Optimierung des Rekonstruktions-Tools TSNI für genregulatorische Netzwerke mittels Parallelisierung und ein Vergleich mit Time Delay ARACNE - OPUS 4
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
ANGEWANDTE BIOWISSENSCHAFTEN
WISSENSCHAFTLICHE BEITRÄGE 2019 Wiss Beitr TH Wildau 23:7 – 10
23. Jahrgang (2019) doi: 10.15771/0949-8214_2019_1
ISSN: 0949-8214 März 2019
Optimierung des Rekonstruktions-Tools TSNI für
genregulatorische Netzwerke mittels Parallelisierung
und ein Vergleich mit Time Delay ARACNE
INFORMATIK / TELEMATIK
Marco Schäfer, Christian Jetschni, Chong Wang und Heike Pospisil*
Zusammenfassung Abstract
OPTISCHE TECHNOLOGIEN / PHOTONIK
Gegenstand dieser Veröfffentlichung ist eine Reimple- The purpose of this work was to optimize a tool for ana-
mentierung des TSNI (Time Series Network Identification)- lyzing biological high-throughput data and to generate
Tools zur Rekonstruktion von genregulatorischen Netz- gene regulatory networks, which are based on ordinary
werken aus Zeitserien von Gen-Expressionsprofilen. Das differential equations. For this task the Time Series Network
auf gewöhnlichen Differentialgleichungen basierende Indentification (TSNI) - tool has been selected. The TSNI-al-
Tool wurde algorithmisch verbessert und an die Verarbei- gorithm has been optimized and transferred into the pro-
tung großer Datensätze angepasst. Durch Ausschöpfung gramming language C. Computationally intensive sections
des Potentials moderner Multi-CPU und GPU-Architektu- were parallelized using modern multi-core CPU and GPU
ren wurden die erforderlichen Berechnungen erheblich architectures as well as functions from the MAGMA-library
beschleunigt. Bei der Überführung des mit Matlab im- for the solution and parallelization of more complex math-
plementierten Tools in die Programmiersprache C wurde ematical problems. The performance of the algorithm was
zudem die Bedienbarkeit verbessert. significantly increased (e.g. the calculation of a network of
more than 11.000 genes was 18.7-fold faster with the im-
Durch Einsatz der GPU-beschleunigten Mathematik-Bib- proved tool compared to the original algorithm). So, large
PRODUKTION UND MATERIAL
liothek MAGMA konnten unter Verwendung eines nor- regulatory networks can now be analysed in a moderate
malen Desktop-Rechners Leistungssteigerungen um das time.
18,7-fache bei 11.000 Genen verzeichnet werden. Darü-
ber hinaus rekonstruierte das verbesserte TSNI weitaus Additionally, more true positve network connections were
mehr biologisch richtige Beziehungen als vergleichbare predicted using the adjusted TSNI - tool.
Anwendungen.
1. Einleitung sich ebenfalls schnell entwickelnden rierung von neuen Erkenntnissen nicht
Computertechnik steht heute weniger nur in der Systembiologie, sondern
VERKEHR UND LOGISTIK
Biologische Systeme wie Gewebe oder die Generierung der Daten, sondern auch in Medizin, Pharmazie und Bio-
Zellen sind über viele Ebenen (metabo- vielmehr die Bewältigung ihrer Aus- technologie. Besonders aus medizini-
lische Netzwerke, Signalwege, genre- wertung und die Schaffung neuen Wis- scher Sicht ist die Kenntnis molekularer
gulatorische Netzwerke) miteinander sens im Vordergrund (Jia et al. 2017). und genetischer Mechanismen wich-
eng verknüpft und hoch organisiert tig, um Krankheiten mit deregulierten
(Karr et al. 2012). Für ein umfassen- Eine große Herausforderung besteht Genen als Ursache besser zu verstehen.
des Verständnis dieser Organisation, darin, schnelle und effiziente Algorith- Dies kann wiederum neue Angriffsziele
ist eine ganzheitliche Betrachtung aller men für die Rekonstruktion von genre- für therapeutische Ansätze liefern (Reiß
Ebenen erforderlich; dies ist Gegen- gulatorischen Netzwerken aus Genex- 2002; Jia et al. 2017). Die Kosten der
stand der Systembiologie (Reiß 2002), pressionsdaten zu unterschiedlichen Forschung in den genannten Bereichen
welche derzeit durch die Fortschritte in Zeitpunkten (Reverse Engineering) zu können durch das Modellieren von
MANAGEMENT UND RECHT
den Sequenziertechniken zunehmend entwickeln, um Hochdurchsatzdaten in genregulatorischen Netzwerken ge-
an Bedeutung gewinnt. Aus diesem akzeptabler Zeit zu analysieren. Dies er- senkt werden, da beispielsweise nicht
Grund und im Zusammenhang mit der möglicht eine deutlich schnellere Gene- getestete laborexperimentelle Ansätze
* korrespondierender Autor
THWildau
Wissenschaftliche Beiträge 2019 7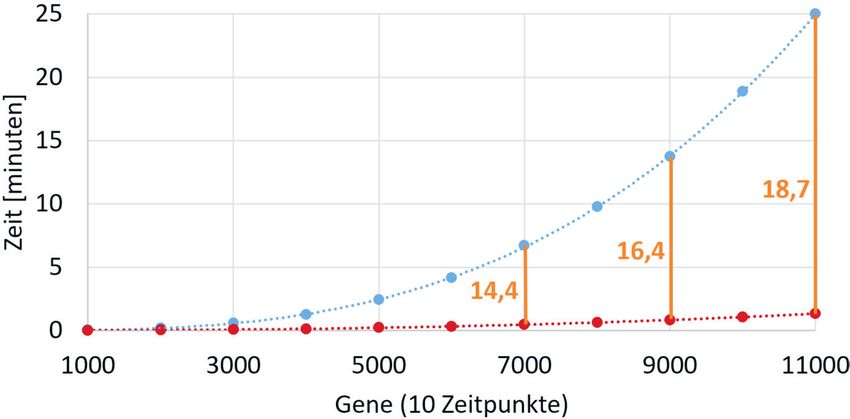
kostengünstig simuliert und zumindest Anschließend wird der Ausdruck vom bis tausenden Kernen für massive Par-
teilweise ersetzt werden (Reiß 2002; zeitkontinuierlichen Raum in den dis- allelisierung bereitstellt (Rauber & Rün-
Karlebach & Shamir 2008). kreten überführt (vgl. Gleichung 3). ger 2012). Zusätzlich greift MAGMA
Die Ausgangsdaten zur Lösung des auf die Pakete LAPACK (Linear Algebra
Ein Software-Werkzeug, das die kom- Gleichungssystem sind dabei ebenfalls PACKage) und OpenBLAS (Basic Linear
plexe Aufgabe der Rekonstruktion von diskret (Bansal et al. 2006). Algebra Subprograms) zurück (Dongar-
genregulatorischen Netzwerken aus ra et al. 2014). Die hybriden Algorith-
Zeitserien von Genexpressionsdaten (3) men der MAGMA-Bibliothek nutzen
realisiert, ist das TSNI (Time Series Net- durch intelligente Zerlegung von Pro-
work Identification)-Tool (Bansal et al. Da das Gleichungssystem nur für den blemen in Tasks variierender Granula-
2006; Jia et al. 2017). Es modelliert gen- Fall M ≥ N + P gelöst werden kann, rität den Leistungsumfang von moder-
regulatorische Netzwerke mittels eines wird zum einen mittels Hauptkompo- nen Multi-CPU und GPU-Architekturen
Ansatzes, welcher auf Differenzialglei- nentenanalyse (engl. Principle Compo- aus (Rauber & Rünger 2012).
chungssystemen beruht. So wird nicht nent Analysis; PCA) eine Dimensionsre-
nur die Topologie eines Netzwerks er- duzierung von N und P durchgeführt. 2.3 Daten
mittelt, sondern auch dessen Dynamik Zum anderen erfolgt mittels kubischer
(d.h. die durch genregulatorische Be- Spline-Interpolation eine künstliche Die für die Weiterentwicklung und Tes-
einflussung hervorgerufenen zeitlichen Vervielfältigung der Zeitpunkte M. tung des Analyse-Tools genutzten Da-
Konzentrationsveränderungen von Nach der PCA wird das Gleichungs- ten stammen von dem gut erforschten
Genprodukten) – und dies sogar im system durch Bildung einer Pseudo- Modellorganismus Arabidopsis thalia-
zeitkontinuierlichen Raum (Bansal et inversen gelöst und abschließend das na. Im Detail sind es Daten einer Stu-
al. 2006). Damit bildet dieses Tools die Ergebnis mittels der bilinearen Trans- die zum Übergang vom vegetativem
biologische Realität deutlich besser ab formation wieder in den zeitkontinu- Wachstum zur Blütenbildung. Dabei
als beispielsweise Boolesche Netzwerke ierlichen Raum überführt (Bansal et al. wurden die Genexpressionsprofile
oder Bayessche Netzwerke. 2006). während der Entwicklung von Arabido-
psis thaliana in einem Zeitraum von 7
2. Methoden und Algorithmen 2.2 Methoden bis 16 Tagen nach der Keimung täglich
aufgenommen (Klepikova et al. 2015),
2.1 Algorithmen Für eine effektivere Berechnung wurde so dass die Expressionsdaten von 10
der ursprünglich für Matlab implem- Zeitpunkten im Abstand von einem
Der Kern von TSNI besteht aus dem tierte Algorithmus in die performante Tag vorlagen. Um die Vorhersagequa-
Differentialgleichungssystem, welches und hardwarenähere Programmier- lität der biologischen Zusammenhän-
in Gleichung 1 dargestellt ist. Dieser sprache C überführt. Zur weiteren ge abschätzen zu können, wird die
Ausdruck beschreibt die Rate ẋl der Syn- Steigerung der Berechnungsgeschwin- Anwendung TD ARACNE (Time Delay
these eines Gens zu einem Zeitpunkt digkeit kam parallele Programmierung Algorithm for the Reconstruction of Ac-
tk in Abhängigkeit von der Konzent- zum Einsatz; dafür wurde die Schnitt- curate Cellular Networks) (Zoppoli et
ration aller anderen Transkripte einer stelle OpenMP (Dagum & Menon al. 2010) vergleichend herangezogen.
Zelle bzw. des Systems. Die Rate eines 1998) verwendet. OpenMP stellt die TD ARACNE ist ein etabliertes und weit
Gens hängt von der Art des Einflusses nötigen Funktionen der Parallelisie- genutztes Tool zur Rekonstruktion von
(inhibierend, verstärkend, oder keiner) rung als Erweiterung von C bereit. Dies genregulatorischen Netzwerken aus
jeden Gens j (1 ≤ j ≤ N, wobei N die umfasst das Zerlegen von Problemen Zeitserien von Genexpressionsprofilen;
Anzahl der Gene bezeichnet) und des- in Teilprobleme, die Synchronisation es verwendet allerdings das Konzept
sen Konzentration xj zum Zeitpunkt tk von Threads sowie die Deklaration von der Transinformation (engl. Mutal In-
ab. Hinzu kommt der Einfluss bil der gemeinsamen und verteiltem Speicher. formation).
Perturbationen P. Die Variable ul reprä- Mit Hilfe von OpenMP konnten die
sentiert die l-te Perturbation zu einem Routinen für kleinere Matrix-Multipli- Für die Abschätzung der Vorhersage-
Zeitpunkt tk (Bansal et al. 2006). kationen parallelisiert werden. qualität der gefundenen Interaktionen
wurde mit einem Datensatz bereits
(1) Größere Operationen wurden mittels beschriebener Proteininteraktionen
der MAGMA-(Matrix Algebra on GPU der Datenbank ATPIN („Arabidopsis
Gleichung 1 lässt sich mit Hilfe der and Multicore Architectures) Bibliothek thaliana protein interaction network“,
Matrizenschreibweise wie in Formel 2 auf der Grafikkarte (engl. Graphics Pro- https://atpin.bioinfoguy.net/cgi-bin/
kompakter darstellen. Dabei steht M cessing Unit; GPU) gelöst (Dongarra et atpin.pl) verglichen. Release 9 enthält
für die Anzahl der Zeitpunkte (Bansal et al. 2014). Sie basiert auf der CUDA1- 96.276 Proteininteraktionen.
al. 2006). Schnittstelle, die die Verwendung ei-
ner GPU über ihren ursprünglichen
(2) Verwendungszweck hinaus ermöglicht
und somit die GPU mit ihren hunderten
1 CUDA ermöglicht die parallele Berechnung auf NVIDIA-Grafikkarten
8 THWildau
Wissenschaftliche Beiträge 2019ANGEWANDTE BIOWISSENSCHAFTEN
2.4 Intrapolation
Da die Zahl der untersuchten Gene
deutlich die Zahl der Messzeitpunkte
übersteigt, ist die Zahl der Freiheitsgra-
de, die sich aus der Differenz zwischen
Genen (N) und Messzeitpunkten (M)
ergibt, zu hoch, um verlässliche Aussa-
gen treffen zu können (vgl. Abschnitt
2.1). Aus diesem Grund wird die Zahl
der Messzeitpunkte mittels Intrapolati-
INFORMATIK / TELEMATIK
on künstlich vergrößert. Eine äquidis-
tante Intrapolation teilt den Zeitbereich
zwischen zwei Messpunkten gleichmä-
ßig auf. Liegen jedoch Messwerte zu Abb. 1) Berechnungsdauer in Minuten gegen Zahl der Gene. In blau ist die Rechenzeit des ursprünglichen Matlab-
Zeitpunkten vor, die nicht paarweise Programms und in rot die des optimierten TSNI-Programms dargestellt. In orange sind die Vielfachen der Leistungs-
zuwächse eingezeichnet. [Testsystem: CPU Xeon 1231 V3 (8x3.7 GHz); GPU GM204-GTX 970 (4 GB, 1664x1.2 GHz);
jeweils denselben Abstand zueinander RAM (16 GB, 1.6 GHz)].
aufweisen, muss eine nicht äquidistan-
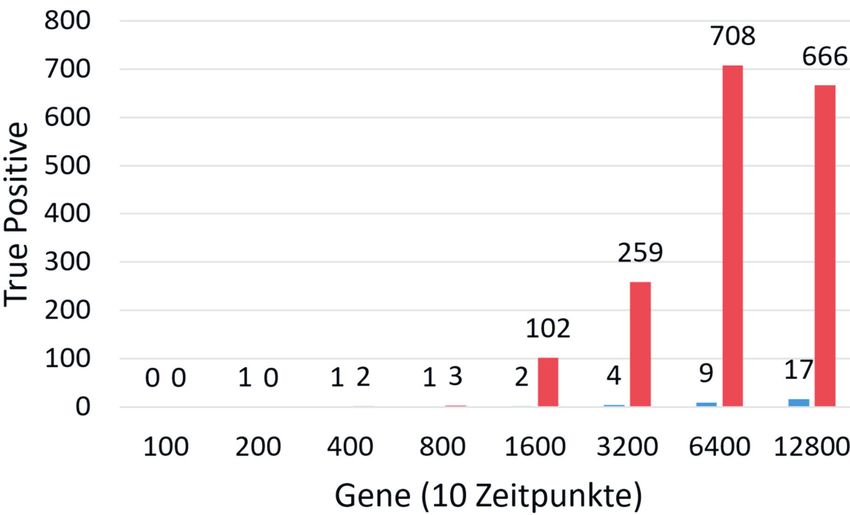
te Intrapolation durchgeführt werden. 3.2 Qualität der biologischen 4. Diskussion
OPTISCHE TECHNOLOGIEN / PHOTONIK
Vorhersage
3. Ergebnisse Das Tool TSNI (Time Series Network
Beim Vergleich der biologischen Kor- Identification) konnte erfolgreich an
Der ursprüngliche Algorithmus wurde rektheit der gefundenen Beziehungen die Bearbeitung von großen Datensät-
bezüglich des Rechenaufwands deut- zwischen Genen lässt sich erkennen, zen mit über 11.000 Genen angepasst
lich optimiert. Dabei wurde OpenMP dass es bei Datensätzen mit 800 oder werden. Gezeigt ist die Auswertung für
zur Parallelisierung von Routinen für weniger Genen kaum Unterschiede 12.800 Gene, um einen Vergleich mit
kleinere Matrix-Matrix-Operationen ge- zwischen den Ergebnissen mit dem dem klassischen TSNI-Tool, welches in
nutzt, bei denen der Kopiervorgang hier vorgestellten Ansatz und dem der Zahl der zu verarbeitenden Gene
auf die GPU keinen Zeitvorteil bringt. Referenztool TD ARACNE gibt (vgl. begrenzt ist, zu ermöglichen. Her-
Bei der Bewältigung der Parallelisie- Abb. 2). kömmliche Tools, wie z.B. TD ARACNE
rung mehrerer rechenintensiverer Ab- und das ursprüngliche TSNI, sind je-
schnitte wurde die MAGMA-Bibliothek Enthält der Datensatz jedoch 1.600 doch nicht in der Lage, solche großen
zur Matrix-Matrix-Multiplikation, für Gene oder mehr, werden deutlich Datenmengen für höhere Organismen
PRODUKTION UND MATERIAL
die Bildung großer Inversen und zur mehr korrekte Beziehungen mit Hilfe (Pflanzen, Tieren und Menschen) mit
Singulärwertzerlegung (engl. Singu- des auf Differenzialgleichungssyste- 20.000 bis 40.000 Genen zu verarbei-
lar Value Decomposition; SVD) einge- men basierenden Ansatzes von TSNI ten. Um neue Erkenntnisse für die Me-
setzt, wobei letztere den Kern der PCA gefunden. Dabei konnte eine bis zu dizin, Biotechnologie oder Systembio-
und der Bildung der Pseudoinversen 79-fach höhere Anzahl an korrekt vor- logie zu erlangen, ist es erforderlich,
darstellt. hergesagten Beziehungen beobachtet die genregulatorischen Netzwerke,
werden. die aus mehreren zehntausend Genen
3.1 Verbesserung der
Algorithmen-Performance
Das Ergebnis der Reimplementierung
des TSNI-Algorithmus in C ist in Abb. 1
dargestellt. Wie zu erkennen ist wird
VERKEHR UND LOGISTIK
mit steigender Datenmenge der Ge-
winn an Berechnungszeit deutlich
vergrößert: Bei 7.000 Genen ist das
verbesserte TSNI 14,4-mal schneller
als der ursprüngliche Algorithmus;
bei 11.000 Genen konnte bereits ein
18,7-facher Gewinn an Rechenzeit er-
mittelt werden. Die Grafik zeigt die Zeit
für bis zu 11.000 Gene, da Matlab bei
Datensätzen >12.000 abbricht.
MANAGEMENT UND RECHT
Abb. 2) Anzahl der biologisch tatsächlich korrekten (True Positive) Beziehungen zwischen Genen gegen die Anzahl
der Gene, die für die Rekonstruktion genutzt wurden. Dabei ist in blau TD ARACNE und in rot TSNI dargestellt. [TSNI
(PCA=2, tol=0.2, threshold=0.05); TD ARACNE (tol=0.2, threshold=0.05)].
THWildau
Wissenschaftliche Beiträge 2019 9bestehen, untersuchen zu können, eiteren wichtigen Schritt zum ganz-
w
was mit dem hier vorgestellten Ansatz heitlichen Verständnis biologischer
nun möglich ist. Auch eine Analyse des Systeme dar.
kompletten Genoms von Arabidopsis
thaliana (mit ca. 42.000 Genen) ist mit LITERATUR
der neu entwickelten Software mög-
Bansal M, Della Gatta G, Di Bernardo D (2006) Inference
lich. Weiterhin wurde die Intrapolation of gene regulatory networks and compound mode of
dahingehend verbessert, dass nun action from time course gene expression profiles. Bioinfor-
matics 22(7):815–822. doi: 10.1093/bioinformatics/btl003
auch nicht äquidistante Zeitpunkte be-
rücksichtigt werden können. Dies be- Dagum L, Menon R (1998) OpenMP: an industry
standard API for shared-memory programming.
deutet, dass zwischen zwei Zeitpunk- Computational Science & Engineering, IEEE 5(1):46–55
ten mehr Zwischenpunkte intrapoliert
Dongarra J, Gates M, Haidar A, Kurzak J, Luszczek P,
werden, wenn der Abstand dieser zwei Tomov S, Yamazaki I (2014) Accelerating Numerical Dense
Zeitpunkte, im Vergleich zu den übri- Linear Algebra Calculations with GPUs.
Numerical Computations with GPUs:1–26
gen Zeitabständen eines Datensatzes
größer ist. Jia B, Xu S, Xiao G, Lamba V, Liang F (2017) Learning gene
regulatory networks from next generation sequencing
data. Biometrics. doi: 10.1111/biom.12682
Darüber hinaus ist die Qualität der bio-
Karlebach G, Shamir R (2008) Modelling and analysis of
logischen Vorhersage der genregulato- gene regulatory networks. Nature reviews. Molecular cell
rischen Netzwerke bei TSNI erheblich biology 9(10):770–780. doi: 10.1038/nrm2503
besser. Jedoch waren mit dem hier vor- Karr JR, Sanghvi JC, Macklin DN, Gutschow MV, Jacobs
gestellten Ansatz viele der Interaktio- JM, Bolival B, Assad-Garcia N, Glass JI, Covert MW (2012)
A Whole-Cell Computational Model Predicts Phenotype
nen der ATPIN-Datenbank nicht ermit- from Genotype. Cell 150(2):389–401.
telbar, da jeweils nur ein Ausschnitt des doi: 10.1016/j.cell.2012.05.044
Gesamtnetzwerks betrachtet wurde. Klepikova AV, Logacheva MD, Dmitriev SE, Penin AA
Desweiteren sind viele der beschriebe- (2015) RNA-seq analysis of an apical meristem time series
reveals a critical point in Arabidopsis thaliana flower
nen Interaktionen nicht in jedem Zu- initiation. BMC Genomics 16:466. doi: 10.1186/s12864-
stand der Organismen vorhanden; es 015-1688-9
kommt beispielsweise nach Störungen Rauber T, Rünger G (2012) Parallele Programmierung,
oder in verschieden Zellzyklusstadien, 3. Aufl. 2013. eXamen.press. Springer, Berlin and Heidel-
berg. ISBN: 978-3-642-13603-0. doi: 10.1007/978-3-642-
unter verschiedenen klimatischen Be- 13604-7
dingungen, bei Befall mit Krankheitser-
Reiß T (2002) Systembiologie: Systeme des Lebens. Bun-
regern etc. zu speziellen funktionellen desministerium für Bildung und Forschung
Wechselwirkungen, die nicht in einem
Zoppoli P, Morganella S, Ceccarelli M (2010) TimeDelay-
einzigen Experiment beobachtet wer- ARACNE: Reverse engineering of gene networks from
den können. Die Verbesserung durch time-course data by an information theoretic approach.
BMC Bioinformatics 11:154. doi: 10.1186/1471-2105-11-
das beschriebene Software-Werkzeug 154
wird anhand der erhöhten Zahl an
Interaktionen im Vergleich zu denen,
die das Tools ARACNE liefert, deutlich. AUTOREN
Marco Schäfer
Die Verbesserungen des Algorithmus Christian Jetschni
und die Parallelisierung haben erhebli- Dr. Chong Wang
Prof. Dr. Heike Pospisil
che Leistungsverbesserungen ergeben, Professur für Life Science Informatics
und die Benutzerfreundlichkeit wurde Arbeitsgruppe “High Performance Computing
in Life Sciences”
erhöht. Es bedarf allerdings noch eini- Technische Hochschule Wildau
ger weiterführender Optimierungen,
da die Anzahl der korrekten Netzwerk- E-Mail für Korrespondenz:
verbindungen noch zu gering ist, was heike.pospisil@th-wildau.de
jedoch an der Qualität der verwende-
ten Daten liegen kann. Wegen der be-
schriebenen Effizienzsteigerung kann
nun aber die Berechnung mit weiteren
Datensätzen ohne Einschränkungen
realisiert werden, so dass eine weitere
Verbesserung der Vorhersagequalität
in den nächsten Schritten in Angriff
genommen werden kann. Der hier
vorgestellte Ansatz unter Nutzung
von Hochdurchsatzdaten stellt einen
10 THWildau
Wissenschaftliche Beiträge 2019Sie können auch lesen

















































