Relevanz der Glykoproteine B, H, L, M und N für die zellgebundene Ausbreitung des humanen Cytomegalovirus - OPARU
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Universitätsklinik Ulm Institut für Virologie Ärztlicher Direktor: Prof. Dr. med. Thomas Stamminger Relevanz der Glykoproteine B, H, L, M und N für die zellgebundene Ausbreitung des humanen Cytomegalovirus Dissertation zur Erlangung des Doktorgrades der Medizin der Medizinischen Fakultät der Universität Ulm Kerstin Adams Neu-Ulm 2019

Amtierender Dekan: Prof. Dr. rer. nat. Thomas Wirth 1. Berichterstatter: Prof. Dr. med. Christian Sinzger 2. Berichterstatter: Prof. Dr. rer. nat. Jan Münch Tag der Promotion: 15.04.2021

Teile dieser Dissertation wurden bereits in folgendem Fachartikel veröffentlicht: Weiler N, Paal C, Adams K, Calcaterra C, Fischer D, Stanton RJ, Stöhr D, Laib Sampaio K, Sinzger C: Role of Envelope Glycoprotein Complexes in Cell-Associated Spread of Human Cytomegalovirus. Viruses 13(4): 614 (2021) https://doi.org/10.3390/v13040614 CC BY 4.0, https://creativecommons.org/licenses/by/4.0/

Inhalt Abkürzungsverzeichnis ........................................................................................................ iii 1 Einleitung .......................................................................................................................... 1 1.1 Klinische Bedeutung von HCMV-Infektionen ..................................................... 1 1.2 Der Replikationszyklus von HCMV ..................................................................... 2 1.3 Glykoprotein-Komplexe der Virushülle von HCMV............................................ 3 1.4 Der HCMV-Stamm Merlin als Modellvirus für die zellassoziierte Ausbreitung.. 4 1.5 Fragestellung ......................................................................................................... 6 2 Material und Methoden ..................................................................................................... 8 2.1 Material ................................................................................................................. 8 2.1.1 Geräte ............................................................................................................... 8 2.1.2 Chemikalien...................................................................................................... 9 2.1.3 Medien und Antibiotika .................................................................................. 10 2.1.4 Puffer .............................................................................................................. 11 2.1.5 Antikörper und Restriktionsenzyme ............................................................... 12 2.1.6 Oligonukleotide und Plasmide ....................................................................... 13 2.1.7 Zell-Linie ........................................................................................................ 15 2.1.8 Bakterien ........................................................................................................ 15 2.1.9 Bakterielle artifizielle Chromosomen ............................................................ 15 2.1.10 Kits ................................................................................................................. 15 2.1.11 Sonstige Materialien ....................................................................................... 15 2.1.12 Software.......................................................................................................... 16 2.2 Methoden ............................................................................................................ 16 2.2.1 Kultivierung primärer Vorhaut-Fibroblasten (HFFs) ..................................... 16 2.2.2 „Seamless“-Mutagenese ................................................................................. 16 2.2.3 Sequenzierung ................................................................................................ 21 2.2.4 Transfektion von BAC-DNA in HFFs zur Virusrekonstitution ...................... 22 2.2.5 Gaussia Luciferase-Messung .......................................................................... 23 2.2.6 Färbung IE-Ag-positiver Fibroblasten ........................................................... 25 3 Ergebnisse ....................................................................................................................... 27 3.1 Genetische Manipulation der BAC-DNA ........................................................... 28 3.2 Auswirkung der Elimination von Glykoproteinen auf die Replikationsfähigkeit von TB40-BAC4-IE-GLuc und Merlin-pAL1502-GLuc ................................... 35 3.3 Reevaluation der Ausschaltung von Glykoprotein N .......................................... 41 3.3.1 Indirekter Nachweis von gN mithilfe von doppelter Fluoreszenzmarkierung .................................................................................. 42 3.3.2 Auswirkung der Insertion weiterer Stopp-Codons in die UL73stop-Mutanten ............................................................................. 44 4 Diskussion ....................................................................................................................... 50 i

5 Zusammenfassung .......................................................................................................... 60 6 Literaturverzeichnis ........................................................................................................ 61 Danksagung ......................................................................................................................... 68 Lebenslauf ........................................................................................................................... 69 ii
Abkürzungsverzeichnis aa: Aminosäure („amino acid“) BAC: bakterielles artifizielles Chromosom bp: Basenpaare BSA: bovines Serumalbumin Cam: Chloramphenicol cART: antiretrovirale Kombinationstherapie („combined antiretroviral therapy“) dd: doppelt destilliert DNA: Desoxyribonukleinsäure („deoxyribonucleic acid“) dpt: Tage nach Transfektion („days post transfection“) E. coli: Escherichia coli EDTA: Ethylendiamintetraessigsäure ER: Endoplasmatisches Retikulum FCS: fötales Kälberserum („fetal calf serum“) gB: Glykoprotein B gH: Glykoprotein H gL: Glykoprotein L GLuc: Gaussia Luciferase gM: Glykoprotein M gN: Glykoprotein N gO: Glykoprotein O HCMV: humanes Cytomegalovirus HFF(F): humaner fötaler Vorhaut-Fibroblast („human fetal foreskin fibroblast“) HIV: humanes Immundefizienz-Virus HPLC: Hochleistungs-Flüssigkeitschromatographie („high performance liquid chromatography“) IE-Ag: „immediate early“-Antigen Kan: Kanamycin kb: Kilobasenpaare o nt: Nukleotid ORF: Leserahmen („open reading frame“) PBS: phosphatgepufferte Salzlösung („phosphate-buffered saline“) PCR: Polymerase-Kettenreaktion („polymerase chain reaction“) iii
PDGFR-α: „platelet-derived growth factor receptor A“ RFLA: Restriktionsfragment-Längenanalyse RLU: relative Lichteinheiten („relative light units“) rpm: Umdrehungen pro Minute („revolutions per minute“) RT: Raumtemperatur SDS: Natriumdodecylsulfat („sodium dodecyl sulfate“) tet: Tetracyclin Tris: Tris(hydroxymethyl)ammonium U: Einheit („unit“) UL: „unique long“ wt: Wildtyp iv
1 Einleitung 1.1 Klinische Bedeutung von HCMV-Infektionen Das humane Cytomegalovirus (HCMV) gehört zu den Herpesviren und persistiert wie alle Mitglieder dieser Familie lebenslang in einem Organismus, nachdem es ihn infiziert hat. Die Seroprävalenz wird durch den sozioökonomischen Status, das Alter, das Geschlecht und die ethnische Zugehörigkeit beeinflusst und schwankt zwischen 45% und 100% (Ludwig und Hengel 2009, Lachmann et al. 2018, Cannon et al. 2010). Da im Fall einer Primärinfektion oder einer Reaktivierung Viren in verschiedene Körperflüssigkeiten ausgeschieden werden können, verbreitet sich HCMV über den Kontakt mit Speichel, Urin, Genitalsekreten, Brustmilch und Blut (Yeager et al. 1981, Cannon et al. 2011). Die HCMV-Infektion führt bei Immunkompetenten meistens nur zu grippeähnlichen Symptomen, die vom Verlauf an eine infektiöse Mononukleose erinnern können (Lancini et al. 2014). Schwere Verläufe treten vor allem auf, wenn das Immunsystem noch nicht voll ausgereift oder beeinträchtigt ist, wie beispielsweise bei Neugeborenen, Transplantat- Empfängern und HIV-positiven Patienten. HCMV kann über die Plazenta auf das ungeborene Kind übertragen werden und unter anderem Blindheit, Schallempfindungs- Schwerhörigkeit und psychomotorische Entwicklungs-Verzögerungen auslösen. Kongenitale HCMV-Infektionen gelten als führende Ursache neurologischer Störungen im Kindesalter (Manicklal et al. 2013, Ludwig und Hengel 2009). Auch Patienten, denen ein solides Organ oder hämatogene Stammzellen transplantiert werden, sind besonders vulnerabel. Primärinfektionen oder Reaktivierungen steigern das Risiko dafür, dass das Spenderorgan abgestoßen wird oder eine Graft-versus-Host-Erkrankung auftritt und begünstigen zudem Superinfektionen mit opportunistischen Keimen, wodurch insgesamt die Mortalität steigt (Sia und Patel 2000, Nichols et al. 2002). Bei HIV-Patienten gilt die HCMV-Retinitis als gefürchtete Komplikation, die zur vollständigen Erblindung führen kann. Sie tritt vor allem auf, wenn die Zellzahl der CD4-positiven Leukozyten im Blut unter 50/µl fällt (Nichols und Boeckh 2000, Mitchell et al. 1999). Durch Fortschritte in der Behandlung, insbesondere durch den Einsatz der antiretroviralen Kombinationstherapie (cART), kann dies heutzutage häufig verhindert werden (Jabs et al. 2010). Erkrankungen durch HCMV betreffen nun vor allem HIV-Patienten, die eine cART nicht vertragen oder ihr Therapieregime nicht einhalten, sowie Patienten, deren Erkrankung erst in einem fortgeschrittenen Stadium diagnostiziert wird (Sugar et al. 2012). 1
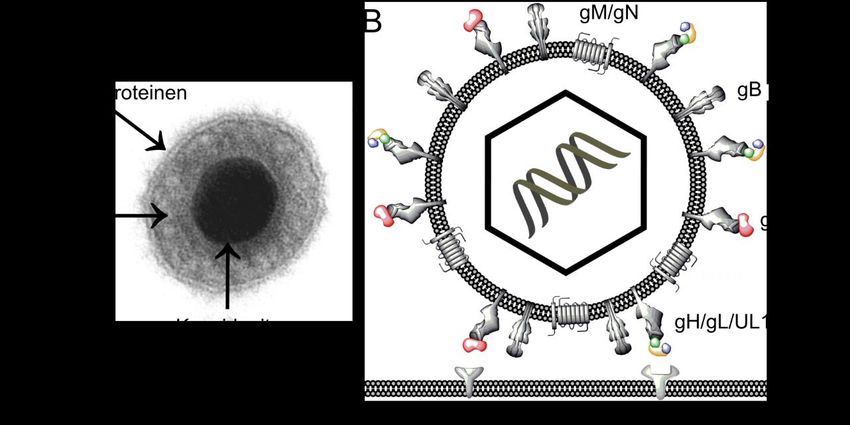
1.2 Der Replikationszyklus von HCMV Nachdem das Virus sich an die Zelle angeheftet hat, fusioniert es und setzt sein Nukleokapsid ins Zytoplasma frei. Ob der Fusionsschritt an der Zellmembran stattfindet (Compton et al. 1992) oder ob das Virus zunächst in ein Endosom aufgenommen wird, mit dessen Membran es dann verschmilzt (Sinzger 2008, Ryckman et al. 2005, Hetzenecker et al. 2016, Abdellatif et al. 2018), ist unklar. Daraufhin wird das Nukleokapsid zum Zellkern transportiert, in dem das virale Genom freigesetzt und repliziert wird (Mettenleiter et al. 2009). Die Viruskapside durchlaufen im Folgenden mehrere Umhüllungsschritte. Zunächst knospen sie an der inneren Kernmembran und erhalten so ihre primäre Hülle (Mettenleiter et al. 2013). Um in das Zytoplasma zu gelangen, verschmelzen sie mit der äußeren Kernmembran, wodurch die erste Hülle verloren geht. Daraufhin erreichen die Kapside das „assembly compartment“. Es handelt sich dabei um eine Struktur neben dem Zellkern, die verschiedene Anteile sekretorischer Zellorganellen enthält, wie etwa des Endoplasmatischen Retikulums (ERs) oder des Golgi-Apparats (Sanchez et al. 2002, Alwine 2012). Dort werden zunächst die Proteine des Teguments um das Kapsid angeordnet. Anschließend wird das Partikel mit zytoplasmatischen Membranen umhüllt, was als „secondary envelopment“ bezeichnet wird. Hierbei entsteht ein umhülltes Virion in einem sekretorischen Vesikel, das daraufhin mit der Zellmembran fusioniert (Schauflinger et al. 2013, Close et al. 2018), wodurch das Virus (Abbildung 1A) aus der infizierten Zelle in die Umgebung freigesetzt wird. Abbildung 1: Humanes Cytomegalovirus. DNA: Desoxyribonukleinsäure, PDGFR-α: „platelet derived growth factor receptor A“. A) Elektronenmikroskopische Aufnahme eines HCMV-Partikels (C. Sinzger, Universitätsklinik Ulm). B) Schematische Darstellung eines HCMV-Partikels. In der Hüllmembran sind verschiedene Glykoprotein-Komplexe eingelagert. 2
1.3 Glykoprotein-Komplexe der Virushülle von HCMV Die Hülle von HCMV enthält verschiedene Glykoprotein-Komplexe (Abbildung 1B): gcI (gB) (Gretch et al. 1988), gcII (gM/gN) (Mach et al. 2000), gcIII (gH/gL/gO, auch Trimer- Komplex genannt) (Huber und Compton 1998) und den Pentamer-Komplex gH/gL/UL128-131 (Ryckman et al. 2008). Die Glykoproteine B, H, L, M und N sind in der Familie der Herpesviren konserviert, was bedeutet, dass verschiedene Spezies über strukturell ähnliche Oberflächenproteine verfügen. Dies deutet darauf hin, dass die Funktionen der genannten Glykoproteine dem Virus unter Selektionsdruck einen Vorteil verschaffen. Sie beeinflussen verschiedene Aspekte der viralen Replikation, wie beispielsweise die Anheftung an die Zielzelle, die Fusion mit der Zellmembran, die Umhüllung neu gebildeter Virionen und nicht zuletzt auch die Abwehr des Immunsystems. Der Trimer-Komplex bindet in Fibroblasten an PDGFR-α (Wu et al. 2018, Stegmann et al. 2017, Kabanova et al. 2016) auf der Oberfläche der Zelle (Abbildung 1B) und schafft so die räumliche Nähe, die notwendig ist, damit das Virus mit der Zellmembran verschmelzen kann. Diese Fusion wird durch gB, gH und gL vermittelt (Vanarsdall et al. 2008). In Endothelzellen bindet vermutlich der Pentamer-Komplex das Virus an die Zellmembran, wobei Neuropilin-2 (Martinez-Martin et al. 2018) und OR14I1 (E et al. 2019) als potenzielle Rezeptoren diskutiert werden. gB und gN sind zudem für die Immunevasion relevant. Sie sind hochgradig glykosyliert und stören das Immunsystem dabei, das Virus zu eliminieren, indem sie mit ihren Zuckerresten die Bindung neutralisierender Antikörper verhindern (Kropff et al. 2012, Burke und Heldwein 2015). gN scheint zudem ebenso wie gM eine Rolle im „secondary envelopment“ zu spielen (Mach et al. 2007, Krzyzaniak et al. 2007). HCMV-Stämme bilden den Trimer- und Pentamer-Komplex in unterschiedlichen Verhältnissen und Mengen auf ihrer Virushülle aus, was den Zelltropismus beeinflusst (Wang und Shenk 2005, Scrivano et al. 2011, Murrell et al. 2013, Li und Kamil 2015). Hierunter versteht man die Fähigkeit des Virus, in bestimmte Zelltypen einzudringen und sich daraufhin zu vermehren. Klinische Isolate können neben Fibroblasten auch Endothel- und Epithelzellen infizieren. Diese Eigenschaft verlieren sie allerdings, wenn sie längerfristig in Zellkulturen passagiert werden (Sinzger et al. 1999). Gleichzeitig ändert sich auch der Ausbreitungsmodus des Virus (Abbildung 2). Initial breiten sich klinische HCMV-Isolate ausschließlich von Zelle zu Zelle aus (Yamane et al. 1983, Sinzger et al. 1999). Durch Mutationen, die sie während der Passage in Zellkultur erwerben, geben sie zusätzlich freie Viren in die Umgebung ab, was als zellfreie Ausbreitung bezeichnet wird (Dolan et al. 2004, Bradley et al. 2009). Es wird angenommen, dass dadurch das 3
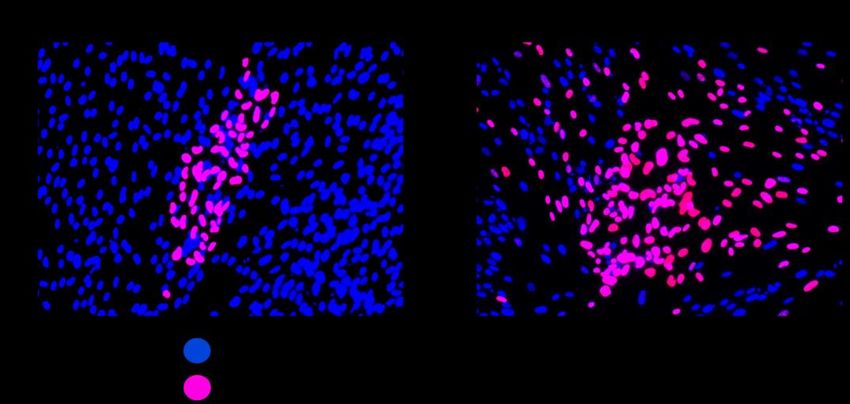
Viruswachstum beschleunigt und die Ausbreitung über große Distanzen gefördert wird (Sattentau 2008, Mothes et al. 2010). Jedoch bietet die ursprüngliche, zellgebundene Ausbreitung dem Virus entscheidende Vorteile im Organismus. Sie ist sehr effizient, da sich bereits mit einer geringen Anzahl von Viren umliegende Zellen infizieren lassen (Zhong et al. 2013). Durch die Nähe der Zellen vermindert das Virus zudem zeitlich und räumlich seinen Kontakt mit dem Immunsystem des Wirts (Gardner und Tortorella 2016). Es ist bisher allerdings nur unzureichend aufgeklärt, wie das zellfreie und das zellgebundene Wachstum im Detail vonstattengehen und ob sie demselben Mechanismus folgen. Abbildung 2: Unterschiedliche Ausbreitungsweisen des humanen Cytomegalovirus. Klinische Isolate und der HCMV-Stamm Merlin verbreiten sich ausschließlich von Zelle zu Zelle. Zellfreie Stämme hingegen produzieren zusätzlich auch freie, infektiöse Viren und geben sie an die Umgebung ab. 1.4 Der HCMV-Stamm Merlin als Modellvirus für die zellassoziierte Ausbreitung Das klinische Merlin-Isolat stammt aus dem Urin eines kongenital infizierten Kinds und wurde nach drei Passagen in Fibroblasten sequenziert (Dolan et al. 2004). Der Stamm Merlin gilt als internationaler WHO-Standard für HCMV (Fryer et al. 2016) und verbreitet sich zellgebunden (Abbildung 3A), was wahrscheinlich dem Ausbreitungsmodus von HCMV in vivo entspricht. Das Genom von Merlin wurde in ein bakterielles artifizielles Chromosom (BAC) kloniert, um reproduzierbare, klonale Vermehrung zu ermöglichen und genetische Manipulationen zu vereinfachen (Stanton et al. 2010). Die Sequenz des BAC- klonierten Genoms wurde mit der ursprünglichen Sequenz des klinischen Isolats abgeglichen, wobei Mutationen in den Gen-Loci RL13 und UL128 entdeckt wurden. Sie begünstigen die zellfreie Ausbreitung (Dolan et al. 2004, Bradley et al. 2009) und wurden repariert. Um funktionale Viren zu rekonstituieren, wurde das Genom daraufhin transfiziert. Bereits nach drei Passagen mutierten die Loci RL13 und UL128 jedoch erneut, 4
was darauf hindeutet, dass sie in der Zellkultur unter starkem Selektionsdruck stehen (Murrell et al. 2016). Um es dennoch zu ermöglichen, genetisch intakte Merlin-Viren zu produzieren, wurde ein Tetracyclin-gesteuertes System eingefügt, mit dessen Hilfe die Genexpression experimentell reguliert werden kann (Yao et al. 1998, Stanton et al. 2010, Gossen et al. 1995). Die Idee dahinter ist, die Expression von RL13 und UL128 gezielt zu inhibieren und dadurch den Selektionsdruck, der auf diesen Loci lastet, zu reduzieren (Wilkinson et al. 2015). Dadurch wird der Zeitraum verlängert, in dem mit einem genetisch intakten HCMV-Virus gearbeitet werden kann, bevor adaptive Mutationen auftreten. Strangaufwärts der Start-Codons von RL13 und UL128 wurden in die Sequenz des reparierten Merlin-BACs Tet-Operatoren eingefügt (Merlin RL13tetO UL128tetO) (Stanton et al. 2010). Gleichzeitig wurde eine Fibroblasten-Zellreihe generiert, die einen Tet-Repressor Abbildung 3: Zellgebundenes Wachstum von Merlin-pAL1502-GLuc und zellfreie Ausbreitung von TB40-BAC4-IE-Gluc. IE-Ag: „immediate-early“-Antigen, HFFs: humane Vorhaut-Fibroblasten. Exempla- rische immunfluoreszenzmikroskopische Aufnahmen transfizierten HFF-Kulturen 35 dpt (A) und 13 dpt (B) in 200facher Vergrößerung. Rotes Kernsignal (Cy3): Expression von viralem IE-Ag (= infizierte Zellen). Blaues Kernsignal (DAPI): IE-Ag-negative Zellen. A) HCMV-Merlin breitet sich wie ein klinisches Isolat rein zellgebunden aus und bildet definierte Herde. B) TB40-BAC4 zählt zu den zellfreien Stämmen und wächst in charakteristischen Herden, deren Form an einen Kometenschweif erinnert. exprimiert (HFFF-tet-Zellen). Wenn Merlin RL13tetO UL128tetO in HFFF-tet-Zellen transfi- ziert wird, bindet der Tet-Repressor der Zellen an die Tet-Operatoren in der viralen DNA. Dadurch werden die betroffenen Gene nicht transkribiert (Merlin RL13- UL128-), obwohl die beiden Gen-Loci intakt sind. Um RL13 und UL128 zu exprimieren (Merlin RL13+ UL128+), gibt es in diesem System zwei Möglichkeiten. Zum einen kann die Repression, die von den HFFF-tet-Zellen ausgeübt wird, durch die Zugabe von Doxycyclin aufgehoben werden. Es gehört zur selben Substanzklasse wie Tetracyclin und bindet den tet-Repressor. Daraufhin ändert dieser seine Konformation (Orth et al. 2000, Das et al. 2016), wodurch die Bindung an die tet-Operatoren aufgehoben wird. Zum anderen kann Merlin RL13tetO UL128tetO – wie in dieser Arbeit – in gewöhnlichen Fibroblasten statt in HFFF-tet-Zellen 5
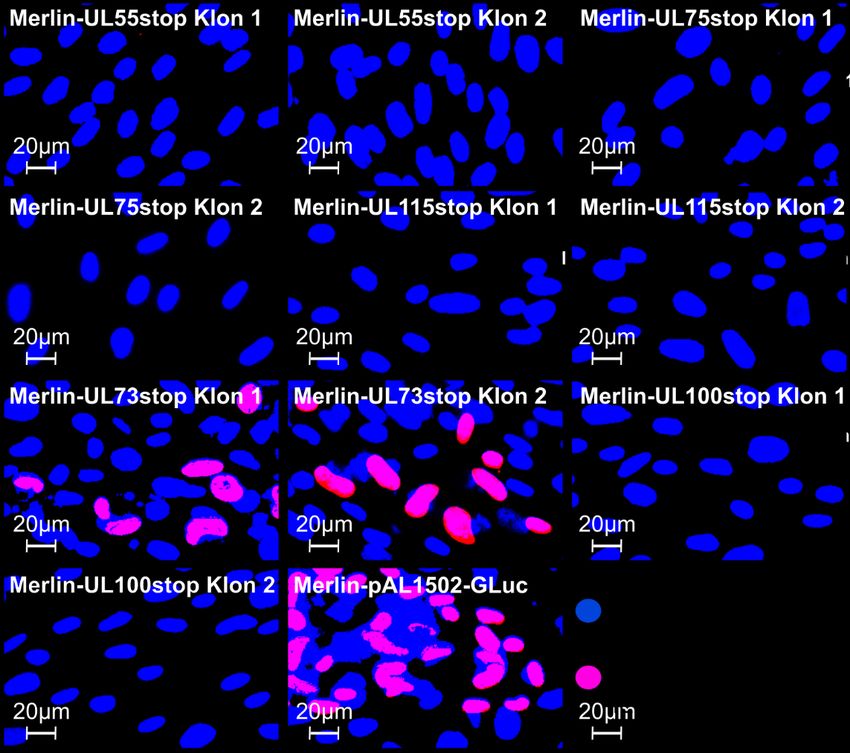
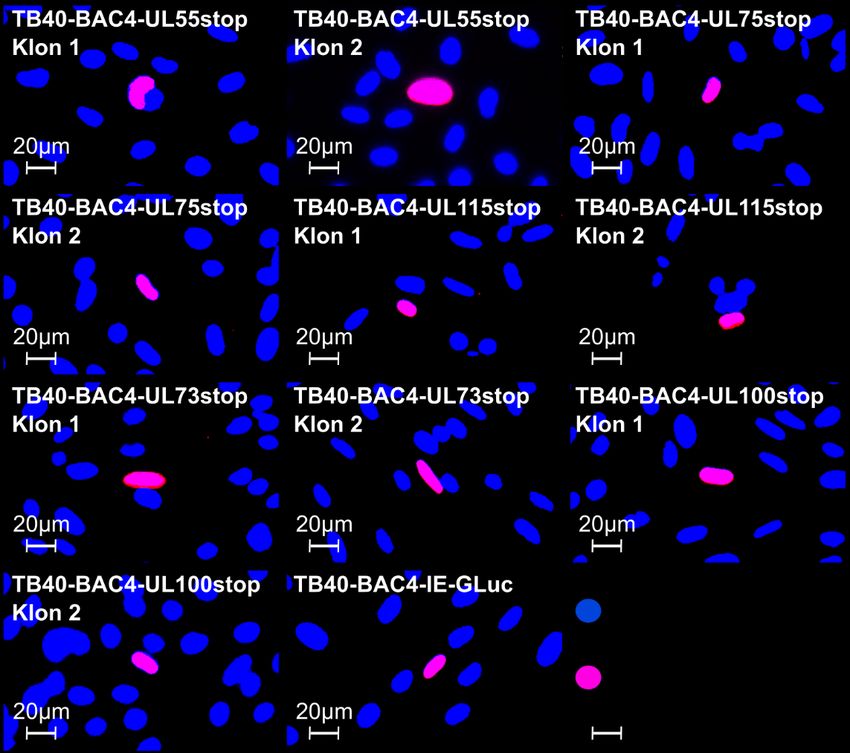
propagiert werden. Da sie keinen tet-Repressor enthalten, der die Expression von RL13 und UL128 verhindern könnte, entsteht dabei Wildtyp-Merlin (Merlin RL13+ UL128+). Das BAC-klonierte, reparierte Merlin-Genom (Merlin-pAL1502-GLuc), das in dieser Arbeit verwendet wurde, enthält zusätzlich eine Gaussia Luciferase-Expressionskassette, die unter der Kontrolle des IE-Promoters steht (Falk et al. 2018). Abbildung 4: Gaussia Luciferase-Expression infizierter Fibroblasten. Schematische, vereinfachte Dar- stellung. Primäre humane Vorhaut-Fibroblasten (HFFs) werden mit viralen Genomen transfiziert. Die Virusreplikation und die Expression der Gaussia Luciferase (GLuc) sind gekoppelt, sodass infizierte Zellen das Reporterenzym bilden. Durch die Umsetzung des Substrats Coelenterazin wird ein Lichtsignal emittiert. 1.5 Fragestellung Das humane Cytomegalovirus (HCMV) kann sich sowohl zellfrei über freigesetzte Viruspartikel als auch zellgebunden direkt von einer infizierten Zelle auf benachbarte Zellen ausbreiten (Abbildung 2). Der zellgebundene Modus ermöglicht es dem Virus, sich auch in Gegenwart neutralisierender Antikörper auszubreiten, die gegen virale Glykoproteine gerichtet sind. Deswegen spielt er vermutlich in vivo eine große Rolle. Die Resistenz der zellgebundenen Ausbreitung gegenüber Antikörpern wirft die Frage auf, ob sich die beiden Infektionsmodi hinsichtlich der beteiligten Virusproteine unterscheiden. Dies ist wichtig, um zielgerichtete Interventionsstrategien gegen die zellgebundene Ausbreitung zu entwickeln. Im genetischen Hintergrund der zellfrei wachsenden Stämme Towne und AD169 wurde bereits gezeigt, dass die Glykoproteine B, H, L, M und N essenziell für die Ausbreitung von HCMV sind (Yu et al. 2003, Hobom et al. 2000, Dunn et al. 2003). In dieser Arbeit soll nun überprüft werden, ob dies auch für einen Stamm gilt, der sich wie ein frisches klinisches HCMV-Isolat rein zellgebunden ausbreitet. Das experimentelle Vorgehen wurde bereits in Weiler et al. 2021 beschrieben. Als genetischer Hintergrund wird eine Version des Stamms Merlin verwendet, die in Gen-Ausstattung und Wachstumsverhalten klinischen Isolaten weitgehend entspricht. Das Genom dieses Stamms liegt als bakterielles artifizielles Chromosom (BAC) in E. coli vor und kann deshalb gezielt genetisch verändert werden. Um die Rolle der genannten Glykoproteine für die 6
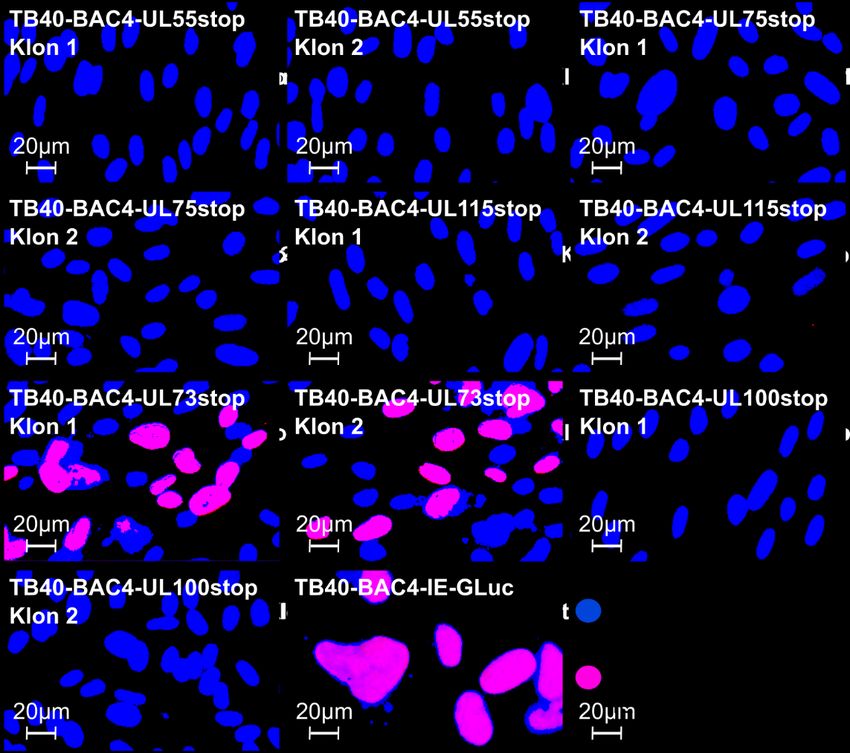
zellgebundene Ausbreitung zu untersuchen, werden sie einzeln durch Punktmutationen ausgeschaltet, die zum Abbruch der Translation führen. Die mutierten Genome werden in Fibroblasten transfiziert, um die entsprechenden Viren zu rekonstituieren und deren Wachstumsverhalten zu untersuchen. Da der verwendete Virusstamm Merlin-pAL1502- GLuc die Gaussia Luciferase unter der Kontrolle des HCMV-„immediate early“-Promoters exprimiert, wird dieses Reporterenzym mit der Kinetik der viralen IE-Gene gebildet und dient folglich als Marker für die Virusreplikation. Nach Transfektion der Virusgenome in Fibroblasten werden regelmäßig Proben des Kulturmediums entnommen, um die Lichtemission zu messen, die bei der Umsetzung des Gaussia Luciferase-Substrats Coelenterazin entsteht, und so den Verlauf der viralen Replikation zu beobachten. Durch Regressionsanalyse der Signalkurve soll das Wachstum quantifiziert werden, um zu erkennen, ob eine Mutation letal ist oder nicht. Zusätzlich werden die Ergebnisse des Gaussia Luciferase-Assays mit einem unabhängigen Verfahren überprüft. Hierzu werden die Versuchsansätze nach Abschluss der Probenentnahmephase fixiert und das IE-Antigen (IE-Ag) angefärbt. Falls sich unter dem Immunfluoreszenz-Mikroskop zu diesem Zeitpunkt Herde aus IE-Ag-positiven Zellen identifizieren lassen, deutet dies darauf hin, dass sich die betreffende Virusmutante in der Kultur vermehrt. Sind die Fibroblasten hingegen IE-Ag-negativ, so ist die Mutation letal. Als Kontrolle werden alle Mutationen auch in den zellfrei wachsenden Stamm TB40- BAC4-IE-GLuc (Abbildung 3B) eingeführt, bei dem alle untersuchten Glykoproteine wie in den Stämmen AD169 und Towne essenziell sein sollten. Die Ergebnisse dieser Arbeit werden dazu beitragen aufzuklären, ob ein zellgebundener HCMV-Stamm für die Ausbreitung dieselben Glykoproteine benötigt wie ein zellfreier. Dadurch werden Rückschlüsse auf den Mechanismus der zellgebundenen Ausbreitung ermöglicht. 7
2 Material und Methoden 2.1 Material 2.1.1 Geräte Thermal-Cycler FlexCycler Analytik Jena Labcycler SensoQuest Fluoreszenz-/Chemolumineszenz-Detektion Fusion SL Peqlab Photometer GeneQuant pro RNA/DNA Calculator GE Healthcare Zentrifugen 5402 Centrifuge Eppendorf Heraeus FRESCO21 Thermo Scientific Heraeus Multifuge X3R Thermo Scientific Gelelektrophorese Agagel Mini Biometra® PowerPAC™ Basic Bio-Rad Laboratories Sub-Cell® GT Bio-Rad Laboratories Mikroplate-Reader Chameleon Hidex Mikroskope Axio Observer.D1 Zeiss Primo Vert Zeiss 8
Inkubatoren B6200 (Bakterien) Kendro Laboratory Products Certomat HK+SII (Bakterien) Sartorius HERAcell 240i CO2 Incubator (eukaryote Zellen) Thermo Scientific Schüttelwasserbad GFL 1083 (Bakterien) Thermolab® Zellkultur-Werkbank HERAsafe Heraeus LaminAir® HB2472 Heraeus Sonstige Geräte Gel Doc™ XR+ Bio-Rad Laboratories Kern EW (Präzisionswaage) KERN & SOHN GmbH MicroPulser™ Bio-Rad Laboratories Pipetboy comfort Integra Biosciences TS1 ThermoShaker Biometra® Vacusafe & Vacuboy Integra Biosciences Vortex Genie® 2 Scientific Industries™ 2.1.2 Chemikalien Acetone AnalaR NORMAPUR VWR Chemicals bFGF (Basic Fibroblast Growth Factor) PeproTech® Coelenterazin PJK GmbH DAPI (4′,6-Diamidin-2-Phenylindol, Arbeitskonzentration: 0,1 ng/μl in PBS) Sigma-Aldrich® DPBS (Dulbecco´s Phosphate Buffered Saline) Gibco® by life technologies™ DNA Ladder (1 kb) New England Biolabs® Inc. DNA Ladder (100 bp) New England Biolabs® Inc. Ethanol Sigma-Aldrich® FCS (Fetal Calf Serum) PAN™Biotech Gel Loading Dye Purple (6x) New England Biolabs® Inc. Glycerol Sigma-Aldrich® H2O (DEPC behandelt) Carl Roth® GmbH + Co. KG 9
Hepes Carl Roth® GmbH + Co. KG Isopropanol AnalaR NORMAPUR VW KH2PO4 (Kaliumdihydrogenphosphat) Merck KGaA L-(+)-Arabinose Sigma-Aldrich® LE Agarose Seakem® Lonza Midori Green Advance DNA stain Nippon Genetics Europe GmbH Midori Green Direct Nippon Genetics Europe GmbH NaCl (Natriumchlorid) Sigma-Aldrich® Na2HPO4 (Natriumhydrogenphosphat) Sigma-Aldrich® Roti®-Phenol/Chloroform/Isoamylalkohol Carl Roth® GmbH + Co. KG TopTaq® Master Mix (2x) Qiagen Trypsin-EDTA (0,05 %) Gibco® by life technologies™ 2.1.3 Medien und Antibiotika Medien zur Bakterienanzucht: LB-Agar (Luria/Miller) Carl Roth® GmbH + Co. KG LB-Medium (Luria/Miller) Carl Roth® GmbH + Co. KG Medien zur Kultivierung eukaryoter Zellen: MEM5 Minimum Essential Medium + GlutaMAX™-I Gibco® by life technologies™ + 0,1 mg/ml Gentamicin Sigma-Aldrich® + 5 % FCS PAN™Biotech MEM complete Minimum Essential Medium + GlutaMAX™-I Gibco® by life technologies™ + 0,1 mg/ml Gentamicin Sigma-Aldrich® + 5 % FCS PAN™Biotech + 0,5 μg/l bFGF PeproTech® DMEM Dulbecco´s Modified Eagle Medium + GlutaMAX™-I Gibco® by life technologies™ 10
DMEM10 Dulbecco´s Modified Eagle Medium + GlutaMAX™-I Gibco® by life technologies™ 0,1 mg/ml Gentamicin Sigma-Aldrich® 10 % FCS PAN™Biotech SOC-Medium Sigma-Aldrich® Antibiotika: Chloramphenicol Carl Roth® GmbH + Co. KG Lagerungskonzentration: 10 mg/ml (in Methanol) Arbeitskonzentration: 25 μg/ml Gentamicin Sigma-Aldrich® Lagerungskonzentration: 50 mg/ml Arbeitskonzentration: 0,1 mg/ml Kanamycin Sigma-Aldrich® Lagerungskonzentration: 50 mg/ml (in H2O dd) Arbeitskonzentration: 50 μg/ml 2.1.4 Puffer TBE-Puffer Rotiphorese® 10x TBE-Buffer Carl Roth® GmbH + Co. KG TE-Puffer TE Buffer (1x) Genaxxon bioscience GmbH 1x PBS 137 mM NaCl Sigma-Aldrich® 2,7 mM KCl Calbiochem® 4,3 mM Na2HPO4 x 12 H2O Sigma-Aldrich® 1,5 mM KH2PO4 Merck KGaA 11
10% Arabinose-Stocklösung (frisch angesetzt) 1 g Arabinose Sigma-Aldrich® ad 10 ml H2O dd steril filtriert 2x Hepes 50 mM Hepes Carl Roth® GmbH + Co. KG 1,5 mM Na2HPO4 Sigma-Aldrich® 280 mM NaCl Sigma-Aldrich® pH eingestellt auf 7,13 und gelagert bei -20°C Glycerolschock-Lösung 20 % Glycerol Sigma-Aldrich® 50 % 2x Hepes (siehe oben) 30 % H2O dd steril filtriert und gelagert bei -20°C MBS-Medium 6 % Lösung 3 (MBS Mammalian Transfection Kit) Agilent Technologies 94 % DMEM Puffer für Restriktionsenzyme CutSmart™ Buffer (10x conc.) New England Biolabs® Inc. NEBuffer for I-SceI (10x conc.) New England Biolabs® Inc. 2.1.5 Antikörper und Restriktionsenzyme Primär-Antikörper Mouse anti-HCMV I.E.A. Argene („E13”) Argene Biosoft Cy™3-conjugated AffiniPure F(ab´)2 Fragment Jackson ImmunoResearch Restriktionsenzyme DpnI (20000 U/ml) New England Biolabs® Inc. EcoRI (20000 U/ml) New England Biolabs® Inc. I-SceI (5000 U/ml) New England Biolabs® Inc. 12
2.1.6 Oligonukleotide und Plasmide Alle Oligonukleotide wurden von der biomers.net GmbH bezogen und in einer Konzen- tration von 50 pmol/μl bei -20°C gelagert. Tabelle 1: Übersicht der verwendeten Primer. for: „forward“, nt: Nukleotid, rev: „reverse“, UL: „unique long“, A: Adenin, C: Cytosin, G: Guanin, L: Lysin, P: Prolin, T: Thymin Bezeichnung Länge Sequenz (5´- 3´) [nt] UL55stop_for 103 GGA ATC CAG GAT CTG GTG CCT GGT AGT CTG CGT TAA CTT GTG AAT CGT CCG TCT GGG TTA AGC GGT TTC CTC ATC TTC TAC AGG ATG ACG ACG ATA AGT AGG G UL55stop_rev 106 GAG TAG CAG AAG TTC CAC GAG TAG AAG ATG AGG AAA CCG CTT AAC CCA GAC GGA CGA TTC ACA AGT TAA CGC AGA CTA CCA CAA CCA ATT AAC CAA TTC TGA TTA G UL55stop_short_for 20 GGA ATC CAG GAT CTG GTG CC UL73stop_for 103 TAA GCA TCG TGG CGG TGG TGT GAT GGA GTG GAA CAC ACT ATG ATT AGG TCT TTT GGT TTA ATC GGT AGT GGC AAG TTC CAA AGG ATG ACG ACG ATA AGT AGG G UL73stop_rev 106 TGC TAG CAG TCG ACG TAT TGT TGG AAC TTG CCA CTA CCG ATT AAA CCA AAA GAC CTA ATC ATA GTG TGT TCC ACT CCA TCA CAA CCA ATT AAC CAA TTC TGA TTA G UL73stop_short_for 20 TAA GCA TCG TGG CGG TGG TG UL73stop_for M3 103 CAA CGT GAT GAG ACC ACA TGC TCA CAA TGA TTT TTA CAA TTG ACA TTG TAC ATC GCA TTAG TAT GAG CTT TCA CTG TCC AGA GGA TGA CGA CGA TAA GTA GGG UL73stop_rev M3 106 TAT TCC ACC AGG CTG CAA AGC TGG ACA GTG AAA GCT CAT ACT AAT GCG ATG TAC AAT GTC AAT TGT AAA AAT CAT TGT GAG CAA CCA ATT AAC CAA TTC TGA TTAG 13
UL73stop_for_short M3 20 CAA CGT GAT GAG ACC ACA TG UL75 stop_for 103 CGC TAT GCG GCC CGG CCT CCC CTT CTA CCT CAC CGT CTT CTA GGT CTA CCT CCT TAG TTG ACT ACC TTC GCA ACG ATA TGG AGG ATG ACG ACG ATA AGT AGG G UL75 stop_rev 106 CTT CGG ATG CGG CGT CTG CGC CAT ATC GTT GCG AAG GTA GTC AAC TAA GGA GGT AGA CCT AGA AGA CGG TGA GGT AGA AGG CAA CCA ATT AAC CAA TTC TGA TTA G UL75 stop_short_for 20 CGC TAT GCG GCC CGG CCT CC UL100stop_for 103 CGT GGA CTT TGA AAG GCT CAA CAT GTC GGC CTA CAA CGT ATG ACA CCT GCA CAC GCC TTA ACT TTT CTT AGA CTC GGT GCA AGG ATG ACG ACG ATA AGT AGG G UL100stop_rev 106 ACA CGG CGT AGC ACA CCA ACT GCA CCG AGT CTA AGA AAA GTT AAG GCG TGT GCA GGT GTC ATA CGT TGT AGG CCG ACA TGT CAA CCA ATT AAC CAA TTC TGA TTA G UL100stop_for_short 20 CGT GGA CTT TGA AAG GCT CA UL115stop_for 103 CTC TCA TCG TGC CGC AGA CTT GAT GTG CCG CCG CCC GGA TTG AGG CTT CTC TTT CTC ATA AGG ACC GGT GGT ACT GCT GTG AGG ATG ACG ACG ATA AGT AGG G UL115stop_rev 106 TGG GCA GCA GAA GGC AAC ACC ACA GCA GTA CCA CCG GTC CTT ATG AGA AAG AGA AGC CTC AAT CCG GGC GGC GGC ACA TCA CAA CCA ATT AAC CAA TTC TGA TTA G UL115stop_short_for 20 CTC TCA TCG TGC CGC AGA CT Kanamycin universal 21 CAA CCA ATT AAC CAA TTC TGA reverse 14
2.1.7 Zell-Linie Primäre humane Vorhaut-Fibroblasten (HFFs) Isolat der Arbeitsgruppe Sinzger, Universitätsklinikum Ulm 2.1.8 Bakterien GS1783-E. coli (Tischer et al. 2010) 2.1.9 Bakterielle artifizielle Chromosomen HCMV-TB40-BACKL7-UL32EGFP-UL100mCherry (Laib Sampaio et al. 2013) Merlin-pAL1502-GLuc (Falk et al. 2018) TB40-BAC4-IE-GLuc (Falk et al. 2016) 2.1.10 Kits DNeasy Blood and Tissue Kit Qiagen MBS Mammalian Transfection Kit Agilent Technologies NucleoBond® Xtra Midi Macherey-Nagel NucleoSpin® Gel and PCR clean-up Macherey-Nagel Phase Lock Gel heavy 5Prime 2.1.11 Sonstige Materialien 3,5 ml Polystyrene-Tubes Sarstedt C-Chip (Hämozytometer) NanoEnTek Cellstar® 6- und 24-well cell culture plates greiner bio-one Cellstar® Cell Culture Flasks greiner bio-one Electroporation Cuvettes (2 mm) Biozym Filtropur S sterile filters (0,20 μm) Sarstedt LUMITRAC 96-well luminescence reader plates greiner bio-one SurPhob® Cell Saver Tips Biozym µ-Slide 8 Well ibidi GmbH 15
2.1.12 Software AxioVision Rel 4.8 Zeiss CLC Main Workbench 8 Qiagen CLC Sequence Viewer 8 Qiagen Corel® PHOTO-PAINT® X6 Corel Corporation Image Lab™ 5.1 Bio-Rad Laboratories Inkscape 0.092.2 Free and open-source software Mendeley Mendeley Ltd. Microsoft® Office 2013 Microsoft Corporation Paint Version 1803 Microsoft Corporation Sigma Plot 11.0 Systat Software GmbH 2.2 Methoden 2.2.1 Kultivierung primärer Vorhaut-Fibroblasten (HFFs) Humane Vorhaut-Fibroblasten wurden in MEM complete kultiviert und alle drei bis vier Tage gesplittet, um eine hohe Teilungsrate beizubehalten. Zunächst wurden die Zellkultu- ren mit Trypsin-EDTA gewaschen, um Rückstände des Mediums zu entfernen. Daraufhin wurden die Fibroblasten etwa 2 min mit Trypsin-EDTA inkubiert, um sie vom Boden der Zellkultur-Flasche zu entfernen. Unter dem Phasenkontrast-Mikroskop wurde kontrolliert, ob sie sich vollständig losgelöst hatten. Anschließend wurden die HFFs in MEM complete aufgenommen und gleichmäßig auf zwei Zellkultur-Flaschen verteilt. Dieser Vorgang zählte als eine Passage. Für die Transfektionen wurden HFFs mit Passagezahlen zwischen 10 und 26 verwendet. Für die Bestimmung der Zellzahl wurden die Fibroblasten mit Trypsin-EDTA gewaschen und abgelöst. Daraufhin wurden sie in Zellkultur-Medium aufgenommen und gründlich durchmischt. 10 µl der Zellsuspension wurden in ein Hämozytometer getropft und unter dem Phasenkontrast-Mikroskop ausgezählt. 2.2.2 „Seamless“-Mutagenese Primerdesign Die Primer für die „seamless“-Mutagenese wurden so designt, wie es von Tischer et al. (2010) beschrieben wurde. Die Länge der Homologien betrug 20 nt, um eine adäquate Bindung an die Rekombinations-Kassette sicherzustellen. Mit dem OligoAnalyzer 3.1 16
(http://www.idtdna.com/calc/analyzer) wurde überprüft, ob die Primer durch ihre Struktur zur Bildung von Haarnadelschleifen neigten. Falls dies der Fall war, wurden sie nachmodifiziert. Alle verwendeten Primer wurden in HPLC-Qualität bei der biomers.net GmbH bestellt (Tabelle 1) Herstellung des Rekombinations-Fragments Da die enthaltenen Homologie-Areale ein hohes Bindungspotential zueinander aufwiesen, wurde das Rekombinations-Fragment in zwei getrennten PCR-Reaktionen synthetisiert (Tabelle 3). In der ersten PCR diente das universelle Plasmid pEPkan-S als Template (Tabelle 2). Mithilfe des Forward-Primers (Tabelle 1) und des Kanamycin-Universal- Reverse-Primers wurde der Bereich der Kanamycin-Kassette und der I-SceI-Restriktions- stelle des Plasmids inklusive einer kurzen HCMV-Sequenz mit den gewünschten Stopp- Codons amplifiziert. Tabelle 2: PCR-Ansätze für die Synthese des Rekombinationsfragments. dd: doppelt destilliert, DNA: Desoxyribonukleinsäure, PCR: Polymerase-Kettenreaktion, Σ: Summe 1. PCR 2. PCR 1 μl Template-DNA 2 μl Produkt der 1. PCR (pEPkan-S; 1 ng/μl) (1:200 in H2O dd) 0,5 μl Primer 5’ 1 μl Primer 5’ („long forward“) („short forward“) 0,5 μl Primer 3’ („kanamy- 1 μl Primer 3’ cin universal reverse“) („long reverse“) 25 μl TopTaq® Mastermix 50 μl TopTaq® (2X) Mastermix (2X) 23 μl H2O (Roth T 143.4) 46 μl H2O (Roth T 143.4) Σ 50 μl Σ 100 μl Die Größe des PCR-Produkts wurde in einer Agarose-Gelelektrophorese (1 % Agarosegel mit Midori Green Advance DNA stain (1:20.000)) überprüft. Zu erwarten war eine Fragmentlänge von 1100 bp. Anschließend wurde das PCR-Produkt mit dem Enzym DpnI bei 37 °C für 2 h verdaut, um die Template-DNA zu entfernen. Daraufhin wurde das Amplikon mithilfe des NucleoSpin® Extract II Kits der Firma Macherey-Nagel aufgereinigt und in 50 μl H2O aufgenommen. Diese Lösung wurde als Template für die zweite PCR 1:200 in H2O dd verdünnt. Der Ansatz (Tabelle 2) wurde auf zwei PCR-Tubes à 50 μl verteilt und das Template mit dem Short-Forward- und dem Reverse-Primer 17
(Tabelle 1) zum fertigen Rekombinations-Fragment synthetisiert (Tabelle 2). Auch die Länge des hierbei entstandenen DNA-Bruchstücks wurde bei etwas mehr als 1 kb erwartet und per Agarose-Gelelektrophorese kontrolliert. Daraufhin wurde das PCR-Produkt mit dem NucleoSpin® Extract II Kit (Macherey-Nagel) aufgereinigt, in 15 μl H2O dd aufgenommen und bei 4 °C bis zur Elektroporation gelagert. Tabelle 3. PCR-Programm für die 1. und 2. PCR zur Synthese des Rekombinationsfragments. ∞: unendlich Temperatur [°C] Zeit [min:s] Zyklen 95 5:00 1 95 0:45 10 51 2:00 68 2:00 95 0:45 25 60 2:00 68 3:00 72 10:00 1 4 ∞ Produktion elektrokompetenter GS1783-E. coli 10 ml LB-Medium (+1:400 Cam) wurden mit GS1783-E. coli, die das gewünschte HCMV-BAC enthielten, beimpft. Diese Vorkultur wurde bei 32°C über Nacht im Schüttler inkubiert. Am nächsten Tag wurde aus 50 ml LB-Medium (+1:400 Cam) und 1 ml der Bakterienlösung eine neue Kultur angesetzt, die für 3 h bei 32 °C und 225 rpm geschüttelt wurde. Daraufhin wurde die Bakteriensuspension einem 15minütigen Hitzeschock im Wasserbad bei 42 °C unterzogen. Sie wurde in vorgekühlte 50 ml Falcons umgekippt und 45-60 min eisgekühlt. Ab diesem Zeitpunkt wurde auf Eis und mit vorgekühlten Geräten gearbeitet, um die Kühlung der Bakterien nicht zu unterbrechen. Die Kultur wurde nun für 5 min bei 4 °C mit 4.000 rpm zentrifugiert. Daraufhin wurde der Überstand verworfen und das Pellet in 50 ml eiskaltem Wasser resuspendiert. Es folgten drei weitere Zentrifugationen bei gleichbleibender Temperatur, wobei sowohl die Umdrehungszahl als auch die Dauer zunahmen (5.000 rpm für 6 min, 6.000 rpm für 7 min, 7.000 rpm für 10 min). Nach jeder Zentrifugation wurde das Bakterienpellet gewaschen. Im Anschluss an den letzten Waschschritt wurde der Überstand vorsichtig abgegossen und das Pellet in der verbliebenen Flüssigkeit resuspendiert. Unmittelbar danach erfolgte die Elektroporation. 18
Elektroporation des Rekombinations-Fragments 100 μl elektrokompetente GS1783-E. coli wurden sanft mit 15 μl des PCR-Produkts vermengt. Die Mischung wurde in eine vorgekühlte Elektroporations-Küvette transferiert und im Biorad Gene Pulser mit den „Ec1“-Standardeinstellungen elektroporiert. Sofort danach wurde 1 ml SOC-Medium zugefügt, um die Transformations-Effizienz zu erhöhen. Die Mischung wurde in ein 2 ml-Eppendorf-Gefäß überführt und bei 32 °C für 2 h im Schüttler inkubiert. Die Bakterien wurden bei 7.000 rpm für 1 min zentrifugiert und 800 μl des Überstandes verworfen. Das Pellet wurde in der Restflüssigkeit resuspendiert und auf einer LB-Agarplatte (+ 1:400 Cam + 1:1000 Kan) ausgestrichen, die anschließend für ca. 24 h bei 32 °C inkubiert wurde. Mini-Präparation der BAC-DNA 10 ml LB-Medium (+ 1:400 Cam + 1:1000 Kan) wurden mit der gewünschten E. coli- Kolonie beimpft und über Nacht bei 32 °C und 225 rpm im Schüttler inkubiert. Am nächsten Morgen wurde die Vorkultur in ein 50 ml-Falcon-Gefäß umgeschüttet und bei 4 °C für 10 min bei 4000 rpm zentrifugiert. Der Überstand wurde abgegossen und das Pellet in 250 μl Lösung I (+ 0,1 [mg/ml] RNase A) durch vorsichtiges Pipettieren gelöst. Danach wurde die Suspension in ein 2 ml-Eppendorf-Gefäß überführt. Nach Zugabe von 250 μl Lösung II wurde das Gefäß zehnmal invertiert, um die Suspension gleichmäßig mit dem Lysepuffer zu vermischen. Während der folgenden fünf Minuten wurden die Bakterien bei Raumtemperatur lysiert, wodurch intrazelluläre Proteine freigesetzt wurden. Diese Reaktion wurde durch die Zugabe von 250 μl Lösung III beendet, die den Lysepuffer neutralisierte. Die ausgefällten Proteine wurden vom Überstand abgeschieden, indem die Suspension fünf Minuten lang mit 14.000 rpm bei Raumtemperatur zentrifugiert wurde. Der Überstand wurde in ein 2 ml-Eppendorf-Phase-Lock-Gel (Heavy)-Gefäß umgegossen und nach Zugabe von 750 μl Phenol/Chloroform mehrfach invertiert. Daraufhin wurde erneut bei 14.000 rpm für 5 min zentrifugiert. Der Überstand wurde mit 1 ml Isopropanol in einem neuen 2 ml-Eppendorf-Gefäß vermengt, um die BAC-DNA zu präzipitieren. Diese wurde bei 14.000 rpm für 20 min pelletiert. Das entstandene Pellet wurde mit 70%igem Ethanol gewaschen und für 10 min bei Raumtemperatur mit 14.000 rpm zentrifugiert. Der Überstand wurde vorsichtig abgesaugt und das DNA-Pellet luftgetrocknet. Daraufhin wurde die DNA in 50 μl TE-Puffer vollständig gelöst und bei 4 °C gelagert, bis sie weiterverarbeitet wurde. 19
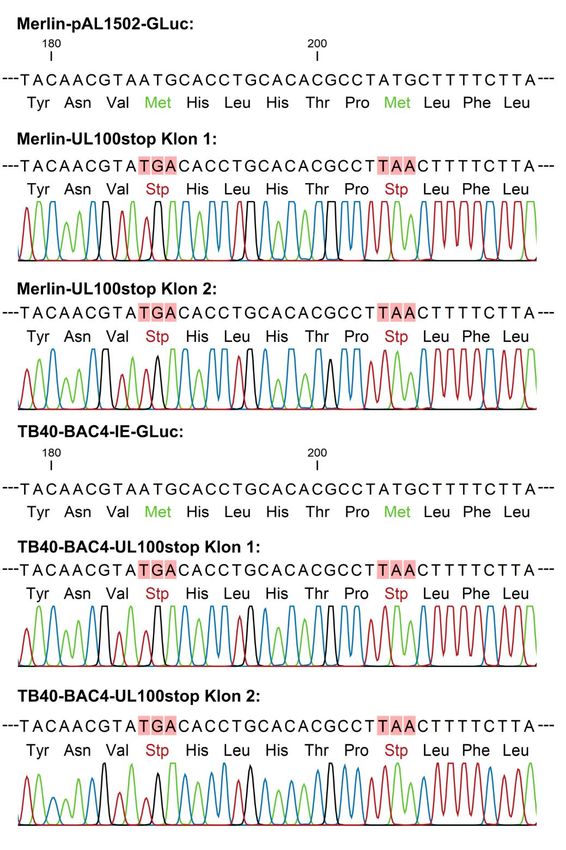
Tabelle 4: Reaktionsansatz für den Restriktionsverdau mit EcoRI und I-SceI. BSA: bovines Serum- albumin, U: Einheit, Σ: Summe 4μl 10X I-SceI Inkubationspuffer 0,7μl EcoRI (20U/μl) 1μl I-SceI (5U/ μl) 0,4μl BSA 33,9 μl H2O Σ 40 μl Inkubation bei 37°C für 2 h Um große, ungewollte Deletionen auszuschließen wurde die mutierte BAC-DNA und die BAC-DNA des korrespondierenden Wildtyps mit I-SceI und EcoRI verdaut (Tabelle 4). Die DNA-Fragmente wurden in einem 1%igen Agarosegel per Elektrophorese aufgetrennt, um zu überprüfen, ob das Bandenmuster der Mutante sich vom Wildtyp unterschied. Entfernung des positiven Selektionsmarkers 10 ml LB-Medium (+ 1:400 Cam + 1:1000 Kan) wurden mit einer elektroporierten E. coli- Kolonie beimpft und über Nacht bei 32 °C und 225 rpm im Schüttler inkubiert. 100 μl der Vorkultur wurden in ein 1,5 ml Eppendorf-Gefäß überführt und bei 7.000 rpm für 1 min zentrifugiert. Der Kanamycin-haltige Kulturüberstand wurde abgekippt, das Pellet in 2 ml LB-Medium (+ 1:400 Cam) resuspendiert und in ein 50 ml Falcon übertragen. Anschließend wurde die Kultur für 2 h bei 32 °C inkubiert. GS1783 E. coli-Bakterien enthalten eine L-Arabinose-induzierbare I-SceI-Expressionskassette, die nun durch Zugabe von 2 ml LB-Medium (+ 1:400 Cam+ 1% Arabinose) aktiviert wurde. Nach weiterer Inkubation für exakt 30 min bei 32 °C wurde durch Erhöhung der Temperatur auf 42°C im Wasserbad für 15 min das Red-Rekombinations-System aktiviert, das unter der Kontrolle eines hitze-sensiblen Promoters steht. Durch die zweite Red-Rekombination und die eingefügten Homologien wurde die Rekombinations-Kassette vollständig entfernt, sodass nur die gewünschte Mutation im BAC blieb. Die Kultur wurde bei 32 °C erneut für 2 h im Schüttler inkubiert. Nach einer 1:1.000-Verdünnung wurden 100 µl der Bakterien- suspension auf LB-Agarplatten (+ 1:400 Cam + 1% Arabinose) ausgestrichen und über Nacht bei 32°C im Brutschrank inkubiert. Überprüfung des Verlusts des positiven Selektionsmarkers Eine Einzelkolonie wurde in 50 µl LB-Medium gelöst und je 5 µl der Suspension auf korrespondierende LB-Agarplatten getropft, wobei eine 1:400 Cam enthielt, die andere 20
1:400 Cam + 1:1000 Kan. So wurde zwischen Klonen differenziert, die ihre Rekombinations-Kassette verloren hatten und dadurch kanamycinsensibel waren und solchen, deren Kanamycin-Resistenzkassette noch vorhanden war. Aus den kanamycinsensiblen Klonen wurde nun BAC-DNA isoliert. Um die exakte Insertion der Stopp-Codons zu überprüfen, wurden die Klone im Bereich der Mutation sequenziert. Daraufhin wurden sie in LB-Medium (+ 1:400 Cam) angezüchtet und als Glycerolstock (700 μl Bakterienkultur + 300 μl 50 %iges Glycerol) bei -80 °C gelagert. 2.2.3 Sequenzierung Alle Sequenz-Analysen wurden bei GATC Biotech erstellt. Hierfür wurden die Nukleotid- Sequenzen, die die Stopp-Codons enthalten sollten, per Kolonie-PCR amplifiziert (Tabelle 5, Tabelle 6). Tabelle 5: Cycler-Programm für die Kolonie- Tabelle 6: Reaktionsansatz für die Kolonie-PCR. PCR. ∞: unendlich PCR. Σ: Summe Temperatur Zeit Zyklen 25 μl TopTaq® Master Mix (2x) [°C] [min:s] 95 5:00 1 0,5 μl Primer 5’ („long forward“) 94 1:00 34 0,5 μl Primer 3’ („long reverse“) 57 1:00 24 μl H2O 72 1:00 + Bakterien 72 10:00 1 Σ 50 μl 10 ∞ Mit einer Agarose-Gelelektrophorese wurde das PCR-Produkt nachgewiesen. Daraufhin wurde es mit dem NucleoSpin® Gel and PCR clean-up Kit (Macherey-Nagel) dem Proto- koll entsprechend aufgereinigt und in 15 μl H2O dd gelöst. Danach wurde die DNA- Konzentration photometrisch gemessen, die Probe gemäß den Vorgaben der GATC Bio- tech (Tabelle 7) vorbereitet und zur Sequenzierung eingeschickt. Tabelle 7: Probenvorbereitung für die Sequenzierung nach Sanger bei GATC Biotech. DNA: Desoxyri- bonukleinsäure. Das DNA-Volumen wurde an die photometrisch bestimmte Konzentration angepasst . 5 μl DNA (20-80 ng/μl) 0,5 μl Primer 5’ („long forward“) ad 10 μl H2O 21
2.2.4 Transfektion von BAC-DNA in HFFs zur Virusrekonstitution 200 ml LB-Medium (+ 1:400 Cam) wurden mit einer E. coli-Kolonie inokuliert und über Nacht bei 32 °C und 225 rpm geschüttelt. Die Kultur wurde bei 45.000 rpm und 4 °C für 10 min zentrifugiert. Die virale BAC-DNA wurde daraufhin gemäß des Herstellerprotokolls des NucleoBond® Xtra Midi Kits (Macherey-Nagel) isoliert, aufgereinigt und in 150 μl TE-Puffer für mindestens drei Stunden auf dem Schüttler gelöst. Anschließend wurde die Konzentration der BAC-DNA im Photometer gemessen. Tabelle 8: Ansatz des EcoRI-Restriktionsverdaus im Rahmen der Midi-Präparation. BAC: bakterielles artifizielles Chromosom, DNA: Desoxyribonukleinsäure, U: Einheit 10X EcoRI Inkubationspuffer 2 μl EcoRI (20 U/μl) 1 μl BAC-DNA 2 µg H2O ad 20 μl Um große, ungewollte Deletionen auszuschließen wurde außerdem ein Teil der BAC-DNA der Mutante sowie BAC-DNA des zugehörigen Wildtypen mit EcoRI verdaut (Tabelle 8). Da nur minimale Veränderungen im Genom vorgenommen worden waren, wurde erwartet, dass die Restriktionsmuster der DNA-Probe der Mutante und des korrespondierenden Wildtypen identisch sein würden. Um aus der viralen BAC-DNA Viren zu rekonstituieren, wurden HFFs mithilfe des MBS Mammalian Transfection Kits der Firma Agilent Technologies transfiziert. Die Fibroblas- ten wurden drei Tage vor der Transfektion in MEM10 gesplittet. Am darauffolgenden Tag wurden sie mit MEM gewaschen und zum Serumentzug in diesem Medium belassen. Am Vortag der Transfektion wurden die Zellen abtrypsiniert, ausgezählt und auf 6-well-Platten in MEM10 ausgesät. Durch den Serumentzug und die Zugabe von FCS-haltigem Medium einen Tag zuvor sollte das Wachstumsverhalten der Fibroblasten zum Zeitpunkt der Trans- fektion optimiert werden. Dadurch sollte die Transfektionseffizienz erhöht werden. Für jede virale BAC-DNA wurden zwei Ansätze à 300.000 Zellen vorbereitet („Transfektions- duplikate“, die folgenden Angaben beziehen sich auf einen Ansatz). Für die Transfektion wurden 2 µg virale BAC-DNA mit H2O (Roth T 143.4) auf ein Gesamtvolumen von 75 µl in Polystyren-Röhrchen aufgefüllt. Es wurden 8,3 µl Lösung 1 hinzugefügt und durch vor- sichtiges Antippen des Röhrchens sanft durchmischt. Daraufhin wurden 83,3 µl Lösung 2 zugetropft und vorsichtig geschüttelt. Während der folgenden Inkubationszeit von 25 min wurden die HFFs einmal mit 3 ml PBS gewaschen. 430 µl MBS-Medium wurden zum DNA-Gemisch hinzu pipettiert und mithilfe von „cell saver“-Pipettenspitzen durchmischt. 22
Die PBS-Lösung wurde von den Zellen abgesaugt, das Reagenz auf die HFFs getropft und bei 37 °C für zwei Stunden inkubiert. Daraufhin wurde die DNA-Lösung entfernt. Die Zel- len wurden mit 1 ml eisgekühlter Glycerolschock-Lösung für exakt 30 s behandelt. Direkt danach wurden die HFFs dreimal mit MEM5 gewaschen und im gleichen Medium über Nacht bei 37 °C inkubiert. Am nächsten Tag wurden die Zellen aus den beiden Transfekti- onsduplikaten in MEM5 gemischt, ausgezählt und mit einer Konzentration von 80.000 HFFs pro Ansatz auf gelatinebeschichtete 24-well-Platten als Duplikate ausgesät. Beide unabhängigen Klone einer Mutante wurden zweimal transfiziert. 2.2.5 Gaussia Luciferase-Messung 24 h vor jeder Probenentnahme wurde das Medium der 24-well-Platte ausgetauscht, um die Vergleichbarkeit der Gaussia Luciferase-Konzentrationen zu gewährleisten. An den Tagen 2, 6, 9, 13, 16, 20, 23, 27 und 30 nach Transfektion wurden 200 µl Gaussia Luciferase- haltiger Kulturüberstand entnommen und bei -20 °C eingefroren. Das entnommene Volu- men wurde durch frisches MEM5 ersetzt. Wenn alle Proben eines Experiments vorlagen, wurde von jeder Probe eine 1:10-Verdünnungsreihe erstellt und je 20 µl auf eine 96-well Lumineszenz-Readerplatte (LUMITRAC, Greiner Bio-One) übertragen. Das Substrat Co- elenterazin (PJK GmbH) wurde in PBS mit 5 mmol/l NaCl auf 0,2 µg/ml verdünnt. 50 µl dieser Lösung wurden im Mikroplatten-Reader (Chameleon, Hidex) automatisch vor der Messung in jeden Ansatz injiziert. Die Gaussia Luciferase setzte nun das Coelenterazin um, wodurch ein Lichtsignal emittiert wurde, das ein Detektor registrierte. Das Ausgangs- signal am Detektor ist proportional zur Intensität des einfallenden Lichts und wird in rela- tiven Lichteinheiten (RLU) angegeben Der Chameleon Plate Reader unterliegt einem De- tektionsmaximum von < 5x107 RLU. Daher wurden Daten, die diesen Grenzwert über- schritten, als unzuverlässig eingestuft und stattdessen über die höheren Verdünnungsstufen der Reihe extrapoliert. Die Expression der Gaussia Luciferase wurde ebenso wie die Virus- replikation durch den IE-Promoter kontrolliert, sodass die Enzymaktivität wahrscheinlich als Maßstab für die Virusreplikation betrachtet werden kann. 23
Abbildung 5: Exemplarische Darstellung der Gaussia Luciferase-Aktivität von Merlin-pAL1502-GLuc und TB40-BAC4-IE-GLuc sowie den UL55stop-Mutanten. Beide unabhängige Klone wurden gleichzeitig transfiziert. In regelmäßigen Abständen wurden Proben entnommen. A) Merlin-pAL1502-GLuc wächst über den gesamten Zeitraum hinweg. Der lineare Verlauf zwischen 9-16 dpt weist auf exponentielles Wachstum in diesem Bereich hin. B+D) Die beiden UL55stop-Klone zeigen nach einer initialen Zunahme der Luciferase- Expression einen Abfall bis in den Bereich des Hintergrunds. C) TB40-BAC4-IE-Gluc erreicht an Tag 20 die maximale Luciferase-Expression. Danach ist die Kultur vollständig durchinfiziert. Der lineare Verlauf zwischen 9-16 dpt weist auf exponentielles Wachstum in diesem Bereich hin. Die Wildtypen wuchsen 9-16 dpt exponentiell. Die Luciferase-Aktivität soll im Folgenden mit einer Funktion f(t) beschrieben werden. Die in diesem Zeitraum enthaltenen Messwerte wurden logarithmiert, um eine lineare Kurve zu erhalten (Abbildung 6): log( ( )) = + 0 , (1) wobei m die Steigung und y0 der Achsenabschnitt der linearen Kurve ist und t die Zeit in Tagen darstellt. Somit lässt sich die Luciferase-Aktivität beschreiben mit ( ) = 10 + 0 (2) Die Wachstumsrate WR gibt die relative Zu- oder Abnahme der Aktivität des Reporterenzyms innerhalb eines Zeitintervalls Δt (üblicherweise Δt = 1 d) an und ist definiert als ( +Δ )− ( ) WR = . (3) ( ) 24
Mit (2) folgt daraus 10 ( +Δ )+ 0 −10 + 0 WR = = 10 Δ − 1. (4) 10 + 0 Somit lässt sich die Wachstumsrate anhand der Steigung m bestimmen, die wiederum durch eine lineare Regression der logarithmierten Luciferase-Aktivität ermittelt wird (Abbildung 6). Jeder Klon wurde zweimal transfiziert und jede Verdünnungsreihe des Gaussia Luciferase-haltigen Überstandes zweimal gemessen, um eventuelle Schwankungen der Messtechnik auszugleichen. Aus den Wachstumsraten wurden der Mittelwert und der Standardfehler des arithmetischen Mittels berechnet. Um statistisch signifikante Unterschiede zwischen den Datensätzen zu identifizieren, wurde eine Einweg- Varianzanalyse („one-way ANOVA“) durchgeführt, an die ein geeigneter post-hoc-Test angeschlossen wurde. Abbildung 6: Exemplarische Darstellung der Ermittlung der Steigung m aus der Gaussia Luciferase- Aktivität. Die Aktivität der Luciferase, die die Zellen in 24 h produzierten, wurde in zwei Transfektionen je zweimal bestimmt. Die lineare Regression zeigt die mittlere Steigung m an. A) Merlin-pAL1502-GLuc. Die Gaussia Luciferase-Aktivität nimmt mit einer mittleren Steigung von m = 0,19 zu. B) Merlin-UL55stop Klon 2. Die Gaussia Luciferase-Aktivität nimmt mit einer mittleren Steigung von m = -0,18 ab. 2.2.6 Färbung IE-Ag-positiver Fibroblasten Die im Folgenden genannten Volumenangaben beziehen sich auf einen Ansatz mit 80.000 Fibroblasten auf einer 24-well-Platte. Zunächst wurde das Kulturmedium entfernt und die Zellen wurden in 80%igem Aceton für 5 min bei Raumtemperatur fixiert. Daraufhin wurden sie mit 500 μl Primär-Antikörper (Mouse anti-HCMV I.E.A. „E13”, gerichtet gegen die IE-Antigene pUL122/pUL123) in PBS überschichtet und für 2 h bei 37 °C inkubiert. Anschließend wurden die Fibroblasten mit 500 μl Sekundär-Antikörper (Cy™3- conjugated AffiniPure F(ab´)2 Fragment Goat anti-Mouse IgG [H+L]) in PBS für 45 min bei 37 °C behandelt. Zum Schluss wurden die Zellkerne mit 1 ml DAPI (0,1 ng/μl) in PBS für 5 min bei Raumtemperatur gefärbt. Zwischen jedem dieser Schritte wurde dreimal mit 25
Sie können auch lesen

















































