Einfluss eines Adenosinrezeptoragonisten und zytosolischem Renin auf den Reperfusionsschaden nach akuter Myokardischämie
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Aus der Klinik und Poliklinik für Innere Medizin B
(Direktor Prof. Dr. med. Stephan Felix)
der Medizinischen Fakultät der Ernst-Moritz-Arndt-Universität Greifswald
Einfluss eines Adenosinrezeptoragonisten und zytosolischem Renin
auf den Reperfusionsschaden nach akuter Myokardischämie
Inaugural - Dissertation
zur
Erlangung des akademischen
Grades
Doktor der Medizin
(Dr. med.)
der
Universitätsmedizin
der
Ernst-Moritz-Arndt-Universität
Greifswald
2013
vorgelegt von:
Katrin Zimmermann
geb. am: 13.02.1986
in: Weimar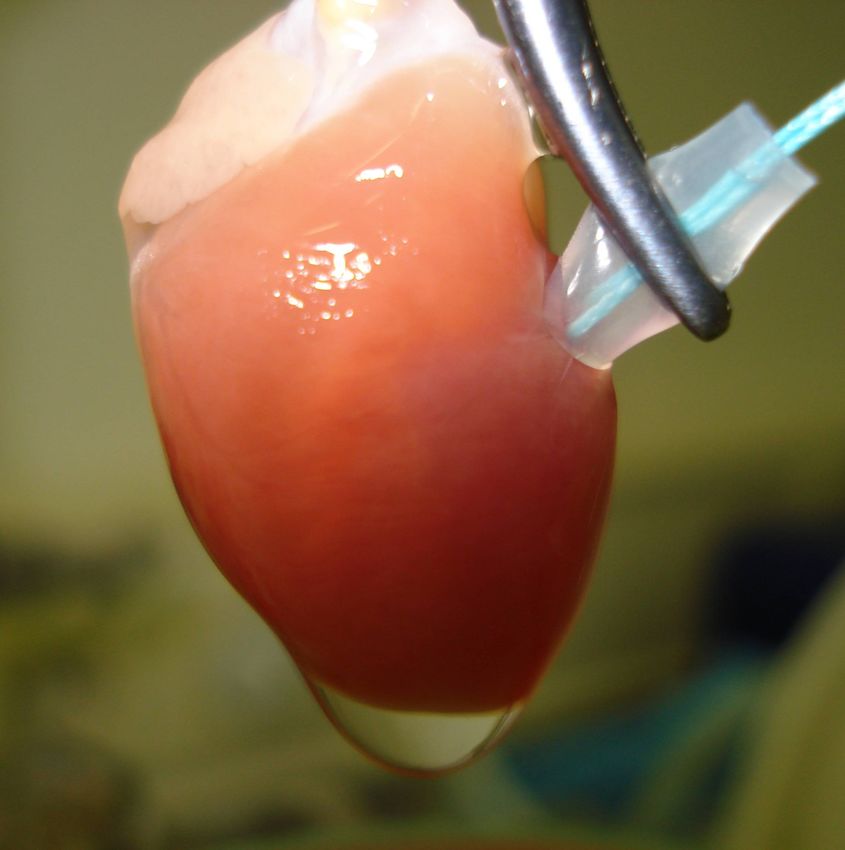
Dekan: Prof. Dr. med. dent. Reiner Biffar 1. Gutachter: Prof. Dr. St. Felix 2. Gutachter: Prof. Dr. K. Werdan Ort, Raum: Greifswald, Seminarraum der Klinik für Innere Medizin A Tag der Disputation: 05.05.2014

INHALTSVERZEICHNIS 3
INHALTSVERZEICHNIS
1. EINFÜHRUNG .................................................................................................................................5!
1.1. Der Myokardinfarkt....................................................................................................................5!
1.2. Die ischämische Prä- und Postkonditionierung .......................................................................12!
1.3. Adenosinrezeptoren (AR) während der Postkonditionierung ..................................................17!
1.4. Expression des intrazellulären zytosolischen Renins im Herzen .............................................18!
1.5. Aufgabenstellung .....................................................................................................................24!
2. MATERIAL UND METHODEN ..................................................................................................25!
2.1. Material ....................................................................................................................................25!
2.1.1. Chemikalien und Reagenzien ...........................................................................................25!
2.1.2. Substanzen und Inhibitoren ..............................................................................................26!
2.1.3. Antikörper ........................................................................................................................27!
2.1.4. Puffer und Lösungen ........................................................................................................27!
2.1.5. Verbrauchsmaterialien .....................................................................................................28!
2.1.6. Software und Geräte .........................................................................................................29!
2.1.7. Versuchstier......................................................................................................................29!
2.2. Methoden ..................................................................................................................................31!
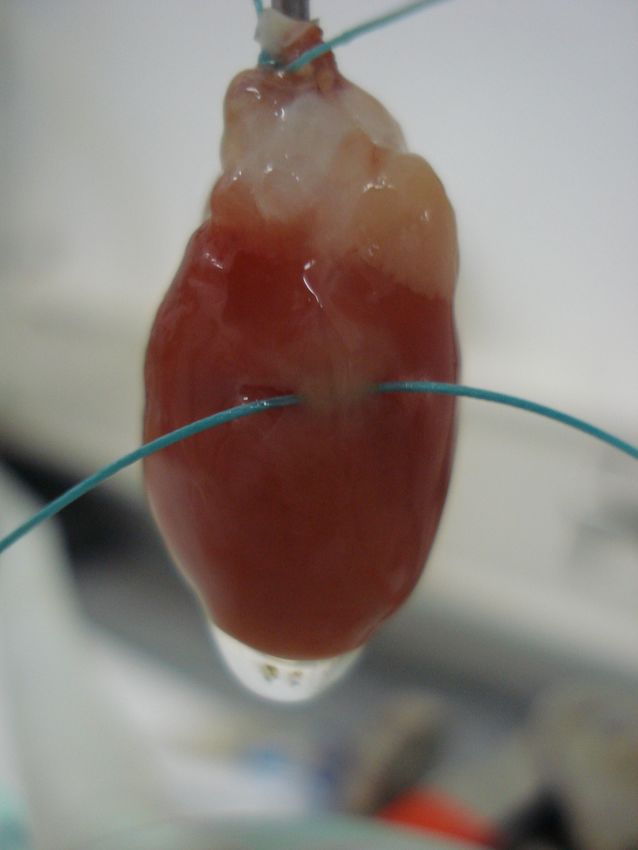
2.2.1. Ex vivo perfundiertes Herz ...............................................................................................31!
2.2.1.1. Operative Technik ....................................................................................................31!
2.2.1.2. Gruppeneinteilung ....................................................................................................33!
2.2.1.3. Infarktgrößenbestimmung ........................................................................................35!
2.2.1.4. Gewinnung transmuraler Biopsien ..........................................................................35!
2.2.2. Proteinanalytische Methoden ...........................................................................................36!
2.2.2.1. Isolation zytosolischer Proteine ...............................................................................36!
2.2.2.2. Bestimmung der Proteinkonzentration ....................................................................36!
2.2.2.3. Natriumdodecylsulfat-Polyacrylamidgelelektrophorese (SDS-Page)......................37!
2.2.2.4. Western Blot Analyse von Proteinen .......................................................................38!
2.2.2.4.1. Nass-Blot Verfahren ........................................................................................38!
2.2.2.4.2. Immundetektion ...............................................................................................39!
2.2.2.4.3. Semiquantitative Auswertung der Bandenintensität ........................................40!
2.2.3. Statistik .............................................................................................................................40!
3. ERGEBNISSE.................................................................................................................................41!
3.1. Pharmakokinetische Wirkung des A2bAR Agonisten Bay 60-6583 .........................................41!
3.1.1. Infarktgrößenbestimmung in isolierten Rattenherzen ......................................................41!
3.1.2. Hämodynamik ..................................................................................................................45!
3.1.2.1. Koronarfluss .............................................................................................................45!
3.1.2.2. Herzrate ....................................................................................................................49!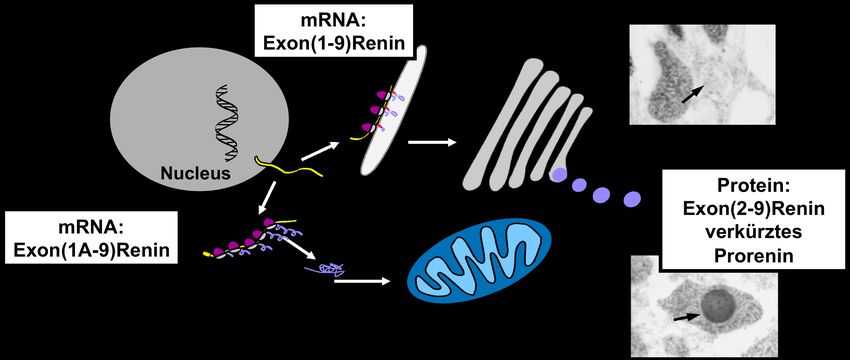
INHALTSVERZEICHNIS 4
3.1.2.3. Druck ........................................................................................................................52!
3.2. Wirkung des zytosolischen Exon(2-9)Renins ..........................................................................56!
3.2.1. Infarktgrößenbestimmung in isolierten Rattenherzen ......................................................56!
3.2.2. Hämodynamik ..................................................................................................................57!
3.2.2.1. Koronarfluss .............................................................................................................57!
3.2.2.2. Herzrate ....................................................................................................................59!
3.2.2.3. Druck ........................................................................................................................60!
3.2.3. Western Blot Analyse isolierter Rattenherzen .................................................................62!
4. DISKUSSION .................................................................................................................................65!
4.1. Methoden ..................................................................................................................................66!
4.2. Rolle der Adenosinrezeptoren während der Reperfusion ........................................................67!
4.3. Rolle des zytosolischen Exon(2-9)Renins in der Reperfusion .................................................71!
5. ZUSAMMENFASSUNG ................................................................................................................76!
6. LITERATURVERZEICHNIS ......................................................................................................78!
7. ANHANG ........................................................................................................................................88!
7.1. Zusammenfassung der erhobenen Daten ..................................................................................88!
7.2. Abbildungsverzeichnis .............................................................................................................91!
7.3. Tabellenverzeichnis ..................................................................................................................94!
7.4. Abkürzungsverzeichnis ............................................................................................................95!
8. EIDESSTATTLICHE ERKLÄRUNG .........................................................................................98!
9. DANKSAGUNG .............................................................................................................................99!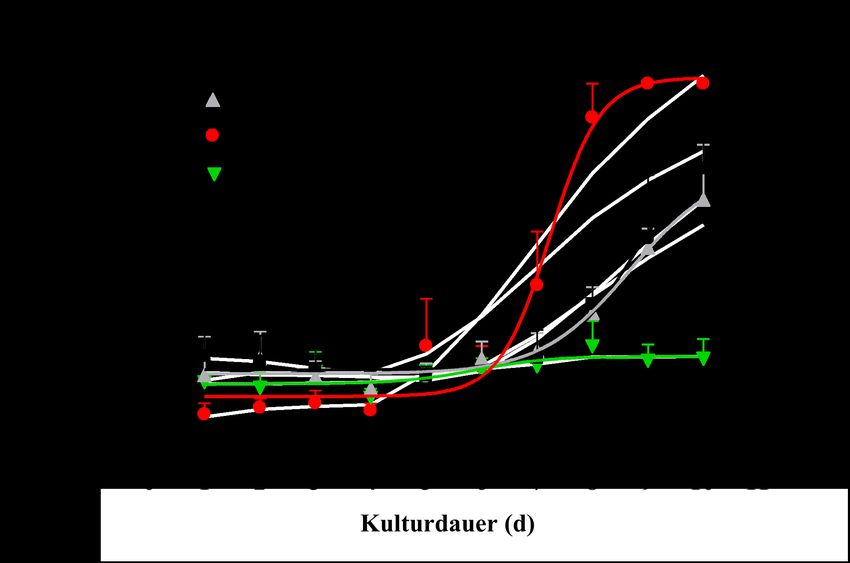
EINFÜHRUNG 5 1. Einführung 1.1. Der Myokardinfarkt Der Myokardinfarkt wird definiert als ischämische Myokardnekrose, die meist auf Grund einer koronaren Herzerkrankung mit hochgradiger Stenose einer Koronararterie entsteht. Es handelt sich um eine akute und lebensbedrohliche Erkrankung des Herzens. In Deutschland ereignen sich ca. 280.000 Herzinfarkte pro Jahr, an denen mehr als 40 % dieser Patienten versterben. Das Herz, ein muskulöses Hohlorgan, sorgt für die Förderung des Blutes durch den Körper. Die Herzkranzgefäße sichern die Sauerstoffversorgung des Herzmuskels. Durch Arteriosklerose der Gefäße und Thrombenbildung kann es zur Verengung oder Verschluss eines oder mehrerer Herzkranzgefäße kommen und somit zu einer Mangeldurchblutung des Herzmuskels führen. Die Hypoxie im Versorgungsgebiet der betroffenen Arterie führt zum Untergang von Myokardgewebe und ist später als bindegewebige Narbe erkennbar. Auslösende Faktoren für einen Herzinfarkt können plötzliche Kraftanstrengungen oder auch Stresssituationen mit stärkeren Blutdruckschwankungen sein. Das klinische Bild zeigt lang anhaltende Angina Pectoris Beschwerden, die durch Ruhe oder Nitroglyzerin kaum beeinflussbar sind. Jedoch 15-20 % der Infarkte gehen ohne Schmerzen einher, insbesondere bei Diabetikern und älteren Menschen sind sogenannte stumme Infarkte zu beobachten. Des Weiteren können Angst, Schwächegefühl und vegetative Begleitsymptomatik auf einen Herzinfarkt hindeuten. Eine entsprechende Diagnostik und sofortige Therapie sollte in diesen Fällen eingeleitet werden. Das wichtigste Therapieziel ist so schnell wie möglich eine Reperfusion des unterversorgten Gewebes zu erreichen. In internationalen multizentrischen Studien konnte gezeigt werden, wie wichtig die `goldene erste Stunde´ ist, da in dieser Zeit die Herzmuskeldurchblutung am ehesten wiederherzustellen ist [1]. Die frühzeitige Reperfusion kann sowohl mechanisch als percutaneous transluminal coronary angioplasty (PTCA) als auch medikamentös als konservative Therapie mit Aktivatoren der Fibrinolyse (Thrombolyse) erfolgen [2]. Bei der PTCA wird ein Katheter über einen peripheren arteriellen Zugang bis zur verschlossenen Koronararterie geführt und diese anschließend durch einen Ballon aufgedehnt. Die PTCA kann mit und ohne Stentimplantation und durch einen drug-eluting stent (DES) erfolgen. Die Thrombolyse erfolgt durch eine intravenöse Applikation fibrinolytisch wirksamer Substanzen wie zum Beispiel Streptokinase, Alteplase oder Reteplase. Da es nach einer erfolgreichen Lyse in 20-25 % zu Reokklusionen kommt, sollte im Anschluss an die Akutbehandlung eine Koronarangiographie erfolgen und gegebenenfalls eine weitere Therapie eingeleitet werden.
EINFÜHRUNG 6 Aus mehreren Studien hat sich gezeigt, dass die PTCA der Thrombolyse überlegen ist, da sie zu einer verringerten Mortalität und zu weniger Reinfarkten führt [3]. Des Weiteren lässt sich durch die medikamentöse Gabe von Clopidogrel, GPIIb/IIIa-Antagonisten sowie durch einen DES die Restenoserate verringern. Eine weitere Therapie ist die Bypass OP, wo ein aortokoronarer Venenbypass angelegt wird. Durch diesen chirurgischen Eingriff wird ein Umgehungskreislauf zwischen der Aorta und der Koronararterie geschaffen. Nach einer erfolgreichen Reperfusion sollten die Komplikationen nach einem Herzinfarkt wichtigstes Therapieziel sein. Hierbei werden Frühkomplikationen (48h) wie zum Beispiel Herzwandaneurysma, arterielle Embolien und Postmyokardinfarktsyndrom unterschieden. Der gefährlichste Zeitraum nach einem Herzinfarkt stellen die ersten 48 Stunden dar, 40 % der Patienten überleben den ersten Postinfarkttag nicht. Die Langzeitprognose ist abhängig von der linksventrikulären Funktionseinschränkung, von Ischämiezeichen (Angina Pectoris, Veränderungen im Belastungs-EKG, Myokardperfusions- szintigraphie), von ventrikulären Herzrhythmusstörungen, von der Anzahl der betroffenen Gefäße und letztendlich von dem Fortbestehen der Risikofaktoren. Die endgültige Größe eines Herzinfarktes ist von verschiedenen Faktoren abhängig. So konnte zum einen gezeigt werden, dass die Größe und Lokalisation von perfundierten Gewebe distal des Koronarverschlusses eine bedeutende Rolle spielt [4, 5]. Des Weiteren ist der durch Kollateralarterien zurückbleibende Blutfluss, die Temperatur und die hämodynamische Situation während der Ischämie wichtig [6, 7]. Dies verdeutlicht, dass die frühzeitige Reperfusion des ischämischen Gewebes die einzige Möglichkeit ist eine Myokardnekrose zu verhindern oder ihre Ausdehnung zu verringern. Es konnte jedoch gezeigt werden, dass die sofortige Wiederversorgung des ischämischen Gewebes mit ausreichendem Blutfluss anderseits zu verschiedenen Dysfunktionen geführt hat. Diese werden im Folgenden erläutert. Zum einen wird vitales Myokardgewebe, das nach einer erfolgreichen Reperfusion anhaltend ventrikuläre Dysfunktionen aufweist als Stunned myocardium bezeichnet. Hier konnte gezeigt werden, dass sich das Gewebe jedoch innerhalb von Stunden bis Wochen vollständig erholt [8]. Auch werden Arrhythmien bei der sofortigen Wiederherstellung des Blutflusses festgestellt. Des Weiteren kann ein vaskulärer Reperfusionsschaden auftreten, vor allem nach einer schweren und lang anhaltenden Ischämie. Dieser wird als ‘‘no-reflow Phänomen’’ beschrieben und besagt, dass nach Eröffnung des Koronargefäßes keine ausreichende Reperfusion des ischämischen Gebietes stattfindet. Dieser vaskuläre Reperfusionsschaden ist verbunden mit ausgeprägten Schäden in der Mikrovaskulatur [9]. Und zuletzt zählt der tödliche Reperfusionsschaden zu den
EINFÜHRUNG 7 Dysfunktionen in der Reperfusion. Hierbei kommt es zum Tod von Kardiomyozyten, die während der Ischämie nur reversibel verletzt wurden und durch die Wiederherstellung des Blutflusses endgültig geschädigt sind [10]. Dieser Vorgang wird als Ischämie/Reperfusionsschaden bezeichnet und wurde erstmals im Jahre 1960 beschrieben [11]. Der Endpunkt des tödlichen Reperfusionsschadens ist die enorme Schädigung von Zytoskelettelementen nach Hyperkontraktur. Dies zeigt zum einen, dass die Reperfusion die einzige Möglichkeit ist ischämisches Gewebe zu retten und zum anderen wird deutlich, dass die notwendige Reperfusion zu schweren und irreversiblen Schäden führt [12, 13]. Im Folgenden werden die Vorgänge geschildert die während der Ischämie/Reperfusion im geschädigten Gewebe ablaufen und zum tödlichen Reperfusionsschaden führen. Während einer Ischämie kommt es zur Unterversorgung der Kardiomyozyten mit Sauerstoff und Nährstoffen. Die für die Zelle notwendige Adenosintriphosphat (ATP) Produktion kann nicht mehr aufrechterhalten werden. Aufgrund des Sauerstoffmangels muss die Zelle auf einen anaeroben Stoffwechsel umstellen. Die Folge ist die Entwicklung einer Azidose durch die Akkumulation von Wasserstoffionen (H+), anorganischen Phosphaten und Laktat. Durch die enorme Anreicherung von H+-Ionen wird der Na+/H+-Austauscher aktiviert und es kommt zu einem Einstrom von Na+-Ionen in die Zelle [14]. Durch den ATP Verlust ist der Na+/K+- Austauscher inaktiv, was den initialen Einstrom von Na+-Ionen zusätzlich erhöht. Ebenso kommt es durch den Verlust von ATP zur Akkumulation von Ca2+-Ionen, da diese nicht mehr ins Sarkoplasmatische Retikulum (SR) aufgenommen werden können [15]. Der Na+/Ca2+- Austauscher ist aufgrund des ATP Mangels in seinem reverse mode aktiviert, daher wird Ca2+ aufgenommen und Na+ abgegeben. Es kommt somit zu einer zusätzlichen Überladung von zytosolischen Ca2+-Ionen in die Zelle. In dieser Situation herrscht in der Zelle eine enorme Anreicherung sowohl von Na+- als auch von Ca2+-Ionen [16, 17]. Die Folge ist ein massives Anschwellen der Kardiomyozyten, welches zur Zerstörung sarkolemmaler Strukturen führt [18]. Bei Wiederherstellung des Blutflusses im ischämischen Myokard erfolgt schließlich die erneute Versorgung der Mitochondrien mit molekularem Sauerstoff und Nährstoffen was zur Aktivierung der Elektronentransportkette führt. Es kommt zu Bildung von ATP. In der frühen Phase der Reperfusion ist der Na+ Gradient noch reduziert und so befindet sich der Na+/Ca2+- Austauscher stets in seinem reverse mode, somit wird weiter Ca2+ in die Zelle aufgenommen, siehe Abb. 1.1. Das SR wird durch das ATP aktiviert und nimmt nun Ca2+ in die intrazellulären Speicher auf, da jedoch viel Ca2+ akkumuliert wird zusätzlich wieder welches frei gelassen. Diese enorme Anreicherung von zusätzlichen Ca2+-Ionen kann zu Oszillationen führen [19]. Weiterhin aktiviert ATP die Myofibrillen und in Anwesenheit von hohen
EINFÜHRUNG 8 zytosolischen Ca2+ Konzentrationen treten unkontrollierte Kontraktionen auf, siehe Abb. 1.1. Letztendlich kommt es zu einer irreversiblen Verkürzung von Zytoskelettelementen in den Kardiomyozyten und führt zum Zelltod nach Hyperkontraktur. Die Hyperkontraktur ist von gewissen Zell-Zell-Verbindungen abhängig. Es konnte gezeigt werden, dass die gap junctions zu einer Ausweitung des Schadens führen [20, 21]. Abb. 1.1: Ionentransportvorgänge während der Ischämie und Reperfusion. links: Während der Ischämie und zu Beginn der Reperfusion bleibt der Na+/Ca2+-Austauscher in seinem reverse mode. Enorme Ca2+ Konzentrationen entstehen. rechts: Die hohen Ca2+ Konzentrationen und das zugeführte ATP führen zu unkontrollierten Kontraktionen (Hyperkontraktur) [19]. Durch das wieder gewonnene ATP kann der Na+/K+-Austauscher aktiviert werden, damit sich ein Na+ Gradient einstellt. Wenn dies geschehen ist, nimmt der Na+/Ca2+-Austauscher seine normale Funktion ein und entfernt das überschüssige Ca2+ gegen Na+ aus der Zelle. Somit kann sich ein normaler Ca2+ Haushalt wieder einstellen [19]. Während der Ischämie sinkt der ph-Wert sowohl intrazellulär als auch extrazellulär stetig ab. Wird das Gewebe wieder reperfundiert normalisiert sich der ph-Wert im Interstitium und ein Gradient zwischen hohen H+-Ionen intrazellulär und normalisierten H+-Ionen extrazellulär entsteht. Nun kann es zur Aktivierung sowohl des Na+/H+-Austauschers als auch des Na+/HCO3−-Symporters kommen die viele H+-Ionen aus der Zelle entfernen, siehe Abb. 1.2. Durch beide Transporter werden zusätzlich Na+-Ionen ins Zytosol aufgenommen. Durch Aktivierung der Na+/K+-Pumpe kann dem entgegengewirkt werden. Ist die Na+/K+-Pumpe nicht in der Lage den massiven Na+ Einstrom entgegenzusteuern, können durch Aktivierung des Na+/Ca2+-Austauschers sekundär die Ca2+ Konzentrationen ansteigen. Die schnelle Mobilisierung der H+-Ionen und die zusätzliche Aufnahme von Ca2+ führt zur Hyperkontraktur [10]. Solange hohe intrazelluläre H+-Ionenkonzentrationen herrschen, bleiben die Myofibrillen inaktiv und eine Hyperkontraktur wird verhindert.
EINFÜHRUNG 9 Abb. 1.2: Vorgänge bei Normalisierung des Gewebe ph-Wertes. Hohe intrazelluläre H+ Konzentrationen verhindern eine Hyperkontraktur. Schnelle Normalisierung des ph-Wertes in der frühen Reperfusion führt zur Hyperkontraktur [10]. Zur Entfernung von überschüssigen Na+-Ionen dient die Na+/K+-Pumpe die sowohl abhängig ist vom ATP als auch von ihrer strukturellen Stabilität welche durch die Proteine Ankyrin und Fodrin aufrechterhalten wird. Zu Beginn der Reperfusion, wie in einigen Studien demonstriert werden konnte, findet durch Calpain eine Proteolyse dieser beiden Proteine statt. Calpain ist eine nichtlysosomale Ca2+ abhängige Protease, die Ankyrin und Fodrin als Substrate hat und inaktiv ist bei niedrigen ph-Wert während der Ischämie [22]. In der Ischämie erhöht sich ebenso die intra- und extrazelluläre Osmolarität. Mit dem Beginn der Reperfusion normalisiert sich die Osmolarität im Interstitium. Somit bildet sich hier ein Gradient zwischen den intra- und extrazellulären Raum. Die Folge ist ein Wassereinstrom nach intrazellulär und das Anschwellen der Zellen, was letztendlich zur sarkolemmalen Zerreißung führen kann, wie in Abb. 1.3 dargestellt [23, 24]. Abb. 1.3: Vorgänge bei Normalisierung der Gewebeosmolarität. Die Reperfusion führt zur Bildung eines Gradienten die Folge ist der intrazelluläre Wassereinstrom, Instabilität und Ruptur der Zelle [10].
EINFÜHRUNG 10 Des Weiteren konnte gezeigt werden, dass erhöhte zytosolische Ca2+ Konzentrationen, eine schnelle ph-Wert Normalisierung und die Bildung von reaktiver Sauerstoff -und Stickstoffspezies (ROS/RNS) zu Beginn der Reperfusion zur Bildung der mitochondrial permeability transition pore (mPTP) führen [25, 26]. Diese Pore ist ein spannungsabhängiger Kanal der inneren und äußeren Mitochondrienmembran, der durch Cyclophilin D (CycD) reguliert wird. Er hat einen Durchmesser von 3 nm und erlaubt sowohl eine schnelle Einstellung des Gleichgewichtes für Ionen und Wasser als auch eine Passage für Moleküle bis zu einer Größe von 1,5 kDa. Unter physiologischen Bedingungen ist die innere Mitochondrienmembran undurchlässig bis auf einige Ionen und Metabolite für spezielle Transportvorgänge. Dies ist notwendig, um den ph-Gradienten und das mitochondriale Membranpotential (Δψm) für die oxidative Phosphorylierung aufrechtzuerhalten. In vielen Studien konnte gezeigt werden, dass es während der Ischämie/Reperfusion zur Bildung der mPTP kommt. Die Folge ist ein Zusammenbruch sowohl des Protonengradienten als auch des Δψm. Die oxidative Phosphorylierung wird entkoppelt [27]. Die Membran wird durchlässig und es strömt Wasser in die Mitochondrien ein. Da die äußere Membran im Vergleich zur inneren Membran durch christae-remodelling nicht dehnbar ist, kommt es zum Einreißen der Membran. Zusätzlich werden Cytochrom C und weitere proapoptotische Substanzen freigesetzt [28]. Es konnte gezeigt werden, dass Inhibitoren der mPTP sowohl in isolierten Kardiomyozyten gegen den durch Hypoxie und Reoxygenierung vermittelten Zelltod in der Reperfusion geschützt haben als auch zu einer Reduktion der Infarktgröße im ex vivo Modell der Ischämie/Reperfusion führen [29-31]. Weiterhin konnte dargestellt werden, dass CycD arme Mäuse verglichen zu wild-typ Mäusen einen Schutz nach Ischämie/Reperfusion aufwiesen [32, 33]. Ein Öffnen der mPTP führt zu erhöhten Ca2+ Konzentrationen und führt zur Hyperkontraktur [34]. Die Reperfusion des ischämischen Gewebes führt gleichzeitig zu einer Schädigung, zum tödlichen Reperfusionsschaden. Entscheidende Einflüsse sind die Kontraktionen, die hohen Ca2+ Oszillationen, die schnelle Normalisierung der intrazellulären Azidose, das Anschwellen der Zellen und eine Ausweitung der Nekrose. Letztendlich kommt es in den einzelnen Kardiomyozyten zu einer Zerstörung des Sarkolemms durch die Hyperkontraktur, als bisher entscheidenden Pathomechanismus. Die Abb. 1.4 zeigt eine Zusammenfassung der Faktoren, die zum tödlichen Reperfusionsschaden führen.
EINFÜHRUNG 11 Abb. 1.4: Übersicht über die Faktoren, die zum tödlichen Reperfusionsschaden führen. + aktivierende Einflüsse, - hemmende Einflüsse [10].
EINFÜHRUNG 12 1.2. Die ischämische Prä- und Postkonditionierung Ein wichtiges Ziel in der Therapie des Myokardinfarktes ist das Ausmaß der Infarktgröße zu reduzieren. Es konnte gezeigt werden, dass sich pharmakologische und interventionelle Strategien in Verbindung mit der notwendigen Reperfusion positiv auf die Reduktion des Infarktes auswirken. Der stärkste Schutz des Herzens vor einem Myokardinfarkt sekundär zur Ischämie/Reperfusion stellt die ischämische Präkonditionierung (IPC) dar, welche erstmals 1986 von Murry et al. beschrieben wurde [35]. An isolierten Hundeherzen konnte gezeigt werden, dass vier Zyklen von jeweils fünf Minuten Koronarverschluss und fünf Minuten Reperfusion vor der 40-minütigen Indexischämie zur Reduktion der Infarktgröße um 75 % im Vergleich zu den Kontrolltieren geführt haben, siehe Abb. 1.5. Abb. 1.5: Ischämische Präkonditionierung. Kurze Intervalle von Ischämie und Reperfusion vor der Indexischämie führen zu einer signifikanten Reduktion der Infarktgröße. Des Weiteren konnte gezeigt werden, dass die IPC nur in Verbindung mit einer rechtzeitigen Reperfusion funktionsfähig ist. Sowohl eine verlängerte Indexischämie von drei Stunden als auch eine Indexischämie, die über zwei Stunden nach der IPC erfolgt, kann den schützenden Effekt der IPC aufheben. So ist der zeitliche Rahmen einer erfolgreichen IPC sehr eng [36]. Eine erfolgreiche IPC konnte sowohl in verschiedenen Spezies, als auch in verschiedenen Organen präsentiert werden und in retrospektiven Analysen wurde gezeigt, dass dieses Phänomen auch auf dem Menschen anwendbar ist [37-39]. Im Folgenden werden nun die Signalwege der IPC genauer erläutert. Hierbei kann unterschieden werden zwischen biochemischen Signalen und rezeptor-abhängigen und rezeptor-unabhängigen Signalen. Es konnte gezeigt werden, dass es während der kurzen Ischämieepisoden zur Freisetzung von endogenen Mediatoren kommt die ihre Wirkung über G-Proteine vermitteln. Die endogenen Mediatoren sind Adenosin [40], Bradykinin [41], Opioide [42] und Prostaglandine [43]. Aber auch die exogene Gabe von Liganden vor der Indexischämie führt zu einem Myokardschutz. Dabei hat sich gezeigt, dass nicht nur die bereits erwähnten Substanzen Adenosin [44], Bradykinin [45] und Opioide [46, 47], sondern auch weitere pharmakologische Substanzen wie Angiotensin [48], Endothelin [49] und α1- adrenerge Stimulantien [50] kardioprotektiv wirken. Abseits von spezifischen Rezeptoren
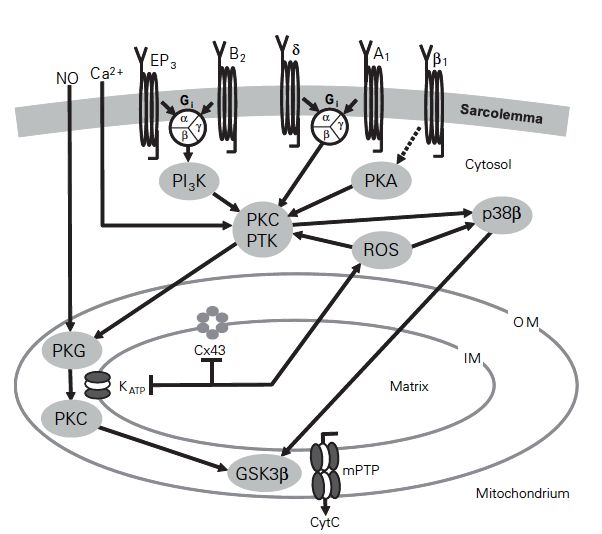
EINFÜHRUNG 13 kann die IPC ebenso durch die exogenen Gabe von ROS, Stickstoffmonoxid (NO) und Ca2+ ausgelöst werden [36, 51]. Die Abb. 1.6 gibt einen Überblick über die Liganden und Signalkaskaden der IPC. Abb. 1.6: Liganden und Signalkaskaden der IPC. A1: Adenosin-A1-Rezeptor, ATP: Adenosintriphosphat, B2: Bradykinin-B2-Rezeptor, β1: β1-Rezeptor, Cx43: Connexin 43, CytC: Cytochrom C, δ: δ-Opioid-Rezeptor, EP3: Prostaglandin-E-Rezeptor3, Gi: inhibitorisches G- Protein, GSK3β: Glykogen-Synthase-Kinase-3β-Isoform, IM: innere Mitochondrienmembran, KATP: ATP- abhängige Kaliumkanäle, mPTP: mitochondriale permeability transition pore, NO: Stickoxid, OM: äußere Mitochondrienmembran, p38β: p38-Mitogen-aktivierte-Kinase-β-Isoform, PI3K: Phosphatidylinositol-3-Kinase, PKA: Proteinkinase A, PKC: Proteinkinase C, PKG: Proteinkinase G, PTK: Protein-Tyrosinkinase, ROS: freie Sauerstoffradikale [36]. Adenosin kann die Rezeptortypen A1 (Gi/o), A2a (Gs), A2b (Gs/q) und A3 (Gi/o) aktivieren [52]. Opioide sind über Gi an µ-, κ- und δ-Rezeptoren gekoppelt, jedoch exprimieren adulte ventrikuläre Myozyten nur κ- und δ- Rezeptoren [53]. Für Bradykinin gibt es zwei Rezeptoren die in Kardiomyozyten exprimiert werden. Zum einen den durch Stress induzierter B1- (Gq/i) und den konstitutiv exprimierten B2-Rezeptor (Gq/i) [54, 55]. Die rezeptorabhängigen Mediatoren Adenosin, Opioid und Bradykinin sind an inhibitorische G- Proteine gekoppelt, wie zum Beispiel Phospholipase C (PLC). Die PLC katalysiert die Umwandlung von Phosphatidylinositol-4,5-bisphosphat (PIP2) zu Inositoltrisphosphat (IP3) und Diacylglycerol (DAG), was zur Aktivierung der Proteinkinase C (PKC) führt. Alle drei Mediatoren haben unterschiedliche Signalkaskaden, welche alle in einer Aktivierung der PKC enden [56]. Des Weiteren konnte gezeigt werden, dass die Signalkaskaden über den Bradykinin- und Opioidrezeptor sehr vielfältig sind und über eine Aktivierung der
EINFÜHRUNG 14 Phosphatidylinositol-3-kinase (PI3K) erfolgen [57]. Diese aktiviert die Proteinkinase B (Akt) und daraufhin wird die endotheliale NO-Synthase (eNOS) stimuliert und NO gebildet [58, 59]. Schließlich kommt es zur Aktivierung der Guanylatzyklase (GC) mit Bildung von cyclischen Guanosinmonophosphat (cGMP). Es folgt die Stimulierung der Proteinkinase G (PKG), die nachfolgend eine Öffnung der mitochondrialen ATP abhängigen Kaliumkanäle auslöst [60, 61]. Dies führt zum Einstrom von K+-Ionen und das mitochondriale Membranpotential wird reduziert. Die Mitochondrien schwellen an und es wird vermehrt ROS gebildet, was letztendlich zur Aktivierung der PKC führt [56]. Die Signalkaskade von Adenosin führt zur direkten Aktivierung der PKC durch das G-Protein [42, 62]. Exogenes NO induziert die IPC über Aktivierung der GC, welche Guanosintriphosphat (GTP) zu cGMP umwandelt und ebenfalls die PKG aktiviert [60]. Des Weiteren hat sich gezeigt, dass ß- Adrenorezeptoren die Proteinkinase A (PKA) aktivieren und eine IPC induzieren [63]. In einer Studie von Vahlhaus et al. konnte gezeigt werden, dass die IPC in Schweinen nur verhindert werden konnte bei einer Blockade sowohl der PKC als auch der Protein- Tyrosinkinase (PTK), in Kaninchen reichte eine Blockade entweder der PKC oder der PTK aus, um den schützenden Effekt durch die IPC aufzuheben [64]. Eine Beteiligung der mitogen-aktivierenden Proteinkinasen p38 und extracellular-signal regulated kinase (Erk 1/2) konnte bei der IPC dargestellt werden [65, 66]. Letztendlich scheint die Aktivierung der PKC der entscheidende Schritt in der IPC zu sein, die darauffolgende Aktivierung der Akt und Erk 1/2 in der frühen Reperfusion konnte bei Hausenloy et al. als Resultat der IPC gezeigt werden [67]. Das Zusammenspiel von Akt und Erk 1/2 führt zur Aktivierung der p70S6 Kinase (p70S6K), welche schließlich die Glykogen-Synthase-Kinase-3β-Isoform (GSK3β) phosphoryliert und damit die Schwelle für das Öffnen der mPTP erhöht [68-70]. Das Öffnen dieser Pore führt zum Einstrom von Ionen und Wasser ins Mitochondrium mit konsekutivem Anschwellen und letztlich Zerstörung des notwendigen Δψm. Die primäre Funktion, nämlich die Bereitstellung von ATP durch die oxidative Phosphorylierung kommt zum Erliegen. Durch Zerstörung der äußeren Membran werden Cytochrom C und weitere proapoptotische Substanzen frei gelassen [71]. Das Öffnen der Pore wird durch die IPC verhindert. Übermäßiges Anschwellen der Zellen führt zur Zerstörung des Sarkolemms und zur Nekrose, auch das konnte durch die IPC unterbunden werden [72]. Letztendlich führt eine bessere Aufnahme von Ca2+ in das SR zu Beginn der Reperfusion zu weniger unkontrollierten Kontraktionen der Myofibrillen. Es konnte gezeigt werden, dass durch die IPC und hierbei durch die Aktivierung der PKG die sarkoplasmatische Ca2+ATPase (SERCA) aktiviert wird, was zu einem niedrigeren Peak an Ca2+ führt [73].
EINFÜHRUNG 15 Zusammenfassend zeigt sich, dass die Signalkaskade der IPC ein großer Komplex aus den oben erwähnten Molekülen und Mechanismen ist. Durch die Ischämie/Reperfusion werden endogene Mediatoren frei gelassen, die über verschiedene Kinasen ein intrazelluläres Signal auslösen, welches ebenso durch exogen hinzugefügte Liganden induziert werden kann. Es zeigt sich, dass es ein Zusammenspiel zwischen den Mitochondrien, dem Sarkolemm und dem SR geben muss. Bis jetzt konnte nicht geklärt werden, was das eigentliche Gedächtnis der IPC darstellt. Vor ein paar Jahren wurde ein weiterer Myokardschutz, die ischämische Postkonditionierung (IPost), von Zhao et al. beschrieben. In Hundeherzen konnte eine deutliche Reduktion der Infarktgröße gezeigt werden, indem drei Zyklen von jeweils 30 Sekunden Koronarverschluss und 30 Sekunden Reperfusion nach einer 60-minütigen Indexischämie durchgeführt wurden, siehe Abb. 1.7 [74]. Der Vorteil der IPost liegt nahe, denn sie führt zu einem Myokardschutz der nach der Ischämie durchgeführt wird und Patienten kommen erst während einer Ischämie ins Krankenhaus. Die IPost konnte ebenfalls in mehreren Studien in den verschiedensten Spezies gezeigt werden [75, 76]. Abb. 1.7: Ischämische Postkonditionierung. Kurze Intervalle von Ischämie/Reperfusion nach der Indexischämie führen zur signifikanten Reduktion der Infarktgröße. Der zeitliche Rahmen für einen ausreichenden Schutz der IPost ist wiederum eng bemessen. So konnte gezeigt werden, dass sowohl eine zeitliche Verzögerung zwischen Reperfusionsbeginn und IPost als auch eine ungeeignete Anzahl der Zyklen von Koronarverschluss und Reperfusion den schützenden Effekt aufheben können [77, 78]. Des Weiteren wurde gezeigt, dass die Zeit der Indexischämie eine Rolle spielt. In Rattenherzen konnte demonstriert werden, dass sowohl eine Indexischämie von 120 Minuten den Myokardschutz der IPost vollständig aufhebt als auch eine Indexischämie von 15 und 30 Minuten mit nachfolgender IPost die Infarktgröße sogar vergrößert [79]. Im Folgenden sollen die Signalwege der IPost genauer erläutert werden. Es konnten einige Elemente demonstriert werden, die ebenso in der IPC eine Rolle spielen. So hat sich gezeigt, dass die endogenen Mediatoren Adenosin [80], Opioide [81] und Bradykinin [82] kardioprotektiv wirken. Die IPost lässt sich ebenfalls nicht nur mechanisch sondern auch
EINFÜHRUNG 16 durch exogen zugeführte pharmakologische Substanzen in der Reperfusion auslösen. Es wurden verschiedene Substanzen getestet, die eine Reduktion der Infarktgröße gezeigt haben, wie zum Beispiel die Adenosinrezeptoragonisten AMP579 [83] und 5’-(N- ethylcarboxamido)adenosine (NECA) [84], GSK-3β-Inhibitoren [81] und der δ-Opioidrezeptor Agonist [D-Ala2,D-Leu5]enkephalin acetat (DADLE) [85]. Des Weiteren konnte eine Beteiligung des mitochondrialen ATP abhängigen Kaliumkanals, der Produktion von ROS und der PKC gezeigt werden [86]. Hausenloy & Yellon demonstrierten, dass spezifische Kinasen, die reperfusion injury salvage kinases (RISKs) am Signalweg der IPost beteiligt sind, zu denen gehören PI3K, Akt, Erk 1/2, p70S6K und eNOS [87]. Auch Tsang et al. konnten die Beteiligung von PI3K, Akt und eNOS während der Reperfusion nachweisen und Darling et al. stellten ebenso die Aktivierung von Erk 1/2 dar [76, 88]. Die Aktivierung von Akt und Erk 1/2, wie oben beschrieben, wurde auch während der IPC beobachtet. Auch zeigt sich hier, dass durch Aktivierung der RISKs die GSK3β phosphoryliert und inhibiert wird und so das Öffnen der mPTP verhindert wird [68, 69]. Wiederum konnte in Schweineherzen gezeigt werden, dass die IPost (drei Zyklen von jeweils 30 Sekunden Reperfusion und 30 Sekunden Ischämie) die Akt und Erk 1/2 phosphoryliert aber in diesem Experiment zu keinem Myokardschutz geführt hat [89]. Dies zeigt, dass die IPC und die IPost gemeinsame Signalwege aufweisen, welche jedoch nicht identisch sind. Im Vergleich zur IPC zeigt sich ein deutlicher Nutzen der IPost in der Durchführbarkeit bei der interventionellen Therapie, PTCA [12, 90]. So konnte in einer Studie mit 30 Patienten, die eine PTCA erhielten, gezeigt werden, dass eine IPost von vier Zyklen von jeweils einer Minute Verschluss (durch einen aufgeblasenen Ballon) und einer Minute Reperfusion zu einer Reduktion der Kreatinkinase geführt hat [91]. Zusammenfassend wird deutlich, dass die Signalkaskaden der IPC und IPost sowohl gewisse Ähnlichkeiten aufweisen als auch zu einem potenten endogenen Myokardschutz führen. Das Zeitintervall für einen potenten Myokardschutz ist bei beiden Phänomenen begrenzt, beide sind auch im Menschen anwendbar und verhindern oder begrenzen den Ischämie/Reperfusionsschaden.
EINFÜHRUNG 17 1.3. Adenosinrezeptoren (AR) während der Postkonditionierung Adenosin ist ein Purin-Nukleosid und kommt in allen Geweben und Körperflüssigkeiten vor. Es besteht aus der Nukleinbase Adenin und dem Zucker β-D-Ribose und ist Bestandteil der energiereichen Verbindungen ATP, Adenosindiphosphat (ADP) und Adenosinmonophoshat (AMP). Unter physiologischen Bedingungen liegt die extrazelluläre Konzentration von Adenosin bei 30-300 nM, kann aber bei Ischämie und somit unter Sauerstoffverlust auf 10µM oder höher ansteigen [92]. Kommt es zu hohen extrazellulären Konzentrationen wird Adenosin in die Zellen transportiert und durch die Adenosinkinase zu AMP phosphoryliert oder durch die Adenosindesaminase zu Inosin degradiert [93]. Im Herzen wird sowohl durch Hypoxie als auch durch einen niedrigen pH-Wert die Adenosinkinase gehemmt. Die Folge ist eine verminderte Aufnahme von Adenosin in die Zellen und somit eine erhöhte extrazelluläre Adenosinkonzentration während der Ischämie [94]. Dies hat viele Auswirkungen am Herzen, so führt es zu negativ dromotropen und negativ chronotropen Effekten, vasodilatierend an den Koronargefäßen und antiadrenerg [95]. Adenosin kann als Hauptagonist an die vier Subtypen A1 (Gi/o), A2a (Gs), A2b (Gs/q) und A3 (Gi/o) der AR binden [96, 97]. Sowohl in der IPC als auch in der IPost konnte die bedeutende Rolle von Adenosin durch Verwendung von Adenosinrezeptoragonisten gezeigt werden [83, 95, 98]. Sowohl der selektive A1AR Antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) als auch der A2aAR Antagonist 8-(13-chlorostyryl)caffeine (CSC) konnten den schützenden Effekt der IPost im Herzen nicht aufheben [99]. Jedoch der nichtselektive Adenosinrezeptorantagonist 8-p-(sulfophenyl)theophylline (SPT) und der selektive A2bAR Antagonist 8-[4-[((4-Cyanophenyl)carbamoylmethyl)oxy]phenyl]-1,3-di(n-propyl)xanthine hydrate (MRS1754) konnten die Schutzwirkung der IPost auflösen [80]. Weiterhin zeigten Philipp et al. eine erfolgreiche Postkonditionierung mit den A1/A2AR Agonisten NECA und konnten durch eine zeitgleiche Gabe des A2bAR Antagonisten MRS1754 demonstrieren, dass die pharmakologische Postkonditionierung inhibiert werden konnte. Somit wird die bedeutende Rolle des A2bAR beim Myokardschutz deutlich. Der niedrig affine A2bAR ist an G-Proteine gekoppelt, was zur Konzentrationserhöhung von zytosolischen cAMP, Inositoltriphosphat und Ca2+ führt [100]. Auch der seit kurzem verfügbare A2bAR Agonist 2- [6-amino-3,5-dicyano-4-(4-hydroxyphenyl)pyridin-2-ylsulfanyl] acetamide Bay 60-6583 schützt das ischämische Kaninchenherz [101]. Schließlich konnte in einer kürzlich veröffentlichen Studie gezeigt werden, dass eine maximale Schutzwirkung erreicht wird, wenn sowohl der A2bAR als auch der A2aAR gleichzeitig während der Reperfusion aktiviert werden [102].
EINFÜHRUNG 18 1.4. Expression des intrazellulären zytosolischen Renins im Herzen Die pharmakologische Postkonditionierung ist ein wichtiger Weg um Myokardschäden nach einem Infarkt zu verhindern. Weiterhin gibt es andere Ansatzpunkte, die versuchen den nekrotischen Zelltod im Herzen zu hemmen. Nekrotische Zellen locken Neutrophile und Monozyten in das geschädigte Myokard und führen schließlich zu Zelluntergängen, Fibrosen und kardialen Dysfunktionen [103, 104]. Das Renin-Angiotensin-System (RAS) spielt nicht nur eine wichtige Rolle in der Blutdruckregulation und der Regulation des Wassers- und Elektrolythaushaltes, sondern auch für die Zellproliferationen, Apoptosen, Entzündungs- und Wachstumsprozesse. [105-107]. Im Herzen führt dieses System sowohl zur Hypertrophie als auch zur Entzündung und Fibrose [108, 109]. Es konnte gezeigt werden, dass die Mortalitätsrate bei Herzkraftversagen durch Angiotensin-converting Enzym- (ACE) Inhibitoren gesenkt werden konnte [110]. Das RAS existiert in zwei verschiedenen Formen, dem zirkulierenden RAS und dem lokalen RAS in verschiedenen Organen mit unterschiedlichen Eigenschaften dieser beiden Systeme [111]. Beide Systeme synthetisieren Angiotensin II. Renin, ein sekretorisches Glykoprotein ist das Schlüsselenzym des RAS. Es spaltet Angiotensin I vom Angiotensinogen ab. Im zirkulierenden RAS wird Angiotensin I vom ACE zum Angiotensin II umgeformt. Hingegen im lokalen RAS werden zur Generierung von Angiotensin II lokale Enzyme benutzt. Es konnte gezeigt werden, dass es sich um Cathepsin D und Chymase handelt [112-115]. Die Existenz und Funktion eines lokalen, spezifischen RAS im Herzen hat immer wieder zu Diskussionen geführt. Intrazelluläre Angiotensin II Effekte konnten ermittelt werden [116], jedoch war es schwierig zwischen Effekten des lokalen, intrakardialen und des systemischen Angiotensin II zu unterscheiden [117]. Wichtige Komponenten des lokalen RAS, wie Angiotensinogen [118, 119], ACE [120, 121], Chymase [122, 123] und Angiotensin I/II [124, 125], konnten leicht identifiziert werden. Die Reninexpression im Herzen war schwierig zu beweisen. Viele Studien bestätigten die Existenz von Renin mRNA im Herzen bei northern blotting, solution hybridization assay und RT-PCR in verschiedenen Spezies [119, 126, 127]. In anderen Studien konnte kein Beweis für eine lokale Reninexpression im Herzen gefunden werden [128]. Des Weiteren wurde versucht die Reninexpression im Herzen durch eine transgene Überexpression des nativen Renin Promotors zu beweisen. Eine erhöhte Reninproduktion im Herzen konnte bei einem Reninkonstrukt aus dem Maus Ren-2-Gen nachgewiesen werden, nicht jedoch bei einem humanen Reninkonstrukt [129, 130]. In anderen Studien konnte gezeigt werden, dass die Reninexpression nicht unter physiologischen, sondern unter pathophysiologischen Situationen involviert ist [131].
EINFÜHRUNG 19 Renin wird überwiegend von der Niere synthetisiert und ausgeschieden. Das genetische Transkript des sekretorischen Renins ist das Exon(1-9)Renin. Als Preprorenin gelangt es, ähnlich jeden sekretorischen Protein, durch den cotranslationalen Transport zum endoplasmatischen Retikulum (ER). Im ER wird das Prefragment abgespalten und es entsteht das Prorenin was in Abb. 1.8 dargestellt ist. Das Prorenin wird zum einen als Renin verarbeitet und abgesondert zum anderen als inaktive Form gespeichert. Abb. 1.8: Entstehung sowohl des sekretorischen Exon(1-9)Renins als und des zytosolischen Exon(1A- 9)Renins. Clausmeyer et al. konnten zeigen, dass zwei verschiedene Renintranskripte vom selben Reningen abgelesen werden können. Dies wird in Abb. 1.9 schematisch dargestellt. Neben dem herkömmlichen Transkript existiert also ein zweites Renintranskript. Diesem Transkript fehlt das Exon 1 und damit die Signalsequenz für den cotranslationalen Transport zum ER. Dieses zweite Renintranskript wurde isoliert und als Exon(1A-9)Renin charakterisiert. Es verschlüsselt ein nicht sekretorisches intrazelluläres Protein [132]. Das Exon(1A-9)Renin Transkript ist somit ein verkürztes Prorenin, dass beginnend vom ersten ATG im Exon 2 abgelesen wird und nicht sezerniert werden kann. Diesem Protein fehlt sowohl das Prefragment vom sekretorischen Renin als auch zehn Aminosäuren vom konventionellen Prorenin [133]. Es wurde daher Exon(2-9)Renin genannt.
EINFÜHRUNG 20 Abb. 1.9: Identifizierung des alternativen Transkriptes nach Clausmeyer et al. [132]. Ähnliche Transkripte konnten in anderen Spezies einschließlich beim Menschen gefunden werden [134-136]. In Rattenexperimenten konnte gezeigt werden, dass dem Exon 2, von dem Exon(1A-9)Renin Transkript, 80 Basenpaare voraus gehen, die vom Intron A abgeleitet werden [132]. Diese Sequenz wird nicht kodiert und hat daher vermutlich eine regulatorische Funktion. Des Weiteren konnten unterschiedliche Expressionen von Renintranskripten in den Geweben der Ratte dargestellt werden, was in Abb. 1.10 dargestellt ist. So zeigte sich in der Niere nur mRNA für das sekretorische Preprorenin. In der Nebenniere konnte sowohl das sekretorische als auch das zytosolische Transkript ermittelt werden. Im Herzen wurde jedoch ausschließlich Exon(2-9)Renin mRNA gefunden. Ebenfalls wurde festgestellt, dass nach vier und fünf Tagen nach einer Ischämie die Expression des zytosolischen intrazellulären Renins Exon(2-9)Renin verglichen zu sham-operierten Kontrolltieren deutlich angestiegen war. Im Gegensatz dazu konnte keine Preprorenin mRNA ermittelt werden [137]. Dies zeigte, dass Exon(2-9)Renin möglicherweise eine Rolle in den Prozessen der Ischämie/Reperfusion spielen könnte.
EINFÜHRUNG 21 Abb. 1.10: Unterschiedliche Expression der Renintranskripte in Rattengeweben nach Clausmeyer et al. [137]. In der Nebennierenrinde konnten Reninproteine sowohl in sekretorischen Vesikeln als auch in Mitochondrien gefunden werden [138, 139]. Mitochondriales Renin muss daher abgeleitet sein vom Exon(2-9)Renin Transkript, denn nur dieses ist lokalisiert im Zytosol und somit verfügbar für den Transport ins Mitochondrium. So konnte gezeigt werden, dass Exon(2- 9)Renin an freie Ribosomen binden und in isolierte adrenale Mitochondrien importiert werden kann [132]. Um weitere Untersuchungen durchzuführen wurde ein transgenes Modell etabliert [133, 140]. Es konnten vier transgene Linien, die Exon(2-9)Renin überexprimieren, unabhängig voneinander generiert werden. Die Expression von Renin im Herzen konnte in allen vier Linien nachgewiesen werden, siehe Abb. 1.11. Die Linien 294 und 307 konnten sich gut entwickeln und wurden weiter untersucht. Die Überexpression des Exon(2-9)Renins führte nicht zur Hypertonie oder zu pathologischen Veränderungen im Herzen. Ebenso wurde festgestellt, dass weder Prorenin noch Renin in den transgenen Ratten verglichen zu Wildtyp Kontrollratten im Plasma erhöht war und es folglich intrazellulär vorhanden sein musste. Somit konnte gezeigt werden, dass sich CX-Exon(2-9) transgene Ratten als Modell zur Analyse der Funktion von Exon(1A-9)Renin in vivo eignen [133]. In der Linie 307 konnte sowohl im Zytosol als auch in der Fraktion der Mitochondrien und Nuclei signifikant erhöhte Renin- und Proreninwerte im Vergleich zu den Kontrolltieren ermittelt werden. Signifikante Unterschiede bei den Reninwerten in der Fraktion des sekretorischen Renins waren nicht zu sehen. Für die Linie 294 konnten erhöhte Proreninwerte in der Fraktion der Mitochondrien und Nuclei gezeigt werden, jedoch keine erhöhten Reninwerte in irgendeiner Fraktion dargestellt werden [133].
EINFÜHRUNG 22 Abb. 1.11: Nachweis der Exon(2-9)Renin überexprimierten Linien bei Etablierung des transgenen Modells in der Western Blot Analyse [133]. Des Weiteren wurden Zellversuche mit embryonalen H9c2 Kardiomyozyten durchgeführt, die mit sekretorischem Exon(1-9)Renin als auch mit zytoplasmatischem Exon(2-9)Renin transfiziert wurden [141]. Es konnte gezeigt werden, dass in Zellen, die Exon(2-9)Renin überexprimierten, erhöhte Konzentrationen von Prorenin und Renin vorhanden waren und dies vor allem in der mitochondrialen Fraktion. Im Gegensatz dazu konnte in Exon(1-9)Renin überexprimierten Zellen erhöhtes Prorenin ermittelt werden in der Fraktion der sekretorischen Vesikeln. Auch wurde gezeigt, dass Prorenin und Renin im Medium von Zellen die Exon(1- 9)Renin überexprimieren erhöht war, jedoch nicht im Medium von Exon(2-9)Renin überexprimierten Zellen. Somit sezernieren nur Exon(1-9)Renin transfizierte Zellen Renin ins Medium [141]. Ebenso konnte in Exon(1-9)Renin transfizierten Zellen festgestellt werden, dass sie einen erhöhten Proteingehalt im Vergleich zu den Kontrollzellen haben und größer sind. In den beiden Renin transfizierten Zelllinien konnten tote Zellen dargestellt werden. Entsprechend ist sowohl das Wachstum der Renin transfizierten Zellen als auch die Proliferation gegenüber den Kontrollen vermindert. Zusätzlich wurde die Apoptose- und Nekroserate in den transfizierten H9c2 Zellen ermittelt. Es konnte gezeigt werden, dass Exon(1-9)Renin Zellen eine verstärkte Nekroserate aufweisen, während Exon(2-9)Renin transfizierte Zellen vor Nekrose geschützt waren. Dies ist in Abb. 1.12 dargestellt.
EINFÜHRUNG 23 Abb. 1.12: Ermittlung der Nekroserate von H9c2, pIRES, Exon(1-9)Renin und Exon(2-9)Renin transfizierten Zellen. Bestimmung des LDH Ratio in vier unterschiedlichen Gruppen [141]. Exon(2-9)Renin transfizierte Zellen zeigten eine deutlich erhöhte Apoptoserate verglichen zu den Kontrollzellen. Zum einen wurde eine signifikante Zunahme apoptotischer Zellen und eine erhöhte Caspase-Aktivität in Exon(2-9)Renin Zellen gefunden, zum anderen konnte gezeigt werden, dass diese Apoptose mitochondrial bedingt ist, da es zu einer Zunahme der Translokation von Phosphatidylserin gekommen war [141]. Intrazelluläres, nicht sekretorisches Exon(2-9)Renin scheint vor nekrotischem Zelltod zu schützen und den apoptotischen Zelltod zu fördern.
EINFÜHRUNG 24 1.5. Aufgabenstellung Ziel dieser Studienarbeit war es mit Hilfe des ex vivo perfundierten Rattenherzes, bei dem durch Verschluss und Wiedereröffnung der linken Koronararterie ein akuter Myokardinfarkt simuliert werden kann, zwei unterschiedliche Aufgaben zu untersuchen. Zum einen sollte sowohl das kardioprotektive Potential des A2bAR Agonisten Bay 60-6583 bei Zugabe in der Reperfusion und zum anderen das kardioprotektive Potenzial der transgenen Exon(2-9)Renin überexprimierten Ratten geprüft werden. Die Postkonditionierung ist vom hohen klinischen Interesse. Jedoch ist es bisher nicht gelungen Therapiemöglichkeiten, die während der Reperfusion einsetzen, in den klinischen Alltag einzubringen. Viele Forschungsuntersuchungen befassen sich mit dem schützenden Signalweg und so auch mit dem Potential neuer möglicher Ansatzpunkte für den Einsatz in der Klinik. Diese Arbeit beschäftigt sich mit der Untersuchung des kardioprotektiven Potentials des A2bAR Agonisten Bay 60-6583 und möglichen Signaltransduktionswegen am ex vivo perfundierten Rattenherz. Des Weiteren sollte das kardioprotektive Potenzial der Exon(2-9)Renin überexprimierten Ratten untersucht werden. Bei einem Myokardinfarkt kommt es auf Grund von Arteriosklerose oder Thrombenbildung in den Herzkranzarterien zu einer Ischämie, die zum Absterben vom Myokardgewebe führt. Dieser Vorgang des Zellunterganges am Herzen wird als Nekrose bezeichnet. Hierbei kommt es zu Zellschwellungen, Membranbrüchen und zu inflammatorischen Prozesse mit der Einwanderung von Leukozyten und Makrophagen. Die Folgen sind Fibrose, Remodelling und Herzinsuffizienz. Dem gegenüber steht die Apoptose. Hier kommt es zur DNA-Fragmentierung bei der die Mitochondrien nicht geschädigt werden. Die Zelle wird in kleine Teile zerlegt und die Zellmembran bleibt intakt. Es kommt zwar auch zum Verlust von Zellen jedoch ohne inflammatorische Prozesse und Fibrose. Daher war es Ziel dieser Studienarbeit eine Infarktgrößenbestimmung an isolierten retrograd perfundierten Rattenherzen, die Exon(2-9)Renin überexprimieren, durchzuführen, um den Zelluntergang durch Nekrose genauer zu untersuchen. Hierfür standen, wie bereits oben erwähnt, die zwei unabhängigen transgenen Linien (294, 307) zur Verfügung welche mit zwei Kontrollgruppen verglichen wurden. Zu definierten Zeitpunkten wurden transmurale Biopsien gewonnen, um zelluläre Signaltransduktionswege während der Ischämie/Reperfusion mittels Western Blot Analyse zu analysieren.
MATERIAL UND METHODEN 25 2. Material und Methoden 2.1. Material 2.1.1. Chemikalien und Reagenzien Chemikalien und Reagenzien Herkunft Acrylamide/bis-Acrylamide (37,5:1) Roth, Karlsruhe A.dest B. Braun, Melsungen AG, Melsungen Agarose Serva, Heidelberg Ammoniumpersulfat (APS) Sigma-Aldrich, Dreisenhofen BCATM Protein assay Kit Pierce, Rockford, USA BSA Rinderserumalbumin, Roth, Karlsruhe β-Mercaptoethanol Roth, Karlsruhe Calciumchlorid Sigma-Aldrich, Dreisenhofen Cell Lysis Buffer Cell Signalling, Danvers, USA 1,4-Dithiothreit (DTT) Roth, Karlsruhe Dimethylsulfoxid (DMSO) Roth, Karlsruhe Entwickler Tetanal, Norderstedt Ethanol 100% Commercial Alcohols, Brampton, Ontario Ethidiumbromid Sigma-Aldrich, Dreisenhofen Ethylendiaminotetraessigsäure (EDTA) Sigma-Aldrich, Dreisenhofen Fixierer Tetanal, Norderstedt Fluorescent Polymer Microspheres, 2-8 Duke Scientific Corp., Fremont, USA µM Glukose Sigma-Aldrich, Dreisenhofen Glycin Roth, Karlsruhe Heparin-Natrium B. Braun Melsungen AG, Melsungen Heparin Sodium Injection Pharmaceutical Partners of Canada INC 4-(2-Hydroxyethyl)piperazine-1- Roth, Karlsruhe ethanesulfonic acid (HEPES) Isopropanol Merck, Darmstadt Kaliumchlorid Sigma-Aldrich, Dreisenhofen
MATERIAL UND METHODEN 26
Kaliumdihydrogenphosphat Sigma-Aldrich, Dreisenhofen
Lämmlipuffer Biorad, Hercules, USA
LumiGloTM Reagent Cell Signalling, Danvers, USA
Milchpulver Roth, Karlsruhe
Magnesiumsulfat Sigma-Aldrich, Dreisenhofen
Methanol Roth, Karlsruhe
Natriumchlorid Sigma-Aldrich, Dreisenhofen
Natriumchlorid 0,9 % B. Braun, Melsungen AG, Melsungen
Natriumhydrogencarbonat Sigma-Aldrich, Dreisenhofen
Paraformaldehydlösung 4 % Sigma-Aldrich, Dreisenhofen
Phosphate buffered saline (PBS) PAA, Pasching, Österreich
Phenylmethylsulforylfluorid (PMSF) Roth, Karlsruhe
Ponceau S Sigma-Aldrich, Dreisenhofen
Salzsäure Merck, Darmstadt
Sodiumdodecylsulfate (SDS) Sigma-Aldrich, Dreisenhofen
N,N,N’,N’-Tetramethylethylenediamine Biorad, Hercules, USA
(TEMED)
Thiopental-Natrium (Trapanal) Altana Pharma, Konstanz
Tris(hydroxymethyl)aminomethan Roth, Karlsruhe
(Tris)
2,3,5-Triphenyltetrazoliumchlorid Sigma-Aldrich, Dreisenhofen
(TTC)
Tween 20 Roth, Karlsruhe
2.1.2. Substanzen und Inhibitoren
Substanzen und Inhibitoren Herkunft
Bay 60-6583 A2bAR Agonist, Bayer
Healthcare, Wupperthal
(2-[6-amino-3,5-dicyano-4-(4-hydroxyphenyl)pyridin-2-
ylsulfanyl] acetamide)
KT 5823 Selektiver PKG Inhibitor, Sigma-
Aldrich, DreisenhofenSie können auch lesen

















































