IC3D-Klassifikation von Hornhautdystrophien1 - The IC3D Classification of the Corneal Dystrophies - Cornea Society
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
S1
IC3D-Klassifikation von Hornhautdystrophien1
The IC3D Classification of the Corneal Dystrophies
DOG
Autoren J. S. Weiss1, H. U. Møller2, W. Lisch3, S. Kinoshita4, A. J. Aldave5, M. W. Belin6, T. Kivelä7, M. Busin8, F. L. Munier9,
B. Seitz10, J. Sutphin11, C. Bredrup12, M. J. Mannis13, C. Rapuano14, G. Van. Rij15, E. K. Kim16, G. K. Klintworth17
Institute Die Institutsangaben sind am Ende des Beitrags gelistet.
Heruntergeladen von: Thieme Verlagsgruppe. Urheberrechtlich geschützt.
Bibliografie Zusammenfassung Abstract
DOI http://dx.doi.org/10.1055/ ! !
s-0029-1245895
Hintergrund: Die in jüngster Zeit verfügbaren ge- Background: The recent availability of genetic
Klin Monatsbl Augenheilkd
netischen Analysen haben die Mängel in der her- analyses has demonstrated the shortcomings of
2011; 228 Suppl. 1: S1 – S39
© Georg Thieme Verlag KG kömmlichen phänotypischen Methode zur Klassifi- the current phenotypic method of corneal dystro-
Stuttgart ∙ New York ∙ kation von Hornhautdystrophien (HD) aufgezeigt. phy classification. Abnormalities in different
ISSN 1431-634X Anomalien in verschiedenen Genen können einen genes can cause a single phenotype, whereas dif-
einzigen Phänotyp verursachen, wogegen verschie- ferent defects in a single gene can cause different
Korrespondenzadresse
Jayne S. Weiss, MD
dene Defekte in einem einzigen Gen verschiedene phenotypes. Some disorders termed corneal dys-
Kresge Eye Institute Phänotypen bedingen können. Einige als korneale trophies do not appear to have a genetic basis.
4717 St Antoine Dystrophien bezeichnete Störungen scheinen kei- Purpose: The purpose of this study was to devel-
Detroit nen genetischen Hintergrund zu haben. op a new classification system for corneal dystro-
MI 48201 Absicht: Ziel dieser Studie war es, ein neues Sys- phies, integrating up-to-date information on phe-
USA tem zur Klassifizierung der Hornhautdystrophien notypic description, pathologic examination, and
Tel.: ++ 3 13/5 77/29 64
zu entwickeln, das gleichzeitig aktuelle Daten phä- genetic analysis.
Fax: ++ 3 13/5 77/50 99
jweiss@med.wayne.edu
notypischer Beschreibung, pathologischer Unter- Methods: The International Committee for Clas-
suchung und Genanalyse miteinbezieht. sification of Corneal Dystrophies (IC3D) was cre-
Methoden: Zur Erstellung einer aktuellen und ated to devise a current and accurate nomencla-
Übersetzte Originalarbeit.
Ursprünglich erschienen in
exakten Nomenklatur wurde das International ture.
Cornea, Volume 27, Suppl. 2, Committee for Classification of Corneal Dystro- Results: This anatomic classification continues to
December 2008 phies (IC3D) gegründet. organize dystrophies according to the level chief-
Ergebnisse: Diese anatomische Klassifikation ly affected. Each dystrophy has a template sum-
führt die Einordnung der Dystrophien nach den marizing genetic, clinical, and pathologic infor-
hauptsächlich betroffenen Hornhautschichten fort. mation. A category number from 1 through 4 is
Jede Dystrophie besitzt ein Schema aus klinischen, assigned, reflecting the level of evidence support-
pathomorphologischen und genetischen Informa- ing the existence of a given dystrophy. The most
tionen. Die Einordnung in die Kategorien 1 – 4 defined dystrophies belong to category 1 (a well-
spiegelt den Wissensstand über die jeweilige defined corneal dystrophy in which a gene has
Dystrophie wider. Die am besten definierten Dys- been mapped and identified and specific muta-
trophien sind in Kategorie 1 zu finden (eine gut tions are known) and the least defined belong to
definierte Hornhautdystrophie, in der ein Gen ent- category 4 (a suspected dystrophy where the clin-
schlüsselt und identifiziert wurde und spezifische ical and genetic evidence is not yet convincing).
Mutationen bekannt sind) und die am wenigsten The nomenclature may be updated over time as
definierten Dystrophien gehören in Kategorie 4 new information regarding the dystrophies be-
(eine mögliche Dystrophie, deren klinischer und comes available.
genetischer Nachweis noch nicht überzeugend er- Conclusions: The IC3D Classification of Corneal
bracht werden konnte). Bei Vorliegen neuer Infor- Dystrophies is a new classification system that in-
mationen kann nach einer gewissen Zeit die No- corporates many aspects of the traditional defini-
menklatur überarbeitet werden. tions of corneal dystrophies with new genetic,
clinical, and pathologic information. Standardized
1
Aus dem Englischen ins Deutsche übersetzt von Birgit templates provide key information that includes a
Scherff und Walter Lisch. level of evidence for there being a corneal dystro-
Weiss JS et al. IC3D-Klassifikation von Hornhautdystrophien… Klin Monatsbl Augenheilkd 2011; 228 Suppl. 1: S1 – S39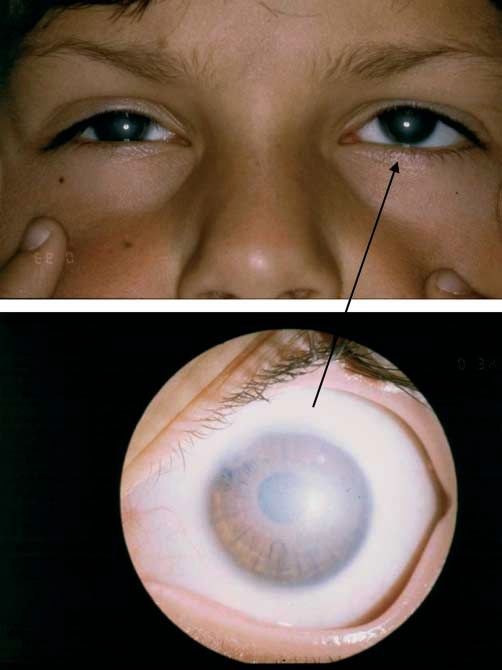
S2
Schlussfolgerungen : Die IC 3D-Klassifikation kornealer Dystro- phy. The system is user-friendly and upgradeable and can be re-
phien ist ein neues Klassifizierungssystem, das sowohl viele trieved on the website www.corneasociety.org/ic3d.
Aspekte herkömmlicher Definitionen kornealer Dystrophien als Key Words: corneal dystrophy, inherited corneal disease, genet-
auch neue genetische, klinische und histopathologische Infor- ic corneal disease, corneal histopathology, gene, mutation, key
mationen berücksichtigt. Die standardisierten Schemata bieten reference, eponym, epithelial basement membrane dystrophy,
Schlüsselinformationen, die eine Grundlage für das Vorhanden- epithelial recurrent erosion dystrophy, subepithelial mucinous
sein einer Hornhautdystrophie beinhalten. Das System ist benut- corneal dystrophy, Meesmann corneal dystrophy, Lisch epithelial
zerfreundlich und erweiterbar und kann auf folgender Webseite corneal dystrophy, gelatinous drop-like corneal dystrophy, Gray-
abgerufen werden: www.corneasociety.org/IC3D (englische Ver- son-Wilbrandt corneal dystrophy, lattice corneal dystrophy, lat-
sion); online-Datenbank Thieme (deutsche Version). tice gelsolin type dystrophy, granular corneal dystrophy 1, gran-
Schlüsselwörter: Hornhautdystrophie (HD), hereditäre korneale ular corneal dystrophy 2, Avellino corneal dystrophy, Reis-
Erkrankung, genetische korneale Erkrankung, korneale Histopatho- Bücklers corneal dystrophy, Thiel-Behnke corneal dystrophy, ma-
logie, Gen, Mutation, Schlüsselreferenz, Eponym, Epitheliale Basal- cular corneal dystrophy, Schnyder corneal dystrophy, Schnyder
membran-Dystrophie (EBMD), Epitheliale Rezidivierende Erosions- crystalline corneal dystrophy, congenital stromal corneal dystro-
dystrophie (ERED), Subepitheliale muzinöse Hornhautdystrophie phy, fleck corneal dystrophy, posterior amorphous corneal dys-
(SMCD), Meesmann-Hornhautdystrophie (MECD), Lisch-epitheliale trophy, central cloudy dystrophy of Francxois, pre-Descemet cor-
Hornhautdystrophie (LECD), Gelatinöse tropfenförmige Horn- neal dystrophy, Fuchs endothelial corneal dystrophy, posterior
hautdystrophie (GDLD), Grayson-Wilbrandt-Hornhautdystrophie polymorphous corneal dystrophy, congenital hereditary endo-
(GWCD), Gittrige Hornhautdystrophie 1 (LCD1), Gittrige Hornhaut- thelial dystrophy 1, congenital hereditary endothelial dystrophy
dystrophie Gelsolin Typ (LCD2), Granuläre Hornhautdystrophie 1 2, X-linked endothelial corneal dystrophy
Heruntergeladen von: Thieme Verlagsgruppe. Urheberrechtlich geschützt.
(GCD1), Granuläre Hornhautdystrophie 2 (GCD2), „Avellino“ Horn-
hautdystrophie, Reis-Bücklers-Hornhautdystrophie (RBCD), Thiel-
Behnke-Hornhautdystrophie (TBCD), Makuläre Hornhautdystro-
phie (MCD), Schnyder-Hornhautdystrophie (SCD), Kongenitale
stromale-Hornhautdystrophie (CSCD), Fleckchen-Hornhautdystro-
phie (FCD), Posteriore amorphe Hornhautdystrophie (PACD), Zen-
tral-wolkenfömige Hornhautdystrophie (François) (CCDF), Prae-
Descemet-Hornhautdystrophie (PDCD), Fuchs-endotheliale Horn-
hautdystrophie (FECD), Hintere polymorphe Hornhautdystrophie
(PPCD), Kongenitale hereditäre Endotheldystrophie 1 (CHED1),
Kongenitale hereditäre Endotheldystrophie 2 (CHED2), X-gebunde-
ne Endothel-Hornhautdystrophie (XECD).
Historie eine ererbte Funktionsstörung verwendet, die Zellen, Gewebe
! oder Organe isoliert oder in Kombination in Mitleidenschaft
Der Begriff Dystrophie stammt aus dem Griechischen (dys = zieht. In der Augenheilkunde wurde der Begriff „Hornhautdys-
falsch, schwierig; trophe = Ernährung) [1] und wurde zum ersten trophie“ für eine Gruppe von erblichen Hornhauterkrankungen
Mal 1884 als eine Erkrankung der Muskulatur in der medizini- eingeführt, die bilateral, symmetrisch, langsam fortschreitend
schen Literatur von Wilhelm Erb (1840 – 1921) erwähnt [2]. sind, ohne Hinweis auf Umwelteinflüsse oder systemische Fak-
1890 veröffentlichte Arthur Groenouw (1862 – 1945) seinen toren [9]. Mit zunehmendem Kenntnisstand wurden Abwei-
klassischen Artikel, in dem er von 2 Patienten mit „noduli cor- chungen bezüglich dieser Definition festgestellt. So besteht bei
neae“ berichtet, davon 1 Patient mit granulärer Hornhautdystro- vielen Patienten mit epithelialer Basalmembran-Dystrophie
phie und der andere mit makulärer Hornhautdystrophie [3]. Zur (EBMD) kein Hinweis auf Vererbung. Einige Patienten mit hin-
gleichen Zeit hat Biber seine Dissertation über die gittrige Horn- terer polymorpher Hornhautdystrophie (PPCD) weisen nur
hautdystrophie veröffentlicht [4]. unilaterale Veränderungen auf. Bei makulärer Hornhautdystro-
In der Zeit vor der Anwendung der Spaltlampe war die Qualität phie findet sich bei einem Teil von Merkmalsträgern eine Im-
der Hornhautuntersuchung begrenzt. Groenouw unterschied an- munantwort auf antigenes Keratansulfat. Außerdem gibt es
fänglich nicht zwischen granulärer und makulärer HD. Eine fa- eine Anzahl erblicher, bilateraler Hornhauterkrankungen, wie
miliäre Disposition wurde nicht erwähnt. Trotzdem wurde in z. B. Cornea plana, die nicht als Hornhautdystrophien klassifi-
der Folgezeit von 2 Hornhautdystrophien gesprochen [5]. Fuchs ziert, sondern den kongenitalen Hornhaut-Anomalien zuge-
[6] verwendete den Begriff Dystrophie als Bezeichnung für Au- rechnet werden.
generkrankungen, die durch Mangelernährung, Fehlen von Hor-
monen, Blut und Nervenversorgung hervorgerufen werden. Spä-
ter wurde die Bezeichnung auch in Publikationen von Wilhelm Literatur zur Hornhautdystrophie
Uhthoff [7] und Yoshiharu Yoshida [8] verwendet. !
Bücklers [10], dessen Name später mit der Reis-Bücklers-Horn-
hautdystrophie in Verbindung gebracht wurde, ist die erste
Definition Hornhautdystrophie Klassifikation von Hornhautdystrophien (HD) zu verdanken. Er
! beschrieb die Unterschiede zwischen granulärer, gittriger und
Obwohl es viele Definitionen für das Wort „Dystrophie“ in der makulärer HD. Obwohl die Dystrophien anhand genetischer
medizinischen Literatur gibt, wird die Bezeichnung meist für Muster, Schweregrad, histopathologischer Merkmale oder bio-
Weiss JS et al. IC3D-Klassifikation von Hornhautdystrophien… Klin Monatsbl Augenheilkd 2011; 228 Suppl. 1: S1 – S39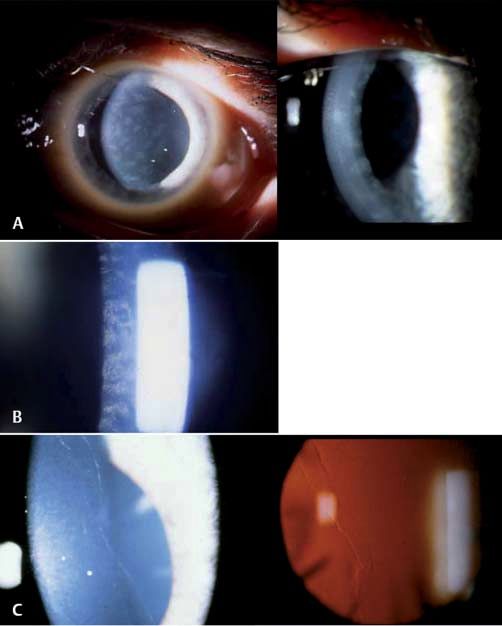
S3
chemischer Charakteristika unterschieden werden können, ba- Symptomatik war eine Nachuntersuchung des Originalstamm-
siert das meist gebräuchliche Klassifizierungssystem auf den kli- baums nicht möglich. Wir sind jedoch heute der Meinung,
nisch sichtbaren Hornhautveränderungen [9]. Klassischerweise dass die dargestellte Entität im Sinne der Reis-Bücklers-HD zu
werden die Dystrophien anhand der jeweilig betroffenen Horn- interpretieren ist, ohne jedoch die Thiel-Behnke-HD mit völli-
hautschicht in epitheliale und subepitheliale Dystrophien, Dys- ger Sicherheit ausschließen zu können.
trophien der Bowman-Lamelle, Stroma- und Endothel-Dystro- Vor und nach der Beschreibung der honigwabenförmigen Horn-
phien unterteilt [11 – 14]. hautdystrophie durch Thiel und Behnke im Jahre 1967 wurden
immer wieder Patienten mit dieser Dystrophie als Merkmalsträ-
ger der Reis-Bücklers-HD interpretiert [19]. Es dauerte mehr als
Mängel bei der Klassifikation von 30 Jahre, bis diese beiden Dystrophien in der Literatur richtig
Hornhautdystrophien interpretiert wurden. Andererseits beschrieben Grayson und
! Wilbrandt [27] eine Familie, die sie als Reis-Bücklers-artige HD
Ein kritischer Blick auf die Literatur über die verschiedenen bezeichnet hatten. Bei dem nachfolgend als Grayson-Wilbrandt-
HD deckt zahlreiche Missverständnisse und Irrtümer auf. In HD bezeichneten Krankheitsbild konnte bisher nicht der Beweis
vielen Publikationen wird beispielsweise die Notwendigkeit ei- erbracht werden, ob es sich um eine eigene Entität oder aber
nes Nachweises von Hornhautkristallen hervorgehoben, um eine Variante der Bowman-Dystrophie handelt.
eine Schnyder-Hornhautdystrophie (SCD) nachzuweisen [15 – Obgleich in der Originalpublikation über die zentral-wolkenför-
16]. Die Analyse zahlreicher Patientenstammbäume mit dieser mige HD von François [28] eine hereditäre Hornhauttrübung be-
Dystrophie zeigt, dass nur 50 % der betroffenen Patienten schrieben wurde, gab es nur wenige andere Publikationen, in
Hornhautkristalle aufweisen [17]. Gleichwohl haben Publika- der eine komplette Familie mit diesem Krankheitsbild vorge-
Heruntergeladen von: Thieme Verlagsgruppe. Urheberrechtlich geschützt.
tionen der vergangenen Jahrzehnte immer wieder irrtümlich stellt wurde [29, 30]. Beide Artikel wurden vor der Einführung
das Vorhandensein von Kristallen als Voraussetzung zur Diag- der Genotypisierung geschrieben, sodass keine genetischen In-
nose von SCD hervorgehoben [18]. formationen zur Verfügung stehen. Die zentral-wolkenförmige
Die Folge war, dass bei einigen Patienten ohne stromale Kris- HD von François ist klinisch nicht von dem degenerativen Be-
talle die Diagnose SCD erst nach Jahrzehnten gestellt wurde fund eines „posterior crocodile shagreen“ zu unterscheiden [31].
[17]. Eine einmal in Lehrbüchern dargestellte Charakterisie- Es ist nicht möglich festzustellen, ob frühere Veröffentlichungen
rung einer seltenen Erkrankung kann in der Folgezeit oft nur über einen einzelnen Patienten mit zentral-wolkenförmiger HD
mit Mühe korrigiert werden. Viele Mythen bleiben aufrecht- François vielmehr im Sinne eines degenerativen „posterior croc-
erhalten, weil nur wenige Augenärzte eine größere Anzahl odile shagreen“ zu interpretieren sind [32]. In Ermangelung
von seltenen und fragwürdigen Hornhautdystrophien gesehen weiterer betroffener Stammbäume oder genetischer Studien,
haben. die eine Vererbung bestätigen, ist es möglich, dass die zentral-
Eine weitere Schwierigkeit in der Literatur besteht in der Ten- wolkenförmige Dystrophie François und der Befund eines „pos-
denz, eine Beschreibung als neues oder seltenes Krankheitsbild terior crocodile shagreen“ identisch sind. Ohne genotypische In-
zu bewerten, bevor eine vollständige Analyse der eventuell formationen ist es nicht möglich, festzustellen, ob seltene oder
neuartigen Erkrankung erfolgt ist. Beispielsweise wurden die neu beschriebene Dystrophien tatsächlich eigene Krank-
histologischen Befunde von Patienten mit Thiel-Behnke-Horn- heitsbilder oder aber phänotypische degenerative Varianten dar-
hautdystrophie als solche von Reis-Bücklers-Hornhautdystro- stellen.
phie interpretiert [19]. In einer anderen Publikation wurde
die Reis-Bücklers-HD als eine seltene Variante der granulären
HD dargestellt [20, 21]. Diese Unstimmigkeiten in der Literatur Genetik
haben die richtige Einordnung der einzelnen HD erschwert. !
Die Entwicklung der genotypischen Analyse hat unser Wissen
über die Hornhautdystrophien sehr erweitert und zusätzliche
Existiert tatsächlich jede einzelne Dystrophie? Ungenauigkeiten in der Nomenklatur aufgezeigt. Die genetische
! Charakterisierung von Hornhautdystrophien erbrachte sowohl
Vor allem vor 1970 wurden neue Formen von Hornhautdystro- genetische Heterogenität durch verschiedene Gene, dem KRT3-
phie anhand ihrer klinischen und teilweise lichtmikroskopi- und KRT12-Gen für die Meesmann-HD, oder aber phänotypi-
schen Symptomatik identifiziert. In einigen Fällen basierte die sche Heterogenität durch ein einziges Gen, dem TGFBI-Gen, das
Beschreibung einer Dystrophie auf dem Bericht einer einzel- unterschiedliche allelische Phänotypien (Reis-Bücklers, Thiel-
nen Familie [22, 23]. Behnke, Granuläre HD Typ 1, Granuläre HD Typ 2, Gittrige HD
In anderen Fällen wurde eine neue Dystrophie als Variante ei- Typ 1) hervorrufen kann.
ner früher beschriebenen Dystrophie falsch eingeordnet. Die Die bisherige rein phänotypische Klassifikation der HD ent-
über Jahre in Artikeln und Lehrbüchern bezeichnete Waarden- spricht infolge der neueren genetischen Informationen nicht
burg-Jonkers-Dystrophie stellt jedoch ein Synonym für die mehr dem modernen wissenschaftlichen Kenntnisstand.
Thiel-Behnke-HD dar [25, 26]. Es ist oft nicht möglich, jede in
Lehrbüchern aufgenommene Form von Hornhautdystrophie als
eigene Entität zu bestätigen oder auszuschließen. Es dauerte Aktuelle Klassifikation der Hornhautdystrophien
oft lange, bis weit verbreitete Missverständnisse aufgeklärt !
werden konnten. So stellte sich zum Beispiel die Frage, ob das Der Wissensstand über Hornhautdystrophien hat sich, gegen-
von Reis und Bücklers ursprünglich dargestellte Krankheitsbild über den Erstbeschreibungen von granulärer, makulärer und
tatsächlich der heute bezeichneten Reis-Bücklers-HD ent- gittriger HD vor 100 Jahren, deutlich erweitert. Das Wort
spricht [22, 23]. Neben der nicht umfassend dargestellten „Dystrophie“ als auch Namensgebungen für die jeweilige HD-
Weiss JS et al. IC3D-Klassifikation von Hornhautdystrophien… Klin Monatsbl Augenheilkd 2011; 228 Suppl. 1: S1 – S39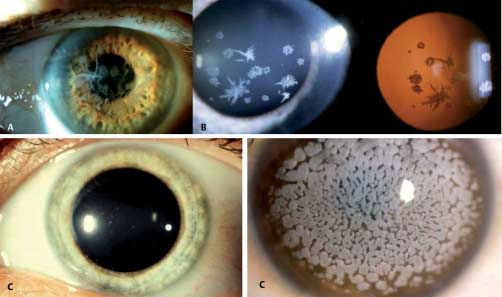
S4
Form wurden kritisch hinterfragt. Das Konzept der bisherigen dung eines internationalen Komitees zu unterstützen, um die
Nomenklatur basierte mehr auf historischen und weniger auf bisherige Nomenklatur der Hornhautdystrophien zu revidieren.
wissenschaftlichen Zusammenhängen. Die populäre Bezeich- Das Ziel war die Bildung einer internationalen Expertengruppe,
nung „Dystrophie“ behielt ihren Stellenwert trotz der Vor- die umfassende persönliche Erfahrungen mit der klinischen, his-
schläge, die Krankheitsbilder als erbliche Erkrankungen der tologischen und genetischen Symptomatik von Hornhautdystro-
Hornhaut zu benennen [33]. phien aufweisen kann. Nach kritischer Bewertung der bisheri-
gen Literatur sollten dabei die jeweiligen Fakten dargestellt und
veraltete sowie ungenaue Informationen entfernt werden. Mit
Änderung der Nomenklatur in anderen der Unterstützung von Dr. Michael W. Belin, dem Präsidenten
medizinischen Fachbereichen der Cornea Society, wurden ophthalmologische Fachgesellschaf-
! ten aller 5 Erdteile kontaktiert, um Hornhautspezialisten, Oph-
Nicht nur in der Augenheilkunde wurde festgestellt, dass die thalmo-Pathologen sowie Genetiker für die Bewältigung dieser
Nomenklatur einzelner Krankheitsbilder nicht mehr zeitgemäß gemeinsamen Aufgabe zu rekrutieren.
ist. Schnelle Fortschritte in der genetischen Typisierung von Das internationale Komitee zur Klassifikation der Hornhaut-
Krankheiten in anderen Fachbereichen führten zu Korrekturen dystrophien (IC3D) hatte seine 1. Zusammenkunft anlässlich
in der Nomenklatur. In einigen Bereichen bestand die Heraus- der Jahrestagung der American Academy of Ophthalmology
forderung, neue Nomenklatursysteme zu entwickeln. Im Jahre im Oktober 2005 in Chicago, gefolgt von Sitzungen im Februar
2001 wurde von der Europäischen Akademie für Allergie und 2006 am World Ophthalmology Congress in São Paulo, im Mai
klinische Immunologie eine neue Nomenklatur für Allergie 2006 bei der Association for Research in Vision and Ophthal-
nach „jahrelanger Diskussion unter zahlreichen Kinderärzten mology in Fort Lauderdale, im Oktober 2006 bei der American
Heruntergeladen von: Thieme Verlagsgruppe. Urheberrechtlich geschützt.
Europas“ vorgestellt [34]. Einer der Autoren führte aus, dass Academy of Ophthalmology in Las Vegas und schließlich im
er über verschiedene Bereiche der Pädiatrie Empfehlungen April 2007 bei der Association of Cataract and Refractive Sur-
aufgestellt und die einzelnen Pädiater in Abständen nach de- geons in San Diego. Dazwischen erfolgte die Online-Diskussion
ren Einschätzung des jeweiligen Vorschlags befragt habe. zwischen den Komitee-Mitgliedern mittels unzähliger E-Mails,
Nachfolgende diesbezügliche Publikationen unterschätzten um das Projekt voranzubringen.
den Wert einer Nomenklatur für atopische und allergische
Krankheitsbilder in Bezug auf Einstufung der jeweiligen indivi-
duellen Erkrankung und der damit durchzuführenden Therapie Merkmale der neuen Nomenklatur
[35]. !
Die Nomenklatur der Muskeldystrophie gab Anlass zu Ausein- Bei der ersten Zusammenkunft wurde über die notwendigen
andersetzungen zwischen Grundlagenforschern und Klinikern Merkmale einer neuen Nomenklatur diskutiert, die genauer, in-
infolge der unterschiedlichen Begriffsbestimmungen. Dubowitz formativer und leichter zu handhaben sein sollte, um damit tat-
wies auf das große Problem bei der Therapie der Muskeldys- sächlich die seit einem Jahrhundert verwendete Nomenklatur zu
trophie hin, das durch eine inadäquate Nomenklatur entsteht ersetzen – eine große Aufgabe, deren Erfolg sich erst mit der
und damit eine negative Auswirkung auf den gesamten Fra- Zeit herausstellen würde. Die neue Nomenklatur sollte den ak-
genkomplex nach sich zieht [36]. Klein schrieb, dass „histo- tuellen klinischen, histologischen und genetischen Wissensstand
risch gesehen, zwei prägende Phasen die aktuellen bzw. sich widerspiegeln, dabei aber leicht ergänzbar sein für aktuelle Er-
weiter entwickelten Klassifizierungsschemata beeinflusst ha- kenntnisse über neue Gene und Mutationen, jedoch benutzer-
ben: 1. Die Definition klinisch-pathologischer Krankheitsbilder freundlich sein durch Anbindung an die alte Nomenklatur.
Anfang des 20. Jahrhunderts und 2. die Anwendung der mole-
kularen Neurogenetik in den letzten 10 – 15 Jahren.“ Er fasste
zusammen, „dass die Mängel der bestehenden Klassifikations- Die IC 3D-Vorlagen („Templates“)
schemata nicht allein auf die komplexe Natur der Krank- !
heitsbilder, sondern auf den Versuch zurückzuführen sind, kli- Die Aufstellung von präzisen und aktuellen Vorlagen für jede
nische, histologische und genetische Charakteristika in einem einzelne Form von Hornhautdystrophie diente als Grundlage
Komplex zusammenzufassen.“ Er führte weiter aus, dass eine für die Erstellung einer neuen Nomenklatur. Jede Dystrophie-
„exakte klinische Diagnose ein wichtiger Schritt für die Klassi- Vorlage beinhaltete eine Zusammenfassung der aktuellen klini-
fikation darstellt, obschon genetische Untergliederungen häu- schen, histologischen und genetischen Befunde, wobei charak-
fig zur Anwendung kommen“ [37]. Der Autor plädierte für teristische klinische Bilder beigefügt wurden. Dieses Konzept
Klassifizierungen, die auf klinisch-histologischen, genetischen ermöglichte es auch, etwaige Fehler in der Literatur zu korri-
sowie molekularen Untersuchungsergebnissen basieren. gieren im Sinne „einer besseren Objektivierung“. Das IC 3D-Ko-
mitee unterzog die bisherige Literatur einer kritischen Über-
prüfung mit besonderer Beteiligung der Mitglieder, die
Die Gründung des Internationalen Komitees zur Klassi- persönliche Erfahrungen mit einem bestimmten Krankheitsbild
fikation der Hornhaut-Dystrophien (IC3D) hatten. Einige wenige Formen von Hornhautdystrophien stüt-
! zen sich lediglich auf eine einzige Publikation oder auf nur
Die wissenschaftliche Sitzung über Hornhautdystrophien anläss- ganz wenige Literaturstellen, sodass von Seiten des IC 3D-Ko-
lich des Welt-Cornea-Kongresses im April 2005 stellte eindeutig mitees über diese Thematik keine oder nur wenig persönliche
fest, dass Probleme bezüglich Nomenklatur nicht nur bei der Erfahrung in die Diskussion eingebracht werden konnte. Die
Schnyder-HD, sondern auch bei vielen anderen Formen von HD vom Komitee eingeschlagene Vorgehensweise erwies sich
bestehen. Dr. Jayne S. Weiss trat an die anderen Direktoriums- grundsätzlich als sehr effektiv.
Mitglieder der „Cornea Society“ mit der Bitte heran, die Grün-
Weiss JS et al. IC3D-Klassifikation von Hornhautdystrophien… Klin Monatsbl Augenheilkd 2011; 228 Suppl. 1: S1 – S39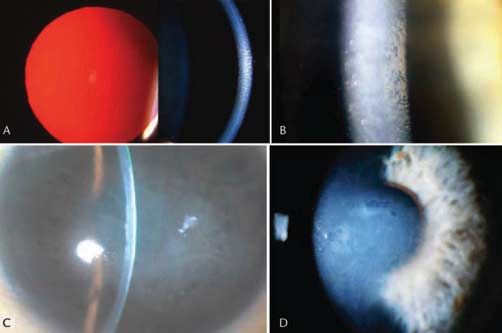
S5
Klassifikation und Entwicklung einer mit dem der nicht kristallinen Schnyder-HD (SCD) völlig iden-
Hornhautdystrophie tisch ist. Das IC 3D-Komitee überprüfte nun die CDCD-Publika-
! tion, ob dieses Krankheitsbild als neue Entität oder aber als
Die größte Herausforderung für das Komitee bestand darin, Variante der SCD einzustufen ist. Eine genetische Untersu-
eine möglichst flexible Klassifikation aufzustellen, in der pro- chung des CDCD-Probanden konnte erfolgen, nachdem das
blemlos zukünftige neue Erkenntnisse, besonders auch auf verantwortliche UBIADI-Gen für die SCD identifiziert wurde
dem Gebiet der Genetik, zu integrieren sind. Der Nachweis ei- [39, 40]. Es konnte eruiert werden, dass der CDCD-Proband
ner eigenständigen Form von Hornhautdystrophie beginnt mit eine spezielle Mutation im UBIADI-Gen aufwies (persönliche
der Beschreibung des klinischen Phänotyps, der sich nach Mitteilung von Jayne S. Weiss), die bei 100 Kontrollpersonen
Möglichkeit die Lokalisation am entsprechenden Chromosom nicht gefunden wurde. Die Mutation im UBIADI-Gen und der
sowie die Identifizierung des Gens und seiner Mutationen an- histologische Hornhautbefund im Sinne von stromalen Vakuo-
schließen sollten. Die Erstbeschreibung einer Hornhautdystro- len mit herausgelöstem Lipidmaterial ergaben den eindeutigen
phie zieht in der Regel weitere Untersuchungen nach sich. Hinweis, dass die sog. „Central discoid corneal dystrophy“ der
Eine zunächst klinisch identifizierte Form von Hornhautdystro- Schnyder-HD zuzuordnen ist. Die Konsequenz bestand darin,
phie führt häufig zu einer Sehverschlechterung mit der Folge diese sog. Kategorie-4-Dystrophie aus der Liste zu entfernen
einer durchzuführenden Keratoplastik. Dadurch können spezi- und in die Literatur der SCD aufzunehmen. Dieses Beispiel de-
fische histologische Befunde für die jeweilige Hornhautdystro- monstriert eindrucksvoll die Bedeutung und den Nutzen des
phie ermittelt werden. Falls eine histologische Untersuchung IC 3D-Klassifizierungssystems. Bei einem Krankheitsbild der Ka-
vorerst nicht möglich ist, sind genetische Analysen insbeson- tegorie 4 können neue Erkenntnisse aufzeigen, ob es sich tat-
dere bei Familien mit einfacher Mendelscher Vererbung anzu- sächlich um eine neue Dystrophie oder aber um die Variante
Heruntergeladen von: Thieme Verlagsgruppe. Urheberrechtlich geschützt.
streben, um die Lokalisation des Krankheitsbilds am jeweiligen einer bereits bekannten Entität handelt.
Chromosom zu bestimmen. Diese Untersuchungen können
schwierig und zeitaufwendig sein, vor allem, wenn mehr als
1 Gen im Sinne einer genotypischen Heterogenität verantwort- Das neue Klassifikationssystem
lich ist oder aber Wechselbeziehungen zwischen genetischen !
und umweltbedingten Faktoren postuliert werden können. Unsere vorgeschlagene Klassifikation der Hornhautdystrophien
Die erweiterte Genanalyse im Sinne von Identifizierung des basiert auf der Anatomie der Hornhaut (www.corneasociety.
jeweiligen Gens und der verschiedenen Mutationen ermög- org/ic3 d), wobei die einzelnen Dystrophien nach der haupt-
licht es, ätiologische Zusammenhänge mit den verschiedenen sächlich betroffenen Schicht unterteilt sind. Wir unterscheiden
phänotypischen Formen eines Krankheitsbilds zu diskutieren. zwischen epithelial und subepithelial, Bowman’scher Lamelle,
Die Identifizierung des Gens stellt einen ersten Schritt dar, stromal sowie Descemet’scher Membran und endothelial. Der
die molekularen Zusammenhänge der Entität eventuell besser Großteil der Dystrophiebezeichnungen ist ähnlich, aber auch
zu verstehen. Dadurch könnten sich auch therapeutische An- abweichend von der früheren Nomenklatur. So wurden die
sätze erschließen. TGFBI-Dystrophien aufgrund gemeinsamer genetischer Eigen-
Das IC 3D-Komitee hat die bisher bekannten Formen von Horn- schaften in einer eigenen Gruppe zusammengefasst.
hautdystrophien in 4 Kategorien unterteilt, um den Evidenz- Jede Vorlage („template“) der jeweiligen Dystrophieform be-
grad für die Existenz der jeweiligen Dystrophie aufzuzeigen. schreibt die klinischen, histologischen und genetischen Eigen-
schaften. Gleichzeitig wird das Krankheitsbild der Kategorie 1,
2, 3 oder 4 zugeordnet, je nach Wissenstand der klinischen,
Kategorien histologischen und genetischen Erkenntnisse. Eine detaillierte
! Zusammenfassung der genetischen Mutationen findet sich im
▶ Kategorie 1: Klinisch und histologisch klar definierte Dystro- Anhang.
phie mit Identifikation des Gens und der Mutationen
▶ Kategorie 2: Klinisch und histologisch klar definierte Dystro-
phie mit bekannter Chromosom-Lokalisation und unbekann- Literatur
ter Gen-Identifikation 1 Warburg M, Møller HU. Dystrophy: a revised definition. J Med Genet
▶ Kategorie 3: Klinisch und histologisch klar definierte Dystro- 1989; 26: 769–771
2 Erb W. Ueber die „juvenile Form“ der progressiven Muskelatrophie und
phie ohne genetische Analyse ihre Beziehungen der sogenannten Pseudohypertrophie der Muskeln.
▶ Kategorie 4: Verdacht auf eine neue oder bereits dokumen- Dtsch Arch Klin Med 1884; 34: 467–519
tierte Dystrophie, wobei die Eigenständigkeit noch nicht be- 3 Groenouw A. Knötchenförmige Hornhauttrübungen „Noduli Corneae.
wiesen ist Arch Augenheilkd 1890; 21: 281–289
4 Biber H. Ueber einige seltene Hornhauterkrankungen: die oberflächli-
Die für eine bestimmte Hornhautdystrophie zugeordnete Kate-
che gittrige Keratitis. Inaugural Dissertation. Zürich: 1980
gorie kann sich aufgrund neuer Erkenntnisse im Laufe der Zeit 5 Møller HU. Granular corneal dystrophy Groenouw type I. Clinical and
ändern. Schließlich sollten alle anerkannten Hornhautdystro- genetic aspects. Acta Ophthalmol 1991; Suppl 198: 1–40
phien einmal der Kategorie 1 zugeordnet werden. So ist die 6 Fuchs E. Dystrophia epithelialis corneae. Graefe’s Arch Clin Exp Oph-
makuläre Hornhautdystrophie ein Beispiel für Kategorie 1. Es thalmol 1910; 76: 478–508
7 Uhthoff W. Ein Fall von doppelseitiger zentraler, punktförmiger, sub-
ist jedoch auch möglich, dass Dystrophien der Kategorie 4 in-
epithelialer „Knötchenförmiger Keratitis“ Groenouw mit anatomi-
folge neuer Erkenntnisse nicht mehr als eigenständige Krank- schem Befunde. Klin Monatsbl Augenheilkd 1915; 54: 377–383
heitsbilder eingestuft werden können und somit komplett aus 8 Yoshida Y. Über eine neue Art der Dystrophia corneae mit histologi-
der Liste herausgenommen werden müssen. So wurde bei der schem Befunde. Graefe’s Arch Clin Exp Ophthalmol 1924; 114: 91–100
„Central discoid corneal dystrophy“ (CDCD) [38] mit der Kate-
gorie 4 festgestellt, dass der phänotypische Hornhautbefund
Weiss JS et al. IC3D-Klassifikation von Hornhautdystrophien… Klin Monatsbl Augenheilkd 2011; 228 Suppl. 1: S1 – S39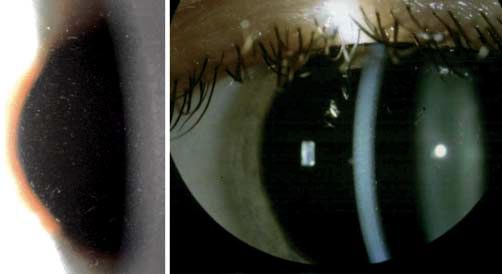
S6
9 American Academy of Ophthalmology. External diseases and cornea. In 38 Aldave AJ, Edward DP, Park AJ et al. Central discoid corneal dystrophy.
Sutphin JE, ed. Basic and Clinical Sciences Course 2007 – 2008. San Cornea 2002; 21: 739–744
Francisco, CA: American Academy of Ophthalmology, 2007: 305–329 39 Weiss JS, Kruth HS, Kuivaniemi H et al. Mutations in the UBIAD1 gene
10 Bücklers M. Die erblichen Hornhautdystrophien. Klin Monatsbl Augen- on chromosome short arm 1, region 36 cause Schnyder crystalline cor-
heilkd 1938; Beiheft 3: 1–135 neal dystrophy. Invest Ophthalmol Vis Sci 2007; 48: 5007–5012
11 Duke-Elder S, Leigh AG. Corneal dystrophies. In Duke-Elder S, ed. Sys- 40 Orr A, Sube MP, Marcadier et al. Mutations in the UBIAD1 gene en-
tem of Ophthalmology. Vol VIII, part 2 London: Kimpton, 1965: 864– coding a potential prenyltransferase are causal for Schnyder crystal-
867 line corneal dystrophy. PLoS ONE 2007; 2: e685
12 Waring GO, Rodrigues MM, Laibson PR. Corneal dystrophies. I. Dystro-
phies of the epithelium, Bowman’s layer and stroma. Surv Ophthalmol
1978; 23: 71–122
13 Klein D, Franceschetti A. Heredo-familiäre Hornhautdystrophien. In
Die IC 3D-Klassifikation (C = Kategorie)
Becker PD, ed. Humangenetik. Vol IV. Stuttgart: Georg Thieme, 1964:
!
80–94
14 Klintworth GK. The molecular genetics of the corneal dystrophies – Epitheliale und subepitheliale Dystrophien
current status. Front Biosci 2003; 8: 687–713 1. Epitheliale Basalmembran-Dystrophie (EBMD) – überwie-
15 Weiss JS. Schnyder’s dystrophy of the cornea. A Swede-Finn connec- gend degenerativ, einige C 1
tion. Cornea 1992; 11: 93–100
2. Epitheliale rezidivierende Erosionsdystrophie (ERED) C 3
16 Weiss JS. Schnyder crystalline dystrophy sine crystals. Recommenda-
tion for a revision of nomenclature. Ophthalmology 1996; 103: 465– (Smolandiensis-Variante)
473 3. Subepitheliale muzinöse Hornhautdystrophie (SMCD) C 4
17 Weiss JS. Visual morbidity in 34 families with Schnyder’s crystalline 4. Meesmann-Hornhautdystrophie (MECD) C 1
corneal dystrophy. Trans Am Ophthamol Soc 2007; 105: 616–648 5. Lisch-epitheliale Hornhautdystrophie (LECD) C 2
18 McCarthy MM, Innis S, Dubord P et al. Panstromal Schnyder corneal
6. Gelatinöse tropfenförmige Hornhautdystrophie (GDLD) C 1
Heruntergeladen von: Thieme Verlagsgruppe. Urheberrechtlich geschützt.
dystrophy. A clinical pathologic report with quantitative analysis of
corneal lipid composition. Ophthalmology 1994; 101: 895–901
19 Kanai A, Kaufman HE, Polack FM. Electron microscopic study of Reis- TGFBI-Dystrophien (Dystrophien der Bowman-Lamelle)
Bücklers dystrophy. Ann Ophthalmol 1973; 5: 953–962 1. Reis-Bücklers-Hornhautdystrophie (RBCD) – Granuläre Horn-
20 Haddad R, Font RL, Fine BS. Unusual superficial variant of granular dys-
hautdystrophie Typ 3 C 1
trophy of the cornea. Am J Ophthalmol 1977; 83: 213–218
21 Møller HU. Interfamilial variability and intra-familial similarities of
2. Thiel-Behnke-Hornhautdystrophie (TBCD) C 1, potenzielle Va-
granular corneal dystrophy Groenouw type I with respect to biomi- riante C 2
croscopical appearance and symptomatology. Acta Ophthalmol 1989; 3. Grayson-Wilbrandt-Hornhautdystrophie (GWCD) C 4
67: 669–677
22 Reis W. Familiäre, fleckige Hornhautentartung. Dtsch Med Wochen-
TGFBI-Dystrophien
schr 1917; 43: 575
23 Bücklers M. Über eine weitere familiäre Hornhautdystrophie (Reis). 1. Gittrige Hornhautdystrophien C 1
Klin Monatsbl Augenkeilkd 1949; 114: 386–397 a) Gittrige Hornhautdystrophie, TGFBI-Typ (LCD): Klassische
24 Waardenburg PJ, Jonkers GH. A specific type of dominant progressive gittrige Hornhautdystrophie, Typ 1 (LCD1) C 1, Varianten
dystrophy of the cornea, developing after birth. Acta Ophthalmol (III, IIIA, I/IIIA, IV) C 1
1961; 39: 919–923
b) Gittrige Hornhautdystrophie, Gelsolin Typ (LCD2) C 1 (Es
25 Thiel HJ, Behnke H. Eine bisher unbekannte subepitheliale hereditäre
Hornhautdystrophie. Klin Monatsbl Augenheilkd 1967; 150: 862–874 handelt sich hierbei nicht um eine echte Hornhautdystro-
26 Wittebol-Post D, Van Schooneveld MJ, Pels E. The corneal dystrophy of phie, wird hier aber aufgeführt, um eine differenzierte Di-
Waardenburg and Jonkers. Ophthalmic Paediatr Genet 1989; 10: agnose zu ermöglichen.)
249–255 2. Granuläre Hornhautdystrophie C 1
27 Grayson M, Wilbrandt H. Dystrophy of the anterior limiting membrane
of the cornea (Reis-Bücklers type). Am J Ophthalmol 1966; 61: 345–
a) Granuläre Hornhautdystrophie, Typ 1 (klassisch) (GCD1)
349 C1
28 François J. Une nouvelle dystrophie hérédo-familiale de la cornée. J Ge- b) Granuläre Hornhautdystrophie, Typ 2 (granulär-gittrig)
net Hum 1956; 5: 189–196 (GCD2) C 1
29 Strachan IM. Cloudy central corneal dystrophy of François. Five cases in
the same family. Br J Ophthalmol 1969; 53: 192–194
30 Bramsen T, Ehlers N, Baggesen LH. Central cloudy corneal dystrophy of Stromale Dystrophien
François. Acta Ophthalmol 1976; 54: 221–226 1. Makuläre Hornhautdystrophie (MCD) C 1
31 Meyer JC, Quantock AJ, Thonar EJ et al. Characterization of a central cor- 2. Schnyder-Hornhautdystrophie (SCD) C 1
neal cloudiness sharing features of posterior crocodile shagreen and 3. Kongenitale stromale Hornhautdystrophie (CSCD) C 1
central cloudy dystrophy of François. Cornea 1996; 15: 347–354
4. Fleckchen-Hornhautdystrophie (FCD) C 1
32 Karp CL, Scott IU, Green WR et al. Central cloudy corneal dystrophy of
François. A clinicopathologic study. Arch Ophthalmol 1997; 115: 5. Posteriore amorphe Hornhautdystrophie (PACD) C 3
1058–1062 6. Zentral-wolkenförmige Dystrophie (François) (CCDF) C 4
33 Kintworth GK. Genetic disorders of the cornea: from research to practi- 7. Prae-Descemet-Hornhautdystrophie (PDCD) C 4
cal diagnostic testing. Graefes Arch Clin Exp Ophthalmol 2005; 33:
231–232
34 Johansson G, Hourihane JO, Bousquet J et al. A revised nomenclature for
Descemet-Membran- und Endothel-Dystrophien
allergy. An EAACI position statement from the EAACI nomenclature 1. Fuchs-endotheliale Hornhautdystrophie (FECD) C 1, C 2 oder
task force. Allergy 2001; 56: 813–824 C3
35 Dreborg S. The implications of nomenclature. Ann Allergy Asthma Im- 2. Hintere polymorphe Hornhautdystrophie (PPCD) C 1 oder C 2
munol 2002; 89: S83–S85
3. Kongenitale hereditäre Endotheldystrophie 1 (CHED1) C 2
36 Dubovitz V. Current and future therapy in muscular dystrophy; need
for a common language between basic scientists and clinicians. Acta
4. Kongenitale hereditäre Endotheldystrophie 2 (CHED 2) C 1
Myol 2004; 23: V–IX 5. X-gebundene Endothel-Hornhautdystrophie (XECD) C 2
37 Klein C. Movement disorders: Classification. J Inherit Metab Dis 2005; (●
▶ Tab. 1)
28: 425–439
Weiss JS et al. IC3D-Klassifikation von Hornhautdystrophien… Klin Monatsbl Augenheilkd 2011; 228 Suppl. 1: S1 – S39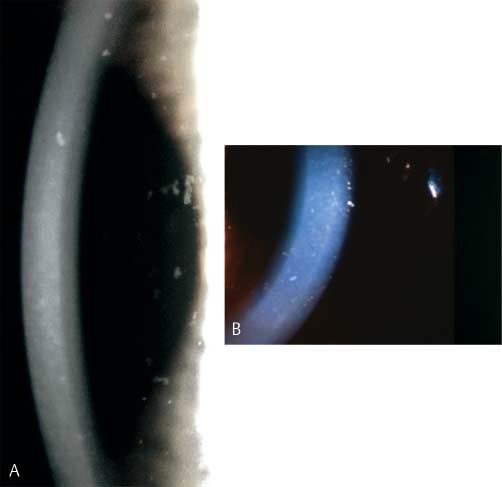
S7
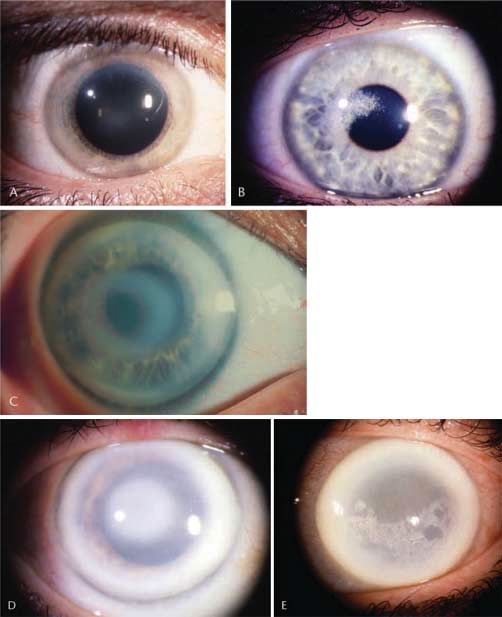
Abb. 1 Epitheliale Basalmembrandystrophie.
A Landkartenförmige Veränderungen („Maps“).
B Intraepitheliale Flecktrübungen („Cogan dots“) +
„maps“. C „Fingerprint lines“ im regredienten Licht.
Tab. 1 Die IC 3D-Klassifikation – Abkürzungen und MIM-Nummer.1 Gen: TGFBI (nur in vereinzelten Fällen)
MIM1- IC3D- MIM1#
Beginn: Im Erwachsenenalter. Selten bei Kindern.
Abkürzung Abkürzung
epitheliale Basalmembran-HD EBMD EBMD 121 820
Klinisches Bild (●
▶ Abb. 1)
epitheliale rezidivierende keine ERED 122 400
Landkarten oder „Maps“: Unregelmäßige Inseln mit verdick-
Erosionsdystrophie
tem, grauem Epithel und umrissenen Grenzen, die vor allem
subepitheliale muzinöse HD keine SMCD keine
Meesmann-HD keine MECD 122 100
die zentrale oder parazentrale Hornhaut befallen. Einzeln
Lisch-epitheliale HD keine LECD keine oder in Kombination mit anderen Zeichen.
gelatinöse tropfenförmige HD GDLD, CDGDL GDLD 204 870 Fleckchen oder Cogan’sche „Dots“: Unregelmäßig rund, oval
oder kommaförmig. Grau-weißliche Trübungen ohne Anfär-
Heruntergeladen von: Thieme Verlagsgruppe. Urheberrechtlich geschützt.
Reis-Bücklers-HD CDB1, CDRB, RBCD 608 470
RBCD bung. Archipelartig in der zentralen Hornhaut. Typischerweise
Thiel-Behnke-HD CDB2, CDTB TBCD 602 082 in Kombination mit anderen Zeichen, insbesondere geografisch
Grayson-Wilbrandt-HD keine GWCD keine angeordneter Linien (maps).
gittrige HD, TGFBI Typ CDL1 LCD 122 200 Fingerabdruck-Linien oder „Fingerprint-lines“: Parallele, finger-
klassische gittrige HD, Typ 1 LCD1
beerartige Linien, in der Regel parazentral. Am besten im re-
gittrige HD, Meretoja Typ Keine LCD2 105 120
gredienten Licht zu sehen. Einzeln oder in Kombination mit
granuläre HD, Typ 1 klassisch CGDD1 GCD1 121 900
anderen Zeichen, insbesondere „maps“.
granuläre HD, Typ 2 CDA, ACD GCD2 607 541
makuläre HD MCDC1 MCD 217 800
Bläschenmuster oder Bron’sche „Blebs“: Subepitheliale, kiesel-
Schnyder-HD keine SCD 121 800 glasartige Veränderungen, im regredienten Licht am besten
kongenitale stromale HD CSCD CSCD 610 048 sichtbar. Einzeln oder in Kombination mit anderen Zeichen.
Fleckchen-HD keine FCD 121 850 Schlechte Haftung der epithelialen Basalzellen an abnormaler
posteriore amorphe HD keine PACD keine Basalmembran wird als Prädisposition für rezidivierende Ero-
Zentral-wolkenförmige HD keine CCDF 217 600 sionen interpretiert.
(François)
Prae-Descemet-HD keine PDCD keine Symptome
Fuchs-endotheliale HD FECD1 FECD 136 800 Entweder symptomfrei oder rezidivierende Erosionen mit
hintere polymorphe HD PPCD1 PPCD 122 000
Schmerzen, Tränenfluss und Sehverminderung. Die zentralen
kongenitale hereditäre Endo- CHED1 CHED1 121 700
Veränderungen können infolge wechselndem irregulärem
theldystrophie 1
Astigmatismus zu Sehstörungen führen.
kongenitale hereditäre Endo- CHED2 CHED2 217 700
theldystrophie 2
X-gebundene Endothel-HD keine XECD keine Verlauf
1
HD = Hornhautdystrophie, MIM = Mendelian Inheritance in Man (Mendel-
Lage und Ausmaß der Veränderungen können mit der Zeit va-
sche Vererbung beim Menschen), OMIM = Online Mendelian Inheritance in riieren.
Man. McKusick VA et al. http://www.ncbi.nlm.nih.gov/sites/entrez.
Lichtmikroskopie
Landkarten („Maps“): Neuformation von intraepithelialem
Epitheliale und subepitheliale Dystrophien multilamellären Material, ausgehend von der Basalmembran.
! Fingerabdruck-Linien („Fingerprint-lines“): Rippenartige intra-
Epitheliale Basalmembran-Dystrophie (EBMD) epitheliale Veränderungen, ausgehend von der Basalmembran.
MIM: # 121 820 Fleckchen („Dots“): Intraepitheliale Pseudozysten mit zytoplas-
matischem Material.
Alternative Namen, Eponyme Bläschenmuster („Blebs“): Unregelmäßige, subepitheliale An-
▶ Map-Dot-Fingerprint-Dystrophie häufung von fokal abgegrenztem fibrillogranulären Material.
▶ Cogan mikrozystische Epitheldystrophie
▶ Vordere Basalmembran-Dystrophie Transmissions-Elektronenmikroskopie
„Maps“: Dicke epitheliale Basalmembran mit 2 – 6 mm dicken
Vererbung multilamellären Abspaltungen Richtung Epithel.
In den meisten Fällen wurde keine Vererbung mitgeteilt. Viele „Fingerprint-lines“: Feine fibrillogranuläre Substanz neben der
sind als degenerativ oder als Folge eines Traumas betrachtet Basalmembran. Die Fibrillen haben einen Durchmesser von
worden. Von familiären Fällen wurde berichtet. etwa 17 nm und das körnige Material von etwa 8 nm.
Genlocus: 5q 31 (eine Mitteilung)
Weiss JS et al. IC3D-Klassifikation von Hornhautdystrophien… Klin Monatsbl Augenheilkd 2011; 228 Suppl. 1: S1 – S39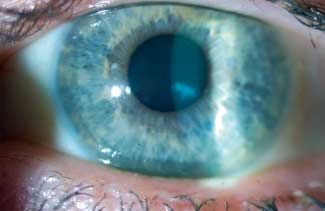
S8
„Dots“: Intraepitheliale Pseudozysten, die degenerative Zellen Abb. 2 Rezidivierende
mit pyknotischen Zellkernen und zytoplasmahaltigem Material epitheliale Erosionsdys-
enthalten. trophie (Smolandiensis-
Variante). Rechtes Auge
„Blebs“: Die Vorderseite der Basalmembran zeigt diskrete Vor-
einer 41-jährigen Frau
wölbungen, die die darüber liegenden basalen Epithelzellen
mit einer zentralen ke-
deformieren. loidartigen Trübung, die
bei der Hälfte der be-
Konfokale Mikroskopie troffenen Familienmit-
„Map-fingerprint-dot“: Intraepitheliale Basalmembran, die glieder zu sehen war.
ohne Verbindung zur unauffälligen Basalzellschicht zu stehen
scheint. Tropfenförmige Konfiguration im Epithel. Ringförmige
Struktur im Bereich der Basalzellen. Klinisches Bild (●
▶ Abb. 2)
Rezidivierende Hornhauterosionen erscheinen in der Regel im
Kategorie 4. – 6. Lebensjahr, gelegentlich aber auch schon im Alter von
Die meisten Fälle sind sporadisch und als degenerativ einzu- 8 Monaten. Sie entstehen durch ein kleines Trauma oder sind
schätzen. Kategorie 1 in seltenen Fällen. spontan. Die Hornhaut kann eine feine subepitheliale Trübung
oder Bläschen zwischen den Attacken aufweisen. In der Smo-
Literatur landiensis-Variante entwickelt die Hälfte der Patienten einzel-
1 Boutboul S, Black GCM, Moore JE, et al. A subset of patients with epithe- ne bzw. mehrere dauerhafte zentrale subepitheliale Hornhaut-
lial basement membrane corneal dystrophy have mutations in TGFBI/ trübungen, die sich frühestens im Alter von 7 Jahren zeigen.
Heruntergeladen von: Thieme Verlagsgruppe. Urheberrechtlich geschützt.
BIGH3. Hum Mutat 2006; 27: 553 – 557
2 Bron AJ, Brown NA. Some superficial corneal disorders. Trans Ophthal-
Das Erscheinungsbild reicht von subepithelialer Fibrose bis zu
mol Soc UK 1971; 91: 13 – 29 prominenten keloidartigen Knötchen.
3 Bron AJ, Tripathi RC. Cystic disorders of the corneal epithelium II. Pa-
thogenesis. Br J Ophthalmol 1973; 57: 361 – 375 Symptome
4 Cogan DG, Donaldson DD, Kuwabara T, Marshall D. Microcystic dystro-
Die meisten Patienten haben Anfälle von Rötung, Photophobie,
phy of the corneal epithelium. Trans Am Ophthalmol Soc 1964; 62:
213 – 225 Epiphora und Augenschmerzen. Einige haben Augenbrennen
5 Guerry D. Fingerprint lines in the cornea. Am J Ophthalmol 1950; 33: und berichten von empfindlichen Augen über mehrere Jahre.
724 – 726 Die Exposition gegenüber Sonnenlicht oder Zugluft, Staub,
6 Laibson PR, Krachmer JH. Familial occurrence of dot (microcystic), Rauch und der Mangel an Schlaf können Anfälle hervorrufen.
map, fingerprint dystrophy of the cornea. Invest Ophthalmol Vis Sci
Bei der Smolandiensis-Variante benötigt schließlich ein Viertel
1975; 14: 397 – 399
7 Laibson PR. Microcystic corneal dystrophy. Trans Am Ophthalmol Soc der Patienten Hornhauttransplantate im Alter von durch-
1976; 74: 488 – 531 schnittlich 44 Jahren. Die Rezidivtrübungen treten innerhalb
8 Lisch W, Lisch C. Die epitheliale Hornhaut-Basalmembran-Dystrophie. von 15 Monaten in der Transplantatperipherie wieder auf, wo-
Klin Monatsbl Augenheilkd 1983; 183: 251 – 255 bei jedoch der zentrale Anteil über viele Jahre klar bleibt.
9 Munier Fl, Korvatska E, Djemai A, et al. Kerato-epithelin mutations in
four 5q31-linked corneal dystrophies. Nat Genet 1997; 15: 247 – 251
10 Pogorelov P, Langenbucher A, Kruse FE, Seitz B. Long-term results of Verlauf
phototherapeutic keratectomy for corneal map-dot-fingerprint dys- Schmerzattacken nehmen allgemein im Laufe der Zeit an Häu-
trophy (Cogan-Guerry). Cornea 2006; 25: 774 – 777 figkeit und Intensität ab und enden meist im Alter von 50 Jah-
11 Rodrigues MM, Fine BS, Laibson PR, et al. Disorders of the corneal epi-
ren. Bei der Smolandiensis-Variante nehmen die zentralen
thelium. A clinicopathologic study of dot, geographic, and fingerprint
patterns. Arch Ophthalmol 1974; 92: 475 – 482
subepithelialen Trübungen zu.
12 Vogt A. Lehrbuch und Atlas der Spaltlampenmikroskopie des lebenden
Auges (1. Teil). Berlin: Springer, 1930: 119 – 121 Lichtmikroskopie
Die Veränderungen bei der Smolandiensis-Variante zeigen kei-
Epitheliale rezidivierende Erosionsdystrophie (ERED) ne Ähnlichkeiten mit denen von EBMD oder Dystrophien der
MIM: # 122 400 Bowman-Lamelle.
Alternative Namen, Eponyme Transmissions-Elektronenmikroskopie: Keine Mitteilung
Hereditäre rezidivierende Erosion der Hornhaut (Franceschetti)
Konfokale Mikroskopie: Keine Mitteilung
Varianten: Dystrophia Smolandiensis
Kategorie: 4, 3 (Smolandiensis-Variante)
Vererbung: Autosomal dominant
Literatur
Genlocus: Unbekannt 1 Hammar B, Björck E, Lagerstedt K, et al. A new corneal disease with re-
current erosive episodes and autosomal dominant inheritance. Acta
Ophthalmol Scand 2009; 87 (6): 659 – 665
Gene: Unbekannt; collagen type VIII alpha 2 (COL8A2), Transfor- 2 Franceschetti A. Hereditäre rezidivierende Erosion der Hornhaut. Z Au-
ming growth factor beta induced (TGFBI), Gelsolin (GSN), Keratin genheilk 1928; 66: 309 – 316
K3 (KRT3) und Keratin K12 (KRT12) wurden bei der Smolandien- 3 Valle O. Hereditary recurring corneal erosions: a family study, with
sis-Variante ausgeschlossen. special reference to Fuchs’ dystrophy. Acta Ophthalmol 1967; 45:
829 – 836
4 Wales HJ. A family history of corneal erosions. Trans Ophthalmol Soc
Beginn: 1. Lebensjahrzehnt NZ 1956; 8: 77 – 78
Weiss JS et al. IC3D-Klassifikation von Hornhautdystrophien… Klin Monatsbl Augenheilkd 2011; 228 Suppl. 1: S1 – S39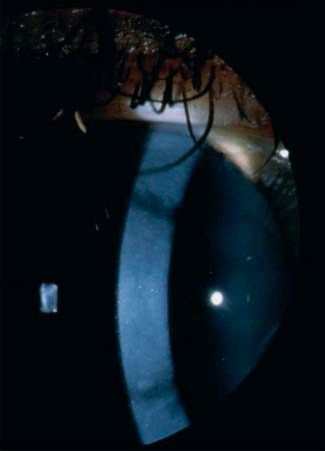
S9
Subepitheliale muzinöse Hornhautdystrophie (SMCD) Mutationen im Bereich der Keratin-Gene
MIM: nicht registriert !
Meesmann-Hornhautdystrophie (MECD) MIM: # 122 100
Alternative Namen, Eponyme: Keine
Alternative Namen, Eponyme: Juvenile hereditäre Epitheldys-
Vererbung: Autosomal dominant trophie
Genlocus: Nicht bekannt Variante: Stocker-Holt-Variante
Gen: Nicht bekannt Vererbung: Autosomal dominant
Beginn: 1. Lebensjahrzehnt Genlocus
▶ Locus 12q13 (KRT3) MIM * 148 043
Klinisches Bild (●
▶ Abb. 3): Bilaterale subepitheliale Trübungen ▶ Locus 17q12 (KRT12) MIM * 601 687 Stocker-Holt-Variante
und Verdichtungen („haze“) über die gesamte Hornhaut mit Be-
vorzugung des zentralen Bereichs. Gene
▶ Keratin K 3 (KRT3)
Symptome: Schmerzhafte Attacken aufgrund rezidivierender ▶ Keratin K 12 (KRT12) Stocker-Holt-Variante
Hornhauterosionen, die während der Pubertät abnehmen (bisher
nur Veröffentlichung einer Familie). Beginn: Frühe Kindheit
Heruntergeladen von: Thieme Verlagsgruppe. Urheberrechtlich geschützt.
Verlauf: Zunehmender Visusverlust während der Pubertät. Klinisches Bild (●
▶ Abb. 4)
Multiple feine Epithelbläschen, die bis zum Limbus reichen
Lichtmikroskopie: Subepitheliales Band im anterioren Bereich können. Sie finden sich am häufigsten in der interpalpebralen
der Bowman-Lamelle, das eosinophilem PAS-gefärbten, Alcian- Zone mit klarem Epithel zwischen den Zysten. Es wurde von
blau-positivem sowie Hyaluronidase-sensitivem Material ent- wirbel- und keilförmigen Epithelmustern berichtet. Die Horn-
spricht. haut kann verdünnt und die Sensibilität verringert sein.
Die direkte Beleuchtung zeigt unterschiedliche diffuse graue
Transmissions-Elektronenmikroskopie: Subepitheliale Ablage- Trübungen in verschiedenen Mustern, die eine deutliche Be-
rungen von feinem fibrillärem Material. grenzung haben. Bereiche der zentralen oder peripheren
Hornhaut können auch frei von Trübungen sein. Die grauen
Immunhistochemie: Kombination von Chondroitin-4-Sulfat Trübungen erscheinen als transparente runde Zysten bei indi-
und Dermatansulfat. rekter Beleuchtung. Die Verschmelzung mehrerer Zysten führt
zu refraktilen schlauchartigen Veränderungen.
Konfokale Mikroskopie: Keine Mitteilung
Stocker-Holt-Variante
Kategorie: 4 Die gesamte Hornhaut zeigt feine, grau gepunktete Epithel-
defekte, die mittels Fluorescein sichtbar werden und feine li-
Literatur neare Trübungen, die auch als wirbelförmiges Muster imponie-
1 Feder RS, Jay M, Yue BY, et al. Subepithelial mucinous corneal dystro- ren können.
phy. Clinical and pathological correlations. Arch Ophthalmol 1993;
111: 1106 – 1114
Verlauf: Langsam fortschreitend
Abb. 3 Subepitheliale
Symptome
muzinöse Hornhautdys- Die Patienten sind in der Regel ohne Symptome oder haben
trophie. Subepitheliale, eventuell geringe Visusbeeinträchtigungen, wobei einige Pa-
besonders zentral aus- tienten über Blend- und Lichtempfindlichkeit klagen. Rezidi-
geprägte Trübungen vierende, schmerzhafte punktförmige Epithelerosionen können
über die gesamte auftreten. In seltenen Fällen kann vernebeltes Sehen als Folge
Kornea (links Übersicht,
von Unregelmäßigkeiten und Vernarbungen der Hornhaut auf-
rechts Spalt).
treten.
Abb. 4 Meesmann-Hornhautdystrophie. A Regre-
dientes Licht: Viele, solitäre Mikrozysten, die im Lid-
spaltenbereich am deutlichsten zu sehen sind.
B Links: Diffuse, graue Trübung in direkter breiter
Beleuchtung; rechts: viele, solitäre Mikrozysten im
regredienten Licht.
Weiss JS et al. IC3D-Klassifikation von Hornhautdystrophien… Klin Monatsbl Augenheilkd 2011; 228 Suppl. 1: S1 – S39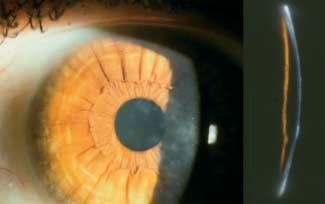
S10
Stocker-Holt-Variante Lisch-epitheliale Hornhautdystrophie (LECD) MIM: nicht
Im Vergleich zur MECD weisen Patienten mit dieser Variante registriert
einen früheren Beginn sowie eine verstärkte subjektive und
objektive Symptomatik auf. Genlocus: Xp 22.3
Lichtmikroskopie Gen: Unbekannt
Das Epithel zeigt immer intraepitheliale Zysten. Diese enthal-
ten PAS-positive und fluoreszierende Zellreste. Das Epithel Alternative Namen, Eponyme: Band- und wirbelförmige mi-
kann verdickt und ungeordnet sein. Verdickte multilaminare krozystische Dystrophie des Hornhautepithels
Basalmembran mit Fortsätzen in Richtung Basalzellen.
Vererbung: X-chromosomal dominant
Stocker-Holt-Variante
Variabel verdicktes Epithel mit intrazellulären Vakuolen und Beginn: Kindheit
Anzeichen von Degeneration. Unterschiedlich verdickte Basal-
membran. Normale Bowman-Lamelle und unauffälliges Stro- Klinisches Bild (●
▶ Abb. 5)
ma. Bei direkter Beleuchtung finden sich graue Trübungen in ver-
schiedenen Mustern: wirbelförmig, radial, bandförmig, gefie-
Transmissions-Elektronenmikroskopie dert und keulenförmig. Bei indirekter Beleuchtung können
Intrazytoplasmatische „peculiar substance“ bestehend aus ei- multiple dicht gedrängte klare Zysten beobachtet werden. Das
ner Ansammlung von fibrogranulärem Material, umgeben von umgebende Epithel ist klar. Kein Unterschied im Phänotyp
Heruntergeladen von: Thieme Verlagsgruppe. Urheberrechtlich geschützt.
schlingenförmigen zytoplasmatischen Filamenten. Runde und zwischen Männern und Frauen.
scharf begrenzte zystische Läsionen (10 – 50 µm breit). Einige
Läsionen mit reflektierenden Punkten im Zytoplasma entspre- Symptome
chen wahrscheinlich den Zellkernen. Asymptomatisch oder Visusbeeinträchtigung, wenn die zentra-
le Hornhaut betroffen ist.
Stocker-Holt-Variante: Keine Mitteilung
Verlauf
Konfokale Mikroskopie: Basalzellen weisen hyporeflektive Zo- Langsames Fortschreiten der Trübung mit möglicher Visusver-
nen von 40 – 150 µm Durchmesser auf mit teilweise zentralen re- schlechterung.
flektierenden Flecken.
Lichtmikroskopie
Stocker-Holt-Variante: Keine Mitteilung Diffuse Vakuolen im Zytoplasma der betroffenen Zellen.
Kategorie: 1 Stocker-Holt-Variante Kategorie 1 Transmissions-Elektronenmikroskopie
Ausgedehnte Vakuolen im Zytoplasma des befallenen Horn-
Literatur hautepithels. Diese sind entweder optisch leer oder enthalten
1 Behnke H, Thiel HJ. Über die hereditäre Epitheldystrophie der Hornhaut schwach osmophiles, teilweise homogenes und auch lamelläres
(Typ Meesmann-Wilke) in Schleswig-Holstein. Klin Monatsbl Augen- Material, welches wohl durch Zerfall und Zusammenballung
heilkd 1965; 147 (5): 662 – 672
2 Burns RP. Meesmann’s corneal dystrophy. Trans Am Ophthalmol Soc
von Vakuolen entsteht.
1968; 66: 530 – 635
3 Fine BS, Yanoff M, Pitts E, et al. Meesmann’s epithelial dystrophy of the Immunhistochemie
cornea. Am J Ophthalmol 1977; 83: 633 – 642 Die immunhistochemisch vereinzelte Anfärbung von Ki67
4 Meesmann A. Über eine bisher nicht beschriebene dominant vererbte
weist nicht auf eine erhöhte mitotische Aktivität hin.
Dystrophia epithelialis corneae. Ber Dtsch Ophthalmol Ges 1938; 52:
154 – 158
5 Stocker FW, Holt LB. A rare form of hereditary epithelial dystrophy of Konfokale Mikroskopie
the cornea: a genetic, clinical and pathologic study. Trans Am Ophthal- Viele einzelne dunkle und gut abgegrenzte Läsionen (50 –
mol Soc 1954; 52: 133 – 144 100 μm) in runder und ovaler Form. Einige Läsionen zeigen re-
6 Thiel HJ, Behnke H. On the extent of variation of hereditary epithelial
flektierende zentrale Punkte, die vermutlich den Zellkernen
corneal dystrophy (Meesmann-Wilke type). Ophthalmologica 1968;
155: 81 – 86 entsprechen.
7 Tuft S, Bron AJ. Imaging the microstructural abnormalities of Mees-
mann corneal dystrophy by in vivo confocal microscopy. Cornea 2006; Kategorie: 2
25: 868 – 870
8 Wittebol-Post D, Van-Bijsterveld OP, Delleman JW. Meesmann’s epitheli-
al dystrophy of the cornea. Biometrics and a hypothesis. Ophthalmolo-
gica 1987; 194: 44 – 49
Abb. 5 Lisch-epitheliale Hornhautdystrophie.
A Graue, wirbelförmige Trübung in direkter sowie
eng zusammengeballte Mikrozysten in indirekter
Beleuchtung. B Wirbelförmige Trübung. C Zusam-
mengeballte, klare Mikrozysten im regredienten
Licht.
Weiss JS et al. IC3D-Klassifikation von Hornhautdystrophien… Klin Monatsbl Augenheilkd 2011; 228 Suppl. 1: S1 – S39Sie können auch lesen

















































