Zellorganellen am exocytotischen und endocytotischen Weg - Dr. Pál Röhlich 1. rER, Golgi-Apparat, Exocytose
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Zellorganellen am exocytotischen
und endocytotischen Weg
1. rER, Golgi-Apparat, Exocytose,
2. Endocytose (Phagocytose, Pinocytose) Lysosom, Autophagie,
vesiculärer Transport,
Dr. Pál Röhlich
prof. emeritus
ÁOK 2018/2019, I. Semester, 19. 20. Sept. 2018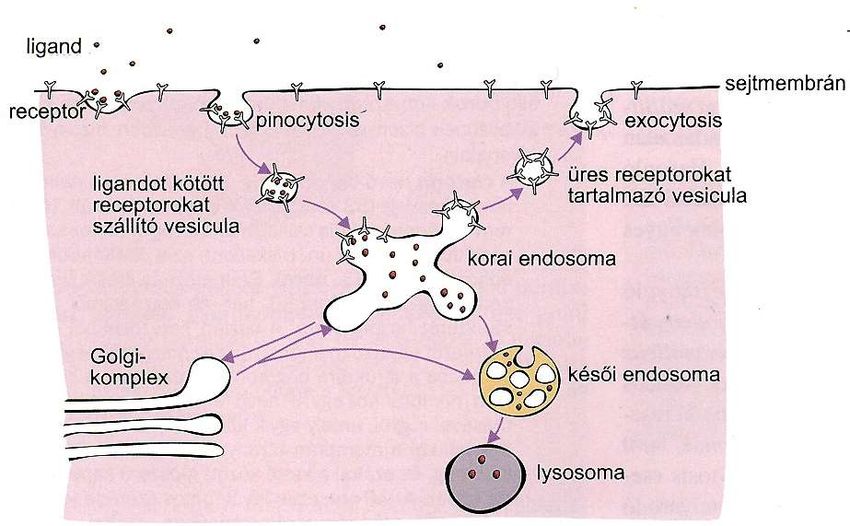
Transkription Translation
(Proteinsynthese)
DNA
Peptidkette
mRNA
Ribosom
Gen
mRNA
Cytoplasma
Zellkern
Die zwei Schritte der
Genexpression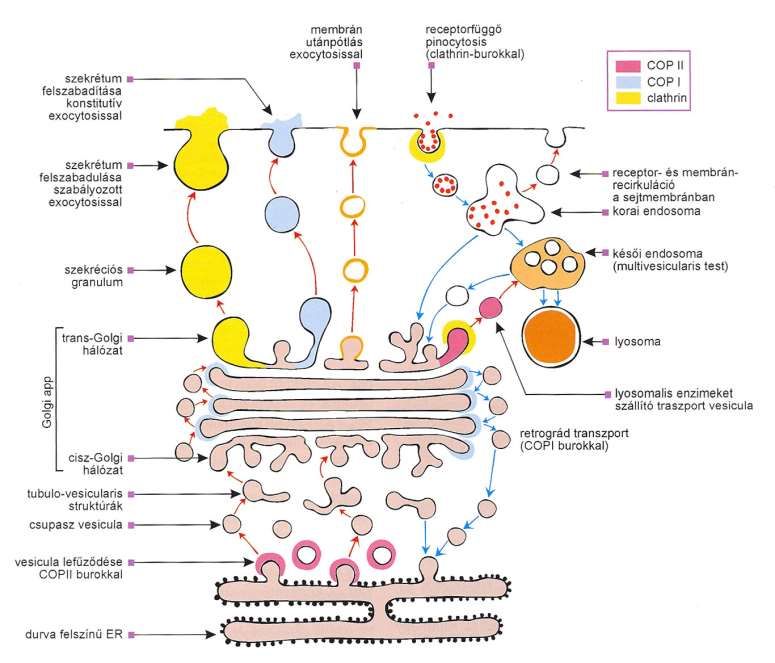
Die Polypeptidkette nach der Translation
Aufnahme der richtigen Raumstruktur in den meisten Fällen spontan während der Translation (die
Aminosäuresequenz bestimmt die endgültige, tertiäre Struktur des Proteins!) oder mit Hilfe (Chaperone: große
Proteinkomplexe mit einem inneren Raum helfen die richtige Raumstruktur aufzunehmen).
Abbau falscher, nicht richtig gefalteter Proteine. Solche Proteine werden erkannt, mit Ubiquitin (kleine
Proteine) markiert, und in großen proteolytischen Komplexen (Proteasomen) abgebaut.
Chemische Modifikationen (posttranslationelle Modifikationen). Z.B. Anknüpfung von reaktiven Gruppen (z.B.
OH-Gruppe), von kürzeren oder längeren Zuckerketten (Glykosylierung), Spaltung der Polypeptidkette an
bestimmten Stellen (gezielte Proteolyse), …
Sortierung der Proteine (targeting). Bedeutung. Lokalisationssignale (kurze Peptidabschnitte bestehend aus
mehreren Aminosäuren), Receptorproteine, die das Signal in den Zielkompartimenten erkennen,
Transportmechanismus.
Proteinsynthese am Ribosom
Cotranslations
Zellkern
-transport
Protein-Sortierung
spätes Endosom, Zellmembran,
bzw. Lysosom extracellulärer Raum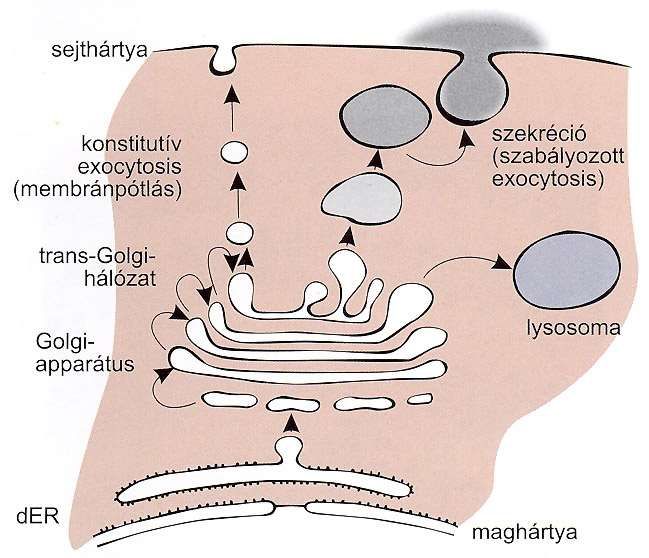
Der Weg durch das endoplasmatische
Reticulum und Golgi-Apparat
Plasmamembran
konstitutivek Exocytose geregelte Exocytose
(Membran-Nachschub)
o Sekretion
Proteine: n
s
t
trans-Golgi-
• Exportproteine (sekretorische Pr.) i
Netzwerk
• Lysosomale Proteine t
u Lysosom
• Integrale Membranproteine Golgi-
Apparat
• ER-Golgi-residente Proteine
Kernhülle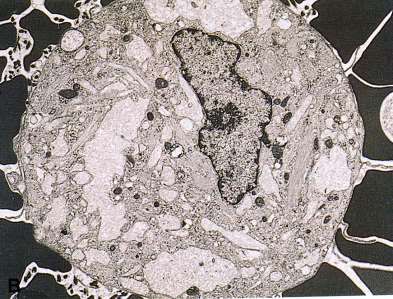
Golgi-Apparat
Der Golgi-Apparat kann mit speziellen Methoden lichtmikroskopisch nachgewiesen werden.
Entdeckung und erste Beschreibung: Camillo Golgi (1843-1926), konnte in 1898 mit einer
Versilberungstechnik in Nervenzellen ein Netzwerk nachweisen („apparato reticolare interno”). Nobel-
Preis1906.
Für eine lange Periode hat man diese
Struktur als Kunstprodukt gehalten.
Netzwerk Dictyosom
Zwei Nervenzellen aus einem
Spinalganglion, Versilberung nach Aoyama
Lichtmikroskopische Darstellung: mit Versilberung, mit
langer Osmiumfixierung, oder mit Immuncytochemie
Lokalisation: meistens in der Nähe des Zellkerns.
Immuncytochemischer Nachweis des Golgi-Apparates an einer
Individuelle Baukomponenten: Dictyosomen.
gezüchteten Bindegewebszelle mit Antikörper gegen ein residentes
Golgi-Protein.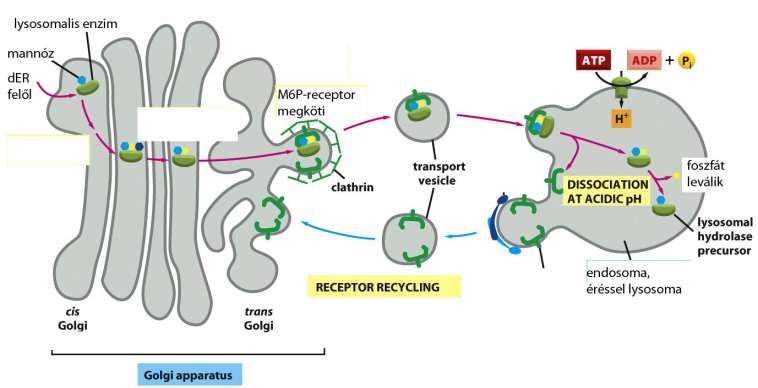
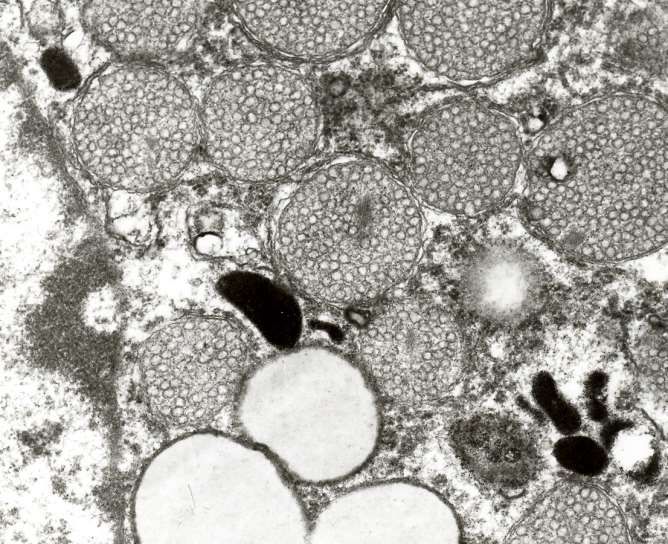
Elektronenmikroskopisches Bild des Golgi-Apparates
Polarität: cis und trans Seite
Kondensierungsvakuol
trans-Golgi Netzwerk (TGN)
rER
cis-Cisterne trans-Cisterne
cis-Golgi Netzwerk
Golgi-Vesikeln 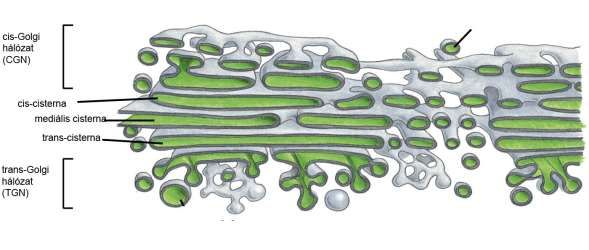
Was geschieht im Golgi-Apparat?
Sortierung: rER Proteine zurück, die
anderen vorwärts.
Mannose-Phosphorylierung der
rER
lysosomalen Proteine (Signal Man-6-P)
vesikulärer Transport rER → Golgi
Abspalten überflüßiger
Mannosen
cis Seite
cis-Golgi
Netzwerk
Anbindung von
Acetylglukosamin cis-Cisterne
mediale Cisterne
Anbindung von Galaktose trans-Cisterne
und Sialsäure
trans-Golgi
Netzwerk
Synthese von (TGN)
Glykosaminoglykanen
trans Seite
Ankopplung von
Sulfatgruppen,
Exportproteine Membranproteine lysosomale Proteine
Sortierung
Richtung Plasmamembran Richtung Lysosom
Glykosylierung (terminaler Abschnitt der N-Glykosylierung, O-
Glykosylierung: Glycocalyx, (Glykoproteine, Glykolipide, Mucine), Synthese
von Glykosaminoglykanen, Proteoglykanen :extracelluläre Matrix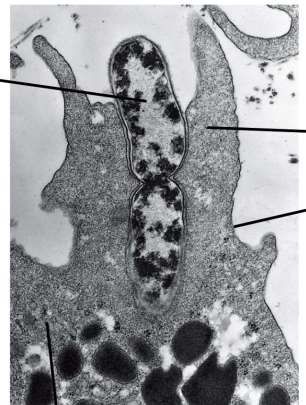
Exocytose:
Öffnen eines Vesikels oder Vakuols an der Zellmembran durch
Fusion der zwei Membranen. Bedeutung.
Sortierung vom trans-Golgi Netz
1. Richtung Plasmamembran
A. Membranproteine Transport dieser Proteine mit den vom
trans-Golgi Netz abgeschnürten Vesikeln. Läuft
kontinuierlich, die Vesikeln öffnen sich an der
Plasmamembran mit Exocytose. Plasmamembran
B. Export- oder Sekretionsproteine. Sekrethaltige Vesikeln
oder Vakuolen (größer) schnüren sich vom trans-Golgi
Netz ab, öffnen sich mit Exocytose an der
konstitutive Exocytose geregelte Exocytose
Plasmamenbran mit Exocytose, ihr Inhalt ist freigesetzt. (Membrannachschub) (Sekretion)
Geregelte Exocytose: Exocytose nur an äussere Signale
(Signalübertragung). zB. Mastzelle.
trans-Golgi
Konstitutive Exocytose: kontinuierlich, ohne spezifische Netzwerk
Signale.
Lysosom
Golgi
Kompllex
2. Richtung Lysosom (bzw. dessen frühe Form, das späte
Endosom)
Lysosome Proteine. Lokalisationssignal: Mannose-6-Phosphat,
rER
Kernhülle
Transportreceptoren: Man-6-P-Receptoren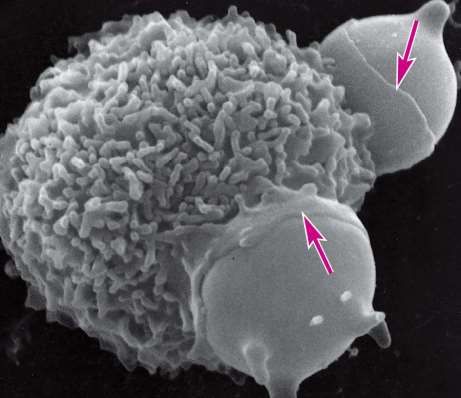
Exocytose in einer sezernierenden Zelle
(Mastzelle)
Fusionsstelle zwischen
Vesikelmembran und Plasmamembran
Exocytose von zwei sekretorischen Sequentielle Exocytose: von aussen nach
Vesikeln innen öffnen sich die sekretorischen Vesikeln
ineinander.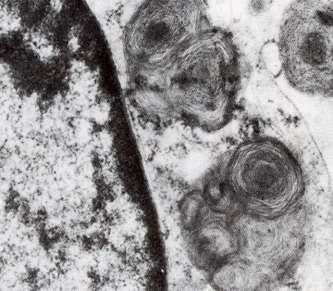
Endocytose: Aufnahme von Stoffen in die Zelle durch Einstülpung der Plasmambran und Abschnürung.
Der endocytierte Stoff ist vom Cytosol mit Membran (abgeschnürte Plasmamembran) getrennt.
I. Phagocytose
Endocytose von großen Teilchen (>0,1-5 μm)
Beispiele: Makrophag, neutrophile Granulocyten (professionelle Phagocyten)
Bakte-
rium
Cytoplasma-
Fortsatz
Plasma-
membran
Erythrocyt
Ein Makrophag phagocytiert zwei Erythrocyten. Phagocytose von Bakterien in eine weiße
Pfeil: Rand des Cytoplasmafortsatzes. Raster EM. Blutzelle (neutrophiler Granulocyt). EM Bild.Phagocytierte Teilchen: Zelltrümmer, Bakterien, veraltete Blutzellen mit geänderter Zelloberfläche, apoptotische Zellen
und apoptotische Körperchen, Fremdpartikel, usw.…
Fc-Ende des IgG-
Moleküls
Phasen der Endocytose: Fc-Receptor
Bakterium
1. Adsorption: Anbindung des Teilchens an die Plasmamembran (meistens mit
Hilfe von Receptoren in der Membran).
Spezifische Receptoren: z.B. Fcγ-Receptoren, Komplement-Receptoren. Beispiel:
Phagocytose der mit Immunglobulin bedeckten Bakterien in einer
Makrophagzelle.
Nicht-spezifische Receptoren: Receptoren, die nicht gewöhnliche
Kohlenhydraten oder eine geänderte Phospholipidmuster an der Zelloberfläche
erkennen und binden (sogenannte Scavanger-Receptoren). Sortierung: lebende
Zellen sind nicht phagocytiert, an ihrer Oberfläche gibt es („freß mich nicht!”) Aktin
Proteine, die hemmende Receptoren an der Phagocyt-Oberfläche aktivieren Mikrofilamente
(Hemmung der Phagocytose).
2. Ingestion: die Zelle mit ihrer Kravattenähnlichen Zellfortsatz
(Pseudopodium, Lamellipodium) fasst das Teilchen um, die freien Ränder
des Fortsatzes schließen sich über das Partikel. Polymerisierung von
Aktin-Mikrofilamenten im Zellfortsatz, ausgelöst durch die Adsorption
des Partikels. Nach der Abschnürung ist das Teilchen in eine Vakuole
eingeschlossen (Phagosom).
Phagosom: Phagosom
(phagocytiertes
Bakterium)
phagocytiertes und mit Membran EM Bild
umgebenes Partikel im Cytoplasma.
Schicksal: Fusion mit Lysosom und Abbau.. Membran
Lysosomen
Lysosom:
EM: membranbegrenztes Zellorganell mit einer
dunklen,feingranulären Substanz im Inneren. Inhalt:
saure hydrolytische Enzyme (>40 verschiedene), pH
Optimum: bei 5. Funktion: Abbau der
Makromoleküle mit Hydrolyse.
EM Bild der Lysosomen,
rote Pfeile Membran des
LysosomsLicht- und elektronenmikroskopischer, enzymhistochemischer
Nachweis der Lysosomen
Mito-
chondrium
β-Glukuronidase (rot) und saure
Phosphatase (braun) in
Lysosomen der Lysosomen
Nierenepithelzellen
Enzymhistochemischer Nachweis der Lysosomen in einer Zelle der
Nebennierenrinde. EM Bild. Nicht-spezifische Esterase.Lysosomale Enzyme: Nukleasen, Proteasen, Glykosidasen, Lipasen, Phosphatasen,
Sulphatasen, …(40 verschiedene Enzyme). Davon mehrere in einem Lysosom.
Saueres pH durch H+ Pumpe (Proton- ATPase, vakuolärer Typ) in der
Lysosomenmembran.
Lysosomenmembran. Ihre Intaktheit (und neutrales pH des Cytosols) schützt die Zelle
von Selbstverdauung durch lysosomale Enzyme. Schutz der Membran gegen Abbau durch
Enzyme: stark glykosilierte Proteine und Lipide in der Membran (die Zuckerketten halten
die Enzyme entfernt von der Membran). Transportproteine und Proton-ATPase in der
Membran.
EM immuncytochemischer
Nachweis eines lysosomalen
Enzyms (Cathepsin D) in der
Leberzelle (schwarze
Goldpartikeln).
Weg der lysosomalen Enzyme vom Golgi-Apparat in das Lysosom
rER, Golgi-App. (Man-6-P Markierung im cis-Golgi Netzwerk, Man6P-Receptoren im trans-Golgi
Netzwerk), vesikulärer Transport in das Lysosom (beim sauren pH des Lysosoms wird das Enzym frei
vom Receptor, auch die Phosphatgruppe löst sich ab). Vesikeln mit den leeren Receptoren kehren
zurück.
lysosomales Enzym
Mannose
kommt mit vesikulärem
Transport vom rER
Bindung an M6P-
Receptoren
Phosphatgruppe
abgetrennt
Endosom, später
Lysosom
Intrazelluläre Verdauung
Verdauung: Lysosom fusioniert mit Phagosom (Phagolysosom), Enzyme bauen die Makromoleküle
des phagocytierten Materials ab, die Monomere (Aminosäuren, Einfachzucker, etc.) werden durch
Membrantransport in das Cytosol transportiert (Energie für den Transport kommt vom
Protongradient).
Phagolysosom
Phagosom
Lysosom (sekundäres
Lysosom)
Verdauung, Monomere sind ins
Cytosol transportiert
Unverdaubare Substanzen können sich als Residualkörper (oder Telolysosomen) anhäufen, und können
von der Zelle mit Exocytose entleert werden. Bei langlebigen Zellen (z.B. Nervenzellen,
Herzmuskelzellen) sind Residualkörper mit akkumulierten, schwer verdaubaren Lipiden und Proteinen
als gelbbraune Lipofuscin-Granulen (oder Alterspigment) im Lichtmikroskop zu sehen. Unverdaubare
Fremdstoffe, wie Kohlepartikeln in Makrophagen der pulmonalen Lymphknoten oder Farbstoffpartikeln
in Makrophagen der Haut nach Tetovierungen bleiben in Lysosomen bis zum Tod der Zelle und werden
nach ihrem Zerfall von anderen Zellen wieder phagocytiert.
Alte Erythrocyten werden von Makrophagen in der Leber, Milz und Knochenmark phagocytiert, das
Eisen von Hämoglobin in eisenbindendes Protein Ferritin aufgenommen, dessen Abbauprodukt
Hämosiderin in Lysosomen dieser Zellen als braune Granulen (Siderosomen) zu erkennen sind.Lysosomale Krankheiten („Speicherungskrankheiten”)
I-Zell Kranheit: das M6P-Signal fehlt von lysosomalen Enzymen (das ankoppelnde Enzym ist defekt), die Enzyme gelangen
nicht in die Lysosomen sonder werden als Exportproteine mit Exocytose in den extracellulären Raum freigesetzt. In den
Zellen akkumulieren viele große Phagosomen (Einschlüsse).
Mucopolysaccharidosen. Glykosaminoglykane (GAG) der extracellulären Matrix sind in Bindegewebszellen wegen
Fehlens des entsprechenden lysosomalen Enzyms (vor allem α-Iduronidase) nicht abgebaut. Die Bindegewebszellen sind voll
mit GAG-enthaltenden Phagolysosomen.
Tay-Sachs Krankheit. Gangliosiden (spezielle Membranlipide) sind wegen Fehlens des entsprechenden lysosomalen
Enzyms (Hexosaminidase A) nicht verdaut. Große Einschlüsse mit unverdauten Gangliosiden in den Zellen, vor allem im
Nervensystem. Schwere neuronale Degeneration.
Gaucher-Krankheit. Fehlen von Glykocerebrosidase in den Lysosomen, Glykocerebrosiden sind in Phagocyten angehäuft.
Gaucher-Krankheit. EM Bild Ausschnitt von einer Nervenzelle in Tay-Sachs
Krankheit. Gangliosid-Einschlüsse. EM Bild.Autophagie
Fokale Verdauung von Cytoplasmaregionen unter ungünstigen Lebensbedingungen, Beseitigung von
überflüssigen Bestandteilen der Zelle.
Eine C-förmige sequestrierende Cisterne schließt eine Cytoplasmaregion um. Zwei Membranen, die
innere Membran verschwindet später.
Der völlig umschlossene Cytoplasmateil ist das Autophagosom, dies fusioniert mit einem Lysosom und
wird abgebaut.
Eine ausgedehnte Autophagie kann zum Sterben der Zelle führen,
viele halten es für die zweite Form des programmierten Zelltodes.
Sequestrierende Cisterne (Pfeile), EM Bild. Autophagosom, EM Bild
Aufnahmen von Prof. J. KovácsII. Pinocytose
Aufnahme von Partikeln kleiner als 0,1 μm, von Makromolekülen, oder gelösten Stoffen mit
Endocytose. Die entstandenen Vesikeln sind etwa 50-100 nm groß.
1. Flüßigkeitspinocytose: die Teilchen sind in gelöstem Zustand und sind im Lumen des endocytotischen
Vesikels aufgenommen.
2. Adsorptive Pinocytose. Die Teilchen sind an die Plasmamembran gebunden (Anreicherung der Teilchen
an der Membranoberfläche!), deshalb effektiv auch bei niedrigen Konzentrationen in der Umgebung.
A. Nicht-spezifische Bindungsstellen (z.B. Glykokalyx, nicht-spezifische Receptoren). Einige
bakterielle Toxine (Diphteria), Viren (z.B. Influenza, VSV, Semliki Forest Virus) sind in dieser
Weise in die Zelle aufgenommen.
B. Spezifische Bindungsstellen (Receptoren): receptorvermittelte Pinocytose (oder
Endocytose).
Gebundene Moleküle (Ligande): Immunglobuline, α2-Makroglobulin, Transferrin, LDL ,
Asialoglikoproteine, …
Receptoren: integrale MembranproteineReceptorvermittelte (clathrinabhängige) Pinocytose
Mechanismus: Abschnürung eines Vesikels von der Plasmamembran mit Hilfe einer Clathrin-Hülle.
Die 3D Umordnung der Hülle zwingt die Membran eine sphärische Form aufzunehmen. Receptorproteine mit der
gebundenen Substanz, Adaptin Proteine, und Clathrin lagern sich aneinander. Das G-Protein Dynamin trennt den Vesikel
von der Plasmamembran ab. Nach Abschnürung löst sich die Clathrin-Hülle ab.
Adaptin Cclathrin
Receptor
Liganden
Abtrennung des
Vesikels von der
Knospung eines Vesikels und Einfangen der Liganden mit der Clathrin-Hülle
Membran mit Dynamin
Ausbildung eines Vesikels mit
Stachelsaum (Clathrinhülle).
EM Bilder.
clathrin-pit (clathrin- unmittelbar vor der clathrin-Vesikel
Grübchen) AbschnürungGrübchen und Vesikeln mit Clathrinhülle unter der Plasmamembran,
vom Inneren der Zelle gesehen, EM Bild, Heuser Technik.
Vesikel mit
Clathrinhülle
Grübchen mit
Clathrinhülle (coated pit)Clathrin-unabhängige Pinocytose
Bildung von glatten Vesikeln durch Invaginationen der Membran. Konstitutiv oder receptorvermittelt.
Zwei Formen:
1. Uncharakteristisch (z.B. Membranrezirkulation), oder
2. Caveolae.
Membran
Caveolae
Spezielle Lipidzusammensetzung (Cholesterin und
Sphingolipide, „membrane rafts”), Cytosol
Caveolin-Hülle an der cytosolischen Seite, Caveoline sind V-
förmige integrale Membranproteine, reichen nur in die
innere Lipidschicht der Membran ein. Andere
Komponenten der Hülle: cavin-Proteine (verantwortlich für
die Krümmung)
Omega-förmige Invaginationen der Plasmamembran, kommen
reichlich in Bindegewebszellen, Fettzellen, Endothelien,
Muskelzellen vor.
Unterschiedliche Funktionen:
können stationär sein (sammeln Signalübertragungsmoleküle
ein)
können sich abschnüren (Pinocytose) z.B. Aufnahme von
Albumin, Transcytose in EndothelzellenDas frühe Endosom ist eine Verteilungsstelle
Plasmamembran
Transportvesikel mit Vesikel mit leeren Receptoren im
Ligand-geladenen Rückkehr (Recirkulation)
Receptoren
frühes Endosom
spätes Endosomtest
multivesicularis (multi-
vesiculäres Körperchen)
Lysosom
Was geschieht mit dem pinocytotischen Vesikel und mit der
aufgenommenen Substanz?
Der Vesikel verliert seine Hülle und fusioniert mit dem frühen Endosom.
Endosom: Membranbegrenztes Zellorganell von variabler Größe und Form. Sein Inhalt wird mehr
und mehr azidifiert durch die Aktivität der Protonpumpen in seiner Membran.
Frühe Endosomen: meistens im marginalen Cytoplasma. pH-Werte 6.5-5, die meisten Liganden
lösen sich von ihren Receptoren bei diesem pH ab (ligand-receptor uncoupling). Wichtige
Sortierungsstelle in Richtungen Plasmamembran, Golgi-Apparat und Lysosom.
Späte Endosomen: tiefer im Cytoplasma, mehr sauer. Lysosomale Enzyme erscheinen durch
vesikulären Transport vom trans-Golgi Netzwerk.
Multivesikuläre Körperchen: bei der Reifung des späten Endosoms sind von seiner Membran
Vesikeln in das Innere abgeschnürt, viele Vesikeln im Lumen. Integrale Membranproteine in ihrer
Membran zum Abbau bestimmt.
Lysosomen: meistens in der Umgebung des Golgi-Apparates, pH 4,5-5, viele hydrolytische
Abbauenzyme, Abbau der Stoffe.Schicksal der Receptoren (verschiedene Möglichkeiten)
• Receptoren kehren in die Plasmamembran zurück mit vesiculärem Transport nach der
Ablösung des Liganden. Vesikeln mit den leeren Receptoren knospen von frühem (teilweise auch
spätem) Endosom ab und nach Erreichen der Plasmamembran fusionieren sie mit der Membran mit
Exocytose: Receptor-Recirkulation (receptor recycling). Damit sind sie in die Plasmamembran
reintegriert. Beispiel: LDL-Receptor.
• Receptoren gelangen weiter in das späte Endosom und sind in Lysosomen abgebaut.
(receptor downregulation). Beispiel: EGF Receptor (epithelialer Wachstumsfaktor mit seinem
Receptor).
Schicksal des Liganden (verschiedene Möglichkeiten)
• Wird vom frühen Endosom bis in die Lysosomen weiter transportiert, wo es abgebaut wird. Oder:
• Im frühen Endosom bleibt das Ligand an seinem Receptor und recirkuliert zusammen mit seinem
Receptor an die Zelloberfläche. Beispiel: Transferrin (ein eisentransportierendes Protein, welches das
gebundene Eisen freisetzt, und dann zusammen mit seinem Receptor in die Plasmamembran für eine
weitere Runde zurückkehrt.Aufnahme von LDL-Partikeln
LDL (low-density lipoprotein):
mit receptorvermittelter Lipoprotein Partikel im Blut, transportiert
Cholesterin
Endocytose Cholesterin und Cholesterinestern. Teilweise
bedeckt mit dem Protein Apoprotein B.
Cholesterin-
Apoprotein B Estern
LDL-Bindungsstelle
extracellulärer Raum
Plasmamembran
Adaptin Bindung
Grübchen mit Clathrin
leere Receptoren
kehren zur
Plasmamembran
zurück
Adaptin-Bindungsstelle fehlt Fusion mit Knospung von
Endosom Vesikeln
Wenn die Adaptinbindungsstelle am LDL- frühes Endosom
Receptor fehlt (Fehler im Gen), kann der
Receptor Adaptin und dadurch Clathrin nicht
binden. Der Aufnahmemechanismus freies Cholesterin
funktioniert nicht, LDL bleibt im Blut. Hohe im Cytosol
Cholesterinkonzentration im Blut führt zu lysosomale Enzyme
Arteriosclerose und Herzinfarkt (familiäre
Hypercholesterinämie).Transcytose
Transport eines Makromoleküls in intakter Form durch die Zelle.
Endocytose → vesiculärer Transport → frühes Endosom → vesiculärer Transport → Exocytose
apicales Membrandomän
Darmlumen
Besonderer Fall der receptorvermittelten
Endocytose:
das aufgenommene Molekül vermeidet den
Abbau in Lysosomen. rückkehrende
Transport-
Vesikeln
Beispiele:
• Aufnahme und Transport vom mütterlichen IgG
durch das Darmepithel des Neugeborenen.
frühes Endosom
• Aufnahme und Transport vom mütterlichen IgG
durch das Epithel der Plazenta in den Foetus.
• Aufnahme und Transport von IgA durch ein
Oberflächenepithel oder Drüsenepithel ins Lumen.
BindegewebsraumVesiculärer Transport
Transport von Substanzen zwischen membranbegrenzten Kompartimenten. Bedeutung.
Abschnüring
des Vesikels Transport Fusion des
Vesikels
Donorkompartiment Zielkompartiment
Aufgaben zu lösen:
•Selektivität: nur bestimmte Substanzen dürfen überführt
Substanzen überführt ins Zielkompartiment: ein werden, Sortierung
Stück Membran (mit Membranproteinen) und • Spezifität: darf nur mit der Membran des
gelöste Substanzen im Lumen des Vesikels. Zielkompartiments fusionieren
• molekulare Identität des Donorkompartiments darf nicht
geändert werden
1. Die Knospung des Vesikels in das Cytosol, Ausbildung der Hülle
Zwei Funktionen der Hülle: 1. Ansammlung von Proteinen zu transportien, und 2. Krümmung der Membran zur Bildung eines Vesikels
Die clathrin-Hülle: Baustein: triskelion, bildet korbähnilche Struktur um den Vesikel (Pentagons und Hexagons). Zwischen clathrin und Receptor
ist ein Adapterprotein die Verbindung.
Adapter
Transport-
Clathrin
Receptor
Abschnürung
triskelion mit Dynamin
clathrin „Korb” Die Knospung des Vesikels und das Einfangen der transportierenden
Moleküle mit der clathrin-Hülle Die COP-Hülle ist aus mehreren Proteinuntereinheiten (Coatomeren) aufgebaut. Die Ausbildung der Hülle beginnt mit der
Verankerung aktivierter G-Proteine (sar1 bzw. Arf) in die Membran, diese sammeln die verschiedenen Coatomere um sich.
Die Membran beginnt zu krümmen, die Coatomere mit ihren Bindungsstellen sortieren die Proteine zum Transport.
Die COPI- und COPII Hüllen bauen sich nach ähnlichen Prinzipien auf, aber binden verschiedene Proteine für den Transport
und transportieren in anderen Richtungen.
Untereinheiten der
COP II Hülle
2. Wanderung des Vesikels. Die Hülle löst sich ab, der Vesikel ist in
kleinerer Distanz mit einfacher Diffusion, bei längeren Strecken mit Hilfe von
Motorproteinen entlang cytoskeletaler Strukturen Richtung Zielkompartiment
bewegt.
residentes
ER Protein
3. Die Verankerung des Vesikels an der Membran des Zielkompartiments
und Fusion. Die Zielmembran wird mit einem molekularen Erkennungsmechanismus
erkannt. Rab Proteine (kleine G-Proteine) in der Vesikelmembran eines bestimmten
Kompartiments markieren die Identität des Vesikels und andere Proteine in der
Membran des Zielkompartiments (rab-Effektoren) sorgen für die spezifische Bindung. Bei
der Erkennung spielen weitere Proteine wichtige Rollen:
Die Fusion: zwei faserige integrale Proteine, eins in der Membran des Vesikels (v-
SNARE) und das andere in der Membran des Zielkompartiments (t-SNARE) binden sich
mit ihren fadenartigen Teilen aneinder und ziehen die zwei Membranen zusammen.
Wassermoleküle werden dadurch vom Zwischenraum der Membranen ausgeschlossen,
die zwei Lipiddoppelschichten lagern sich eng aneinander, die Lipide sind umorganisiert,
und die Trennwand bricht durch. Nach der Membranfusion sind v- und t-SNAREs
voneinander durch ein NSF Protein getrennt.
Regelung der Fusion zB. bei der Exocytose von synaptischen Vesikeln in Nervenendigungen:
durch Entfernung eines hemmenden Proteins.Membran Receptorabhängige
Nachschub mit Pinocytose mit Clathrin-
Exocytose Hülle-
Freisetzung des
Sekretes mit
konstitutiver
Exocytose
Zellmembran
Freisetzung des
Sekretes mit Receptor- und Membran-Recirkulation an
geregelter die Zellmembran
Exocytose
Frühes Endosom
Spätes Endosom
Sekretorisches
Granul
Trans-
Golgi
Netzwerk
Lxsosomale Enzyme transportierendes
Vesikel
Retrogader Transport mit COP I
Cis-Golgi Vesikel
Netzwerk
Tubulovesiculäre
Strukturen
Nackte Vesikel
Vesikelabschnürung
mit COPII Hülle
Rauhes ERKapiteln in Lehrbüchern:
Lüllman-Rauch: Histologie, Kapiteln 5.1, 5.2, 5,3
Lehrbuch der molekularen Zellbiologie, 3. Auflage, Kapiteln 7.2, 15.2, 15.3, 15.4, 15,5
Quellen der verwendeten Illustrationen:
Röhlich: Szövettan, 3. und 4. Auflagen, Semmelweis Verlag, Budapest
Alberts – Johnson – Lewis – Raff – Roberts – Walter: Molecular biology
of the cell. 5. Auflage, Garland Science
Röhlich: eigene Präparate und/oder Aufnahmen, bzw. Zeichnungen
Campbell – Reece: Biologie, Spektrum – Fischer
Robbins: Basic Pathology, 7. Auflage, Saunders, 2003Sie können auch lesen

















































