißt Relaxation (siehe Kapitel 3.3). Gleichzeitig wird in der Empfängerspule, welche sich in der xy-Ebene befindet, durch die Präzession der Kernspins ein Induktionsstrom gemessen. Dieses Signal wird als freier Induktionsabfall (free induction decay, FID) bezeichnet (Abb. 4). Die Amplitude des FID beschreibt die Abnahme der Quermagnetisierung My in Abhängigkeit von der Acquisitionszeit (aq), seine Frequenz stellt alle beobachteten Larmorfrequenzen in der Probe dar. Durch die Fourier-Transformation wird der FID von der Zeitdomäne in die Frequenzdomäne umgerechnet. Abb. 4: Schematische Darstellung des Free Induction Decay (FID). 11
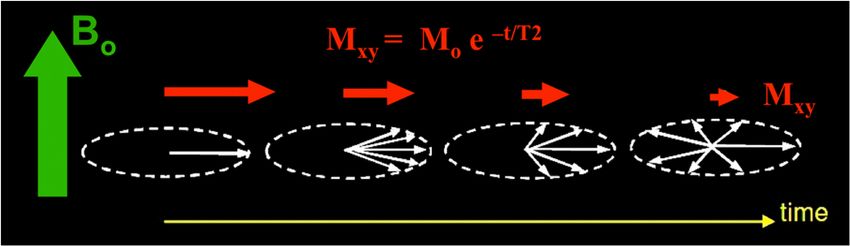
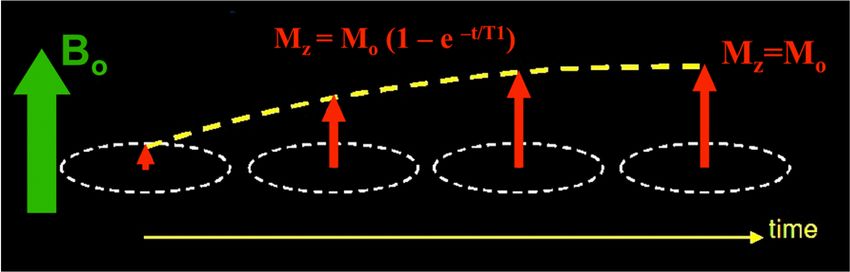
Zur Durchführung eines NMR-Experiments müssen die jeweiligen Messparameter (acquisition parameters) festgelegt werden, um die gewünschten Ergebnisse zu erzielen. Die wichtigsten Messparameter sind in Tabelle 1 zusammengefasst. Tab. 1: Wichtige Messparameter. Parameter Bedeutung ns Anzahl der Scans p1 [µs] Pulsdauer des Pulses im 1. Kanal (F1) pl1 [dB oder W] Leistung des HF-Pulses im 1. Kanal o1p [ppm] transmitter offset („Mitte“ des Spektrums) 3.3 Relaxation Es werden zwei Mechanismen der Relaxation unterschieden. Die longitudinale Relaxation (Spin- Gitter-Relaxation) beschreibt die Rückkehr zur Längsmagnetisierung M0 und wird anhand der T1- Relaxationszeit charakterisiert. Dabei handelt es sich um einen thermischen Prozess, es wird Energie in Form von Wärme an die Umgebung des Spins abgegeben. Die T1-Relaxationszeit liegt bei Protonen üblicherweise im Bereich von wenigen Sekunden, während sie bei 13C-Kernen Werte bis zu 100 Sekunden annehmen kann. Abb. 5: Zeitabhängige Entwicklung der Magnetisierung in z-Richtung während der T1- Relaxation, aus: http://mriquestions.com/what-is-t1.html. Die transversale Relaxation (Spin-Spin-Relaxation) beschreibt das Auffächern der Spins in der xy-Ebene und wird durch die Relaxationszeit T2 beschrieben. Die Ursache für den Verlust der Phasenkohärenz ist die Spin-Spin-Wechselwirkung. Die T2-Relaxationszeit liegt typischerweise in der Größenordnung von einigen 100 Millisekunden. 12
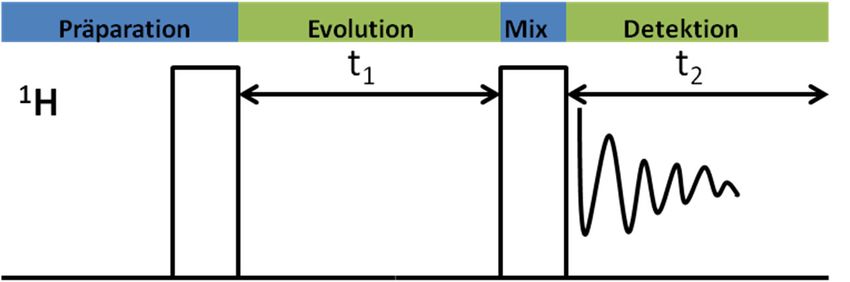
Abb. 6: Zeitabhängiger Phasenverlust der Spins in der xy-Ebene während der T2-Relaxation, aus: http://mriquestions.com/what-is-t2.html. 3.4 Zweidimensionale NMR-Spektroskopie Zur Aufklärung unbekannter Strukturen komplexer organischer Moleküle reichen die her- kömmlichen, eindimensionalen 1H- und 13C-Spektren häufig nicht aus, sodass die Anwendung zweidimensionaler Techniken erforderlich ist. Jedes zweidimensionale NMR-Spektrum beinhaltet eine direkte (horizontale Achse, F2) und eine indirekte Dimension (vertikale Achse, F1). Die direkte Dimension ergibt sich analog zu den eindimensionalen Experimenten aus der Aufnahme des FIDs während der Acquisitionszeit (t2). Für die Erzeugung der indirekten Dimension wird ein variabler Zeitabschnitt in der gewählten Pulsfolge (Evolutionszeit, t1) eingebracht. Dieser Zeitabschnitt wird während des Experiments systematisch verändert (inkrementiert). Anschließend erfolgt eine zweidimensionale Fourier- Transformation der Spektrenmatrix nach der Acquisitions- und der Evolutionszeit (Abb. 8), welche das zweidimensionale Frequenzspektrum liefert. Die dritte Dimension ist die Signalintensität. Abb. 7: Schematischer Ablauf eines 2D-NMR-Experiments am Beispiel des COSY-Experiments. t1 t1 FT (t2) FT (t1) F1 t2 F2 F2 Abb. 8: Schematische Darstellung der zweidimensionalen Fourier-Transformation. 13
Die in den gebräuchlichen zweidimensionalen Spektren abgebildeten Zusammenhänge basieren auf skalaren (über die Bindung) oder räumlichen Wechselwirkungen zwischen benachbarten Kernen einer oder verschiedener Kernsorten (z.B. 1H/1H bzw. 1H/13C). In den nachfolgenden Tabellen sind die wichtigsten zweidimensionalen NMR-Experimente zusammengefasst. Tab. 2: Übersicht über die wichtigsten 2D-Flüssig-NMR-Experimente 2D-Experiment Informationsgehalt COSY skalare Kopplung zwischen benachbarten 1H-Kernen Wechselwirkung von 1H-Kernen über den Raum (KEINE skalare NOESY Kopplung!) Wechselwirkung von X-Kernen* (z. B. 13C) mit den direkt an dieses HSQC oder HMQC Atom gebundenen 1H-Kernen (1J- Kopplung) Wechselwirkung von X- mit 1H-Kernen über mehrere Bindungen (2J HMBC und 3J) *) X-Kerne bzw. Y-Kerne können in der Sprache der NMR-Spektroskopie allgemein für alle NMR-aktiven Spezies außer den Protonen stehen. 14
4 Praktikumsversuch Flüssig-NMR 4.1 Vorbereitung des Spektrometers Bei der NMR-Messung in Lösung werden ausschließlich deuterierte Lösungsmittel verwendet. Zur Referenzierung der chemischen Verschiebung ist diesem Lösungsmittel ein interner Standard zugesetzt. Das NMR-Röhrchen wird mit einem sauberen Tuch abgewischt und in dem Spinner platziert, wobei die korrekte Höhe mithilfe des Sample-Racks festgelegt wird. Anschließend wird das NMR- Röhrchen im Spinner über den air-lift in den Probenkopf des Spektrometers überführt. Bevor das gewünschte NMR-Experiment durchgeführt wird erfolgt das Locken und Shimmen des Spektrometers, um die zeitliche bzw. örtliche Homogenität des externen -Feldes zu gewährleisten. 4.2 Versuchsdurchführung Nach dem Antestat erfolgt zunächst eine Einweisung in die Handhabung des Spektrometers und die Betriebssoftware Topspin®. Anschließend wird ein 1H-NMR-Spektrum von Zimtsäure-n- Propylester (Abb. 9) aufgenommen und es werden die Pulsdauer des 90°-1H-Pulses sowie die T1- Relaxationszeitmessung für zwei Protonensignale dieser Substanz diskutiert. Die restlichen Spektren des organischen Spektrensatzes bekommen Sie sowohl für Zimtsäure-n- propylester als auch für die unbekannte Substanz als Dateien zur Verfügung gestellt. Die Prozessierung der FID-Dateien sollen Sie für das Protokoll zu Hause mithilfe der Software MNOVA selbständig durchführen (Hinweise zum Download der Spektren und Installation der Software siehe Abschnitt 1). Abb. 9: Struktur von Zimtsäure-n-propylester. + 4.3 Pulsdauerbestimmung Zur Bestimmung der Pulsdauer wird bei gegebener Leistung (pl1) die Dauer des Pulses (p1) systematisch variiert, sodass man einen sinusartigen Verlauf der Signalintensitäten erhält (Abb. 10). Aus dieser Darstellung wird die Dauer des 360°-Pulses anhand des zweiten Nulldurchgangs der Sinuskurve bestimmt. Um den 90°-Puls zu erhalten, muss die so bestimmte Pulsdauer durch vier dividiert werden. 15
p1 in µs Abb. 10: Sinusartiger Verlauf der Signalintensitäten bei der Bestimmung der Pulsdauer. 4.4 T1-Messung Die Bestimmung der T1-Relaxationszeit erfolgt mithilfe des Inversion-Recovery-Experiments, des- sen allgemeine Pulsfolge in Abbildung 11 dargestellt ist. Abb. 11: Pulsfolge des Inversion-Recovery-Experiments zur Bestimmung der T1-Zeit. Die Gleichgewichts-Magnetisierung M0 wird durch den 180°-Puls zunächst in –z-Richtung ausgelenkt. Während der variablen Delay-Zeit relaxiert diese z-Magnetisierung mit der Ge- schwindigkeitskonstante , wobei die nach Ende dieser Zeit noch vorhandene z-Mag- netisierung Mz durch den 90°-Puls auf die y-Achse gedreht wird und somit ein messbares Signal ergibt. Die Intensität des Signals ändert sich dabei mit in charakteristischer Weise. In Abhängigkeit der Delay-Zeit ergibt sich ein exponentieller Anstieg der Signalintensitäten, welcher mit der folgenden Gleichung beschrieben wird: 1 (5) Aus den bei verschiedenen -Werten bestimmten Signalintensitäten bzw. Flächen (Integrale) kann die gesuchte Relaxationszeit mithilfe des T1/T2-Relaxationsmoduls der Software Topspin® nach Gleichung (5) ermittelt werden. 16
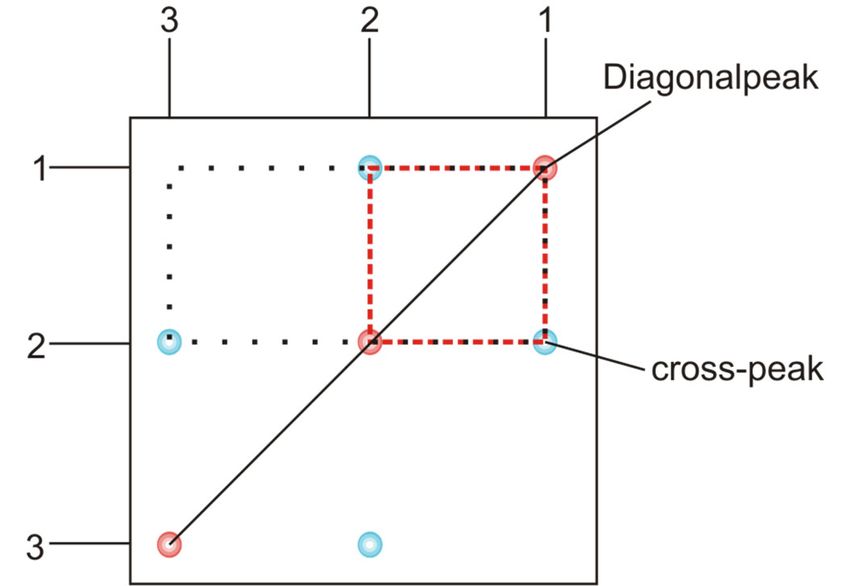
4.5 1D-Spektren-Auswertung Für jede zu messende NMR-Probe wird ein eindimensionales NMR-Spektrum aufgenommen. Die Signalstärke ist dabei abhängig von der natürlichen Häufigkeit, dem gyromagnetischen Verhältnis des Kerns sowie dem Flip-Winkel. Für organische Proben werden in der Regel 1H- und 13C- Spektren angefertigt. In beiden Spektren gibt die Position der Signale, die chemische Verschiebung, Auskunft über die Wechselwirkung des Kerns mit dem Magnetfeld und damit über funktionelle Gruppen oder räumliche Anordnung. Im Falle von 1H-Spektren wird auch eine Aufspaltung beobachtet, die durch die J-Kopplung, eine Wechselwirkung von magnetisch nicht äquivalenten Kernen über die Bindung, entsteht. Im 1H- Spektrum tritt dabei nur die 1H-1H-Kopplung auf, da 13C-Kerne zu selten sind. Im 13C-NMR- Spektrum wäre die 1H-13C-Kopplung sichtbar, sie wird jedoch durch die 1H-Entkopplung während der Aufnahme ausgemittelt. Protonen-Spektren können (fast) quantitativ über ihre Integrale ausgewertet werden. Das Verhältnis der Peakintegrale zueinander ist dabei (fast) gleich dem Protonenverhältnis. Zusammen mit der chemischen Verschiebung lassen sich einfach die Zahl und Art der Protonen in der vorgelegten Substanz bestimmen. Im organischen Spektrensatz findet sich zudem ein APT-Spektrum. Es handelt sich hierbei um ein 1H-entkoppeltes 13C-Spektrum, in dem CH- und CH3-Gruppen positive und CH2-Gruppen sowie quartäre Kohlenstoffe negative Signale zeigen. Die eindimensionalen Spektren von Zimtsäure-n-propylester werden mit Ihnen im Praktikum besprochen. 4.6 2D-Spektren-Auswertung Zweidimensionale NMR-Spektren ermöglichen die Aufklärung komplexer Strukturen. Über sie lassen sich 1H-13C und 1H-1H-Paare zuordnen, welche Informationen über die Konnektivität oder räumliche Anordnung der Atome im Molekül liefern. Es wird in homonukleare und heteronukleare Spektren unterschieden. In homonuklearen Spektren werden die Wechselwirkungen von Kernen gleicher Art (z. B. 1H) gemessen. Dabei werden in den Frequenzdomänen F2 (horizontal) und F1 (vertikal) die gleichen Kerne aufgetragen. In heteronuklearen Spektren werden hingegen zwei unterschiedliche Kernarten (z. B. 1H und 13C) miteinander korreliert. Homonukleare Spektren zeichnen sich durch eine Diagonale aus, in denen der Kern mit sich selbst korreliert (vgl. Abb. 12). Ergibt sich eine Korrelation zu dem Signal eines anderen Kerns, tritt ein cross-peak auf. Im organischen Spektrensatz sind das COSY- und das NOESY-Spektrum Vertreter für homonukleare Spektren. In beiden Fällen werden Protonen-Signale miteinander korreliert. Beim COSY-Spektrum erfolgt die Korrelation über die 2J- oder 3J-Kopplung, d. h. magnetisch nicht äquivalente Protonen, die zwei oder drei Bindungen voneinander entfernt sind, ergeben cross-peaks im Spektrum. Im NOESY-Spektrum erfolgt die Kopplung zweier magnetisch nicht äquivalenter Kerne über dipolare Wechselwirkungen, d. h. über den Raum. Ein gutes Mittel um cross-peaks zwischen zwei Protonen abschätzen zu können, ist es den Abstand sowie die räumliche Orientierung der beiden Protonen im Molekül zu betrachten. Je größer der Abstand zweier Protonen voneinander ist, desto schwächer ist ihr Signal im NOESY-Spektrum. 17
Abb. 12: Beispielhafte Darstellung eines zweidimensionalen, homonuklearen Spektrums, in dem die Diagonale und die cross-peaks markiert wurden. Heteronukleare Spektren zeigen keine Diagonale. Ihre Auswertung kann direkt über die Korrelationssignale erfolgen. HSQC- und HMBC-Spektren, wie sie im organischen Spektrensatz vorkommen, korrelieren Protonen mit den direkt gebundenen (HSQC) oder mit den über mehrere Bindungen entfernten (HMBC) Kohlenstoffen. Im HSQC-Spektrum wird die 1J-Kopplung zwischen 1H- und 13C-Kernen genutzt, wodurch ersichtlich wird, welches Proton an welches Kohlenstoffatom gebunden ist. Quartäre Kohlenstoffatome sind nicht sichtbar. Dahingegen wird im HMBC-Spektrum die 2J- und 3J-Kopplung genutzt, was es auch ermöglicht quartäre Kohlenstoffatome sichtbar zu machen und ihre C-C-Konnektivität im Molekül aufzuklären. Im Praktikum werden Sie beispielhaft die 2D-Spektren von Zimtsäure-n-propylester auswerten. 4.7 Hinweise zur Anfertigung des Protokolls Das Protokoll soll wie folgt aufgebaut sein: 1 Einleitung: schriftliche Beantwortung der folgenden Fragen 1. Welche Funktion haben die einzelnen Komponenten eines NMR-Spektrometers? 2. Wozu ist Locken und Shimmen notwendig? 3. Bei welcher chemischen Verschiebung (in ppm) erscheinen die 1H-Signale von Methanol, wenn bei einem 300 MHz-Spektrometer eine Frequenzdifferenz von 693 Hz und 1134 Hz zu TMS gemessen wurde? 4. Wie hängt das Signal-zu-Rausch-Verhältnis S/N von der Anzahl der Scans ab? 5. Wie viele Signale erwarten Sie für das 1H-NMR-Spektrum von Pyridin? Warum sieht man keine 13C-13C-Kopplung im 13C-Spektrum der Verbindung? 6. Welche Experimente werden zur Bestimmung von T1 bzw. T2 genutzt und wie funktionieren sie? 7. Welche Größenbeziehung besteht zwischen der Dauer von T1 und T2 und warum? 18
8. Wie ist der Unterschied der T1 Relaxation zwischen 1H-Signalen einer Methylgruppe und Aromaten zu erklären? 9. Welche Informationen können aus den Experimenten APT, DEPT sowie dem 1H- gekoppelten 13C-NMR-Spektrum entnommen werden? 10. Was ist der „organische Spektrensatz“ und was bedeuten die Abkürzungen der entsprechenden Pulssequenzen? Welche Information liefert welches Spektrum? 2 Auswertung der Spektren von Zimtsäure-n-propylester: Hinweise siehe 4.8, bitte KEIN zusätzlicher Text! 3 Auswertung der Spektren Ihrer unbekannten Substanz: Hinweise zur Angabe der Signale siehe 4.8. Beschreiben Sie hier zusätzlich kurz wie Sie zur Lösung der Struktur gekommen sind. Sind die aus den NMR-Spektren erhaltenen Informationen ausreichend? Wie müssen die anderen Methoden des Praktikums miteinbezogen werden? 4.8 Hinweise zur Spektreninterpretation Jedes Spektrum sollte vollständig interpretiert werden, d. h. alle relevanten Informationen im Spektrum (chemische Verschiebung, Signalintensität, Multiplizität, Kopplungskonstante, Zu- ordnung) müssen aufgelistet werden. Die korrekte Auswertung für 1D 1H- bzw. 13C-Spektren soll wie im Beispiel erfolgen: 1 H-NMR (CDCl3, int. TMS, 300 MHz): δ= 2.95 (s, 3H, N-CH3); 3.47 (dd, 2H, 3JHH = 7.6 Hz, 2 JHH = 8.7 Hz, CH2-NH); 3.94 (dd, 2H, 3JHH = 7.6 Hz, 2JHH = 8.7 Hz, CH2-NH); 5.48 (dd, 1H, 3J HH = 8.1 Hz, Ph-CH); 7.40, (m, 5H, Ph). Bei der Angabe der H- und C-Atome ist eine entsprechende Nummerierung bzw. Kennzeichnung der Atome in der Zielstruktur möglich (z. B. HA, CA oder H-1, C-1). Für 2D-Spektren ist die Verwendung von Tabellen (Tab. 4) sinnvoll. Tab. 4: Beispielhafte Auswertung von homonuklearen (COSY; links) und heteronuklearen (HSQC; rechts) Spektren. H-n/H-m δ [ppm] C-n/H-m δ [ppm] H-1/H-2 2.3 / 2.4 C-1/H-1 10.3 / 2.3 H-2/H-5 2.4 / 5.3 C-3/H-2 56.7 / 2.4 19
Organische Massenspektrometrie (MS) 1 Ziel des Praktikums (1) Kennenlernen eines Massenspektrometers, eines Sektorfeldgerätes mit Elektronenstoß-Io- nisation (EI) (2) Aufnahme von EI- Massenspektren unter Anleitung (3) Übungen zur EI- Spektreninterpretation. Das Praktikum wird unter Zuhilfenahme Ihrer Vorlesungsmitschriften selbstständig vorbereitet. In einem Antestat wird Ihr Wissen zu folgenden Sachverhalten geprüft: Grundlagen der Massenspektrometrie, Aufbau eines Massenspektrometers und ablaufende Prozesse, Sekundärelektronenvervielfacher Aussehen eines Massenspektrums, Begriffe Electron Impact Ionisation, Aufbau der Quelle (Regeln der) Spektreninterpretation, Isotopie, entstehende Spezies, Regeln der Fragmen- tierung Dabei dienen die gestellten Aufgaben als Orientierung. Die Lösungen der im Skript gestellten Übungsaufgaben sind gleichzeitig Bestandteil des Protokolls. 2 Einleitung Das Grundprinzip der EI- Technik ist der Beschuss der im Hochvakuum (10-5-10-7 mbar) iso- lierten, gasförmigen Moleküle mit Elektronen hoher kinetischer Energie (meist 70 eV, um eine optimale Ionenausbeute und reproduzierbare Fragmentierung zu erzielen). Dabei wird aus dem Molekül ein Elektron herausgeschlagen und das Molekülion M∙+ erzeugt, ein Radikalkation (o- pen-shell ion, odd-electron ion). EI ist eine „harte“ (energiereiche) Ionisationsmethode. Zur Erzeugung von Radikalkationen aus organischen Molekülen reichen prinzipiell Energien von 8-14 eV. Der Energieüberschuss führt deshalb bereits in der Ionenquelle zur Fragmentierung der Radikalkationen: M+. → A+ + B. Fragmentierung in Ion (closed-shell) und Radikal M+. → A. + B+ M+. → C+. + D; A+ → F+ + G Neutralverlust (open-shell ion) Radikalische Spaltungen treten nur aus Radikalkationen auf (open-shell-Ionen), daraus entstan- dene Kationen (closed-shell-Ionen, odd-electron) fragmentieren unter Neutralabspaltung (even- electron rule), so dass auch Folgefragmentierungen möglich sind. Neutralabspaltungen können aber auch direkt aus dem Molekülradikalkation auftreten. 3 Grundlagen der Spektreninterpretation 3.1 Isotopie der Elemente und Informationen aus Isotopenpeaks Informieren Sie sich über die Einteilung der Elemente nach 1. Häufigkeit der Isotope und 2. Isotopenabstand. 20
Die Häufigkeit der Moleküle mit einem Molekulargewicht über dem Molekulargewicht des monoisotopischen Moleküls hängt von der Anzahl der vorhandenen Atome und von der relati- ven Häufigkeit der Isotope in den beteiligten Elementen ab. (Orientierungsfrage: welche Häu- figkeit tragen 6 C-Atome auf der M+1 Stelle bei?) Daher kann man aus der Häufigkeitsverteilung der Isotope mit entsprechenden Massenabstand (M+1, M+2; ...) auf Art und Anzahl der im Molekül vorhandenen Elemente schließen. Tabelle 1: Isotopenverteilung wichtiger Elemente, bezogen auf das häufigste Isotop. Element M M+1 M+2 Masse % Masse % Masse % H 1 100 2 0.015 ‐ ‐ C 12 100 13 1.1 ‐ ‐ N 14 100 15 0.37 ‐ ‐ O 16 100 17 0.04 18 0.21 F 19 100 ‐ ‐ ‐ ‐ Si 28 100 29 5.1 30 3.4 S 32 100 33 0.8 34 4.5 Cl 35 100 ‐ ‐ 37 32.0 Br 79 100 ‐ ‐ 81 98 I 127 100 ‐ ‐ ‐ ‐ Berechnung der Isotopenverteilung Die Isotopenverteilung eines Moleküls kann nach einem vereinfachten Ausdruck berechnet werden: (a + b)n a ist die relative Häufigkeit des leichten Isotops b ist die relative Häufigkeit des schweren Isotops n ist die Anzahl der Atome des betrachteten Elements im Molekül Die Verteilung ergibt sich dabei aus den Summanden. Bei Si und S können Überlagerungen mit dem 13C- oder anderen Mustern leicht zum Verwischen führen. Aufgabe 1: Berechnen Sie das Isotopenmuster von CS2. 3.2 Fragmentierungsreaktionen Ein Massenspektrum ist die Ergebnisanalyse einer Reihe von Zerfallsreaktionen eines Ions. In der nachfolgenden Aufgabe werden wichtige Fragmentierungs- und Umlagerungsreaktionen genannt, die nach der Elektronenstoß- Ionisation auftreten. Aufgabe 2: Formulieren Sie Reaktionsgleichungen für die aufgeführten Fragmentierungsreaktionen mit den in Klammern gegebenen Verbindungen und geben Sie möglichst alle mesomeren Grenzstrukturen der ent- stehenden Produkte an (stabilstes Produkt). Identifizieren Sie die wahrscheinlichste Stelle der Ionisie- rung im Molekül. Verwenden Sie für die Formulierung des Ionisationsprozesses und der Fragmentie- rung das Konzept der lokalisierten Ladung und charakterisieren Sie jede der Fragmentierungen als Ra- dikal- oder Neutralabspaltung: Radikal induzierte Spaltungen - (Alkyl-) Spaltung (2-Methylpentan) - α-Spaltung aktivierter Bindungen: Heteroatom (4-Phenylbutan-2-on) Allylspaltung (4-Phenylpent-1-en) Benzylspaltung (Phenylbutan-2-on) 21
- McLafferty-Umlagerung, McL (Trimethylsilylacetat) - Retro-Diels-Alder-Reaktion, RDA (Cyclohexen) Ladungsinduzierte Spaltungen - Induktive Spaltung (3-Fluoroiodobenzol) - Eliminierung, H-Umlagerung (4-Chlorhexen) - Neutralverlust (Phenol) Aufgabe 3: Welche Fragmentierungen sind für dieses Molekül möglich? Geben Sie die ausführlichen Reaktions- gleichungen an, verwenden Sie das Konzept der lokalisierten Ladung und benennen Sie die entspre- chende Fragmentierung. 3.3 Herangehensweise an die Spektreninterpretation Im Nachfolgenden wird die allgemeine Herangehensweise bei der Interpretation eines Massen- spektrums skizziert. Eine korrekte Zuordnung charakteristischer Ionen im Spektrum ist zur Ab- sicherung des Strukturvorschlags und der Summenformel unerlässlich. Bei der Spektreninter- pretation wird bereits vorhandenes Wissen über den Analyten immer mit einbezogen. 1. Charakterisieren Sie den Molekülionenpeak. Welche Heteroatome vermuten Sie bzw. kön- nen Sie ausschließen (Isotopenmuster!)? 2. Erstellen Sie einen oder mehrere Strukturvorschläge durch Interpretation großer und cha- rakteristischer Schlüsselbruchstücke bzw. Massendifferenzen, besonders für den Ba- sispeak. Überlegen Sie sich mögliche Fragmentierungen dieser Strukturvorschläge, ver- gleichen Sie diese mit dem Spektrum und achten Sie dabei auf „fehlende“ Ionen. Regeln für die EI- Spektreninterpretation Regel 1: Zwischen Molekülion und Fragmentionen müssen als Folge der Fragmentierung des Molekülions chemisch sinnvolle Massendifferenzen bestehen. Regel 2: Einmal gebildete closed-shell- Ionen A+ oder B+ gehen keine erneute Radikalspaltung mehr ein, sondern zeigen nur noch Neutralabspaltungen. (even electron rule) Regel 3: Bei konkurrierenden Homolysen bestimmt meist die Produktstabilität den bevorzug- ten Reaktionsweg, der größere Alkylrest wird abgespalten. Regel 4: Enthält ein Molekül 1, 3, 5, ... Stickstoffatome, ist seine Molmasse ungeradzahlig. Enthält ein Molekül 0, 2, 4,... Stickstoffatome, ist seine Molmasse geradzahlig (Stickstoff- regel). 22
Regel 5: Homolysen (Radikalabspaltungen) führen zur Bildung von Primärfragmenten mit nicht geradzahliger Massendifferenz zum Ausgangsion. Verluste intakter Moleküle (Um- lagerungen, Neutralabspaltungen) führen zu geradzahligen Massendifferenzen zum Mole- külion. Zusammen mit einer ungeradzahligen Anzahl Stickstoff im abgespaltenen, elektrisch neutralen Fragment kann eine Umkehrung dieser Regel eintreten (durch H2NR. bzw. NH3- Verluste). Aufgabe 4: Es sind relative Intensitäten aus dem Peaklisting zweier EI-Massenspektren gezeigt. Welches Molekül könnte sich hinter dem Spektrum verbergen? Erklären und zeigen Sie das Zustandekommen der charak- teristischen Fragmente (mind. 2 davon anhand einer ausführlichen Gleichung)! m/z rel. Int. [%] m/z rel. Int. [%] 64 43.3 29 12.4 66 ~0.4 43 100 67 15.4 45 14.6 69 5.8 61 15.3 99 100 70 11.8 101 42.2 73 4.9 103 ~0.3 88 7.0 134 69.4 89 0.51 135
- Einführen der Schubstange - Beobachtung der Peakintensitäten - gegebenenfalls manuelles Heizen der Schubstange unter Kontrolle der Signalintensitäten (zwischen 1 und 10 Mill. counts) - Herausziehen der Schubstange, Schließen des HV-Ventils - „Stop“ und „Close Acquisition“ - Entfernen der Schubstange, Entfernung des Probentiegels Hinweise zur Anfertigung des Versuchsprotokolls Erstellen Sie bitte in jeder Gruppe ein Protokoll über den durchgeführten Versuch. Beginnen Sie mit einer kurzen Darstellung der Aufgabe, keine theoretische Einleitung. Achten Sie auf eine systematische und übersichtliche Formatierung (Inhaltsverzeichnis, Legenden, Überschrif- tenhierarchie). - Lösen Sie schriftlich die im Skript gestellten und im Antestat besprochenen Aufgaben. - Interpretieren Sie ausführlich das Spektrum, das vom Zimtsäurepropylester aufgenommen wurde (Sie erhalten von jedem erzeugten Spektrum ein pdf). Orientieren Sie sich an Punkt 3.3., Herangehensweise an die Spektreninterpretation. Ordnen Sie prägnante Fragmentionen anhand von ausführlichen Zerfallsgleichungen (mind. 4) entsprechenden Strukturen zu. Für das Zeichnen chemischer Strukturen können Sie neben ChemDraw auch z.B. die im Netz frei herunterladbaren Programme Accelrys Draw, Marvin Sketch, ACD/ChemSketch oder BKChem benutzen. - Diskutieren Sie ebenso ausführlich das Massenspektrum Ihrer Verbindung. Nutzen Sie dazu den Strukturvorschlag, den Sie sich aus allen Analysen (IR, UV, NMR) abgeleitet ha- ben. Ordnen Sie prägnante Fragmentionen anhand von ausführlichen Zerfallsgleichungen (mind. 3) entsprechenden Strukturen zu. Nutzen Sie auch die Information, die Sie aus den Isotopenmustern gewinnen können. Waren die mit der Massenspektrometrie erhaltenen In- formationen zur Strukturaufklärung ausreichend oder benötigten Sie weitere Methoden, und wenn ja, welche? 24
Sie können auch lesen

















































