Genetik der Osteoporose - Lothar Seefried - MVZ Aschaffenburg
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Genetik der Osteoporose
Lothar Seefried
MCW www.orthopaedie.uni-wuerzburg.de
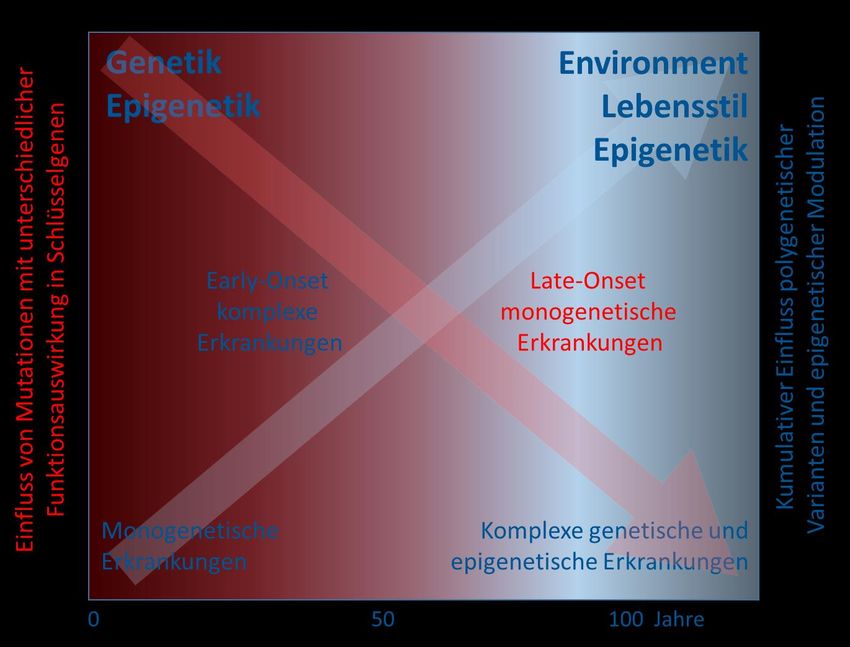
Fließender Übergang zwischen monogenetischer Skeletterkrankung und multifaktorieller Osteoporose Seefried, Jakob, Internist 2021, Seltene osteologische Erkrankungen und ihre Therapie Mäkitie et al., Front Endocrinol 2019, New Insights Into Monogenic Causes of Osteoporosis
Osteogenesis imperfecta / Osteogenesis imperfecta WNT1 / LRP assozierte Early X-Chromosomale Hypophosphatasie / HPP X-Chromosomale Bindegewebsschwäche;
OI, Typ 1-4 (klassisch) / Typ 5 Varianten Typ 6-20 Onset Osteoporose Osteoporose Hypophosphatämie / XLH Marfan-Syndrom, Ehlers-
Danlos Syndrom, Loeys-
Dietz-Syndrom
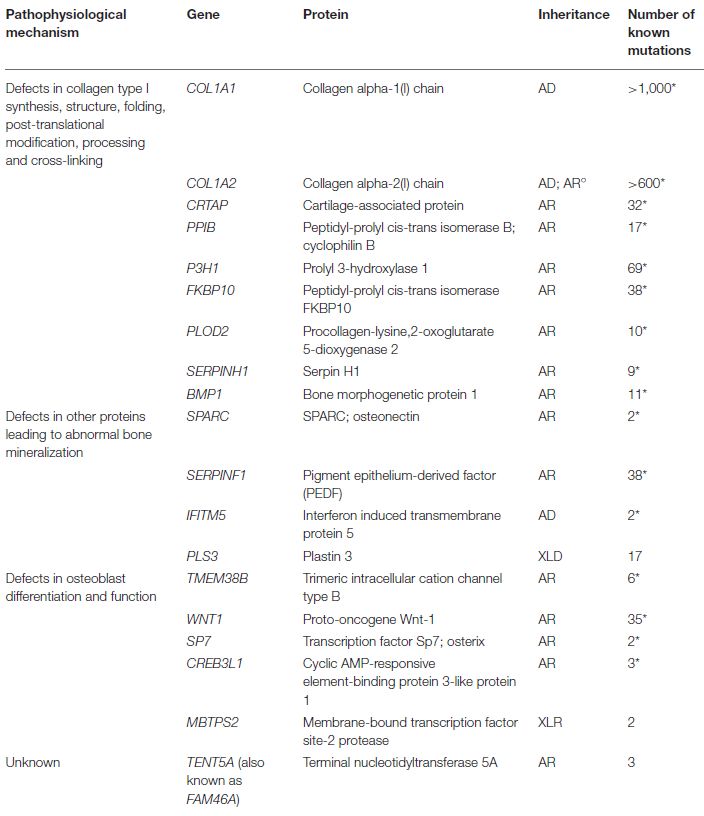
COL1A1 / COL1A2 / IFITM5 u.a. SERPIN F / H, CRTAP, WNT1 / LRP5 / LRP 6 PLS-3 ALPL-Gen PHEX-Gen FBN1, COL5A1/2, COL1A1/2, ,
Genetik
(Typ 5) BMP1, FKBP10 TGF-β R1/2, TGF-β 2/3
Autosomal dominant Autosomal rezessiv Autosomal rezessiv X-chromosomal dominant, Autosomal rezessiv / X-chromosomal dominant, MFS / LDS Autosom dom.,
Vererbung Männer i.d.R. schwerer dominant M:F 1:2 EDS nach Form teils autosom.
betroffen dominant, teils rezessiv
Störung Kollagensynthese / - Kollagen-Reifungsstörung, Formationsdefizit durch Gestörte Knochenformation, AP-Defizienz, Substrat- Erhöhung FGF-23, Reduktion sekundäre Osteoporose
prozessierung, Verminderte defiziente Hydroxylierung vermindertes wnt Signalling, ggf zus. Unkoordinierte Akkumulation (PPi, PLP), TmP/GFR, renaler durch verminderten
Pathophysiologie Knochenmasse bei fokal (CRTAP,, PPIB, P3H1), Faltung gestörte Osteoblastäre Resorption Mineralisationsdefizit, Phosphatverlust, verminderte Krafteintrag, DD primäre
suffizienter / gesteigerter (FKBP10) Differenzierung und Aktivität Vitamin B6 Umbaustörung 1α-Hydroxylase Aktivität Störung der Knochenstruktur
Mineralisierung
Spondyloepiphysäre andere Formen der andere Formen der andere Formen der Rheumatologische Vitamin D abhängige Osteogenesis imperfecta
Dysplasien, Osteogenesis imperfecta, sek. Osteogenesis imperfecta, sek. Osteogenesis imperfecta, sek. Erkrankungen, Fibromyalgie / Rachitis/Osteomalazie, (overlap)
Klinische Mucopolysaccharidose 4A / Osteoporose Osteoporose Osteoporose Myastenie, Multiple Sclerose andere Formen von
Differentialdiagnosen Morquio, Early-onset / X- / Chronic Fatigue Syndrom, Phosphatdiabetes / Fanconi-
chromosomale Osteoporose, Chron. Entzündliche Syndorm, Achondroplasie /
Ehlers-Danlos-Syndrom, Darmerkrankungen Hypochondroplasie
unspezifisch, ggf. PINP / CTx / unspezifisch unspezifisch, ggf. Osteocalcin unspezifisch AP ↓, PLP ↑, PEA/Krea Phosphat (Serum) ↓, FGF- unspezifisch
Labor NTx ↑ ↓ (Urin) ↑, ggf. PINP / CTx / 23↑, 1,25-D3 ↓, Urin-
NTx ↓ Phosphat ↑ (relativ)
WK-Sinterungen, Frakturen WK-Sinterungen, Frakturen WK-Sinterungen WK-Sinterungen, Frakturen Diaphysäre Pseudofrakturen/ Diaphysäre (Pseudo- Extremitätenfrakturen
Frakturen der Röhrenknochen der Röhrenknochen der Röhrenknochen Looser‘sche Umbauzonen, )frakturen,
Femur, Metatarsalia
Kleinwuchs, Deformitäten Röhrenknochen Heterotope Ossifikationen, Kleinwuchs, Skoliose
Achsabweichungen n. bei comp. Het. Konstellation; Tendinosis calcarea Achsabweichungen infolge
Skelettale
Frakturen, Platt-/Keilwirbel heterozygote Pat Rachitis, Frühe Arthrose,
Auffälligkeiten
IFITM5: Hypertropher Kallus phänotypisch unauffällig Osteo-/Spondylo-
/Enthesiophyten
Überdehnbarkeit tbd tbd tbd Schmerzen, Schwäche, Schwäche Überdehnbarkeit, Gefäß-
Muskeln/Gelenke
Erschöpfung /Klappeninsuffizinez
Lumbal + femoral ↓(↓) Lumb. /fem. teils nur mäßig Lumbar ↓↓, Femoral ↓ Lumb. /fem. teils nur mäßig Lumbal: normal - ↑, Femoral Lumbal normal - ↑, Femoral
Knochendichte / DXA
erneidrigt erneidrigt normal normal
Blaue Skleren, Parodontitis, ggf. Zahnabszessse, Hörverlust, vegetative Symptomatik,
Extraskeletale
Dentinogenesis imperfecta, Kalzifikationen: Cornea, Nephrokalzinose Schmerzen
Charakteristika
Höstörungen Conjunctive, Nephrokalzinose
Gute Daten für BP, teilweise BP unterschiedlich gut Hinweise auf Nutzen Daten für BP und TPTD Asfotase alfa für "päd. Onset" Burosumab (Anti-FGF-23);
TPTD, Anti-Sclerostin wird geeignet osteoanaboler Therapie + Skelett verfügbar; TPTD / alternativ Substitution mit
Therapeutische
erprobt Anti-Sclerostin möglich; BP Phosphat (Reducto spezial /
Besonderheiten
und Dmab, relativ KI / keine Calcium Sandoz) + Calcitriol
guten DatenBei welchen Osteoporosepatienten sollte man eine molekulargenetische Diagnostik erwägen Außergewöhnlich niedrige Knochendichte, die durch das Risikoprofil nicht adäquat erklärt werden kann Multiple Frakturen die durch die Knochendichte nicht stimmig erklärbar ist Multiple Frakturen auch im unmittelbaren familiären Umfeld Multiple Frakturen bereits in der Kindheit Auffällige Stigmata im Sinne einer Skelettdysplasie (Kleinwuchs, Deformierungen der langen Röhrenknochen, Deformierungen der Wirbelsäule, Morphologische Auffälligkeiten im Kopf/Gesichtsbereich) Zusätzliche extraskelettale Veränderungen als Hinweis auf eine syndromale Erkrankung mit Knochenbeteiligung
Implikationen einer korrekten molekulargenetischen Diagnostik Teilweise spezifische Therapieoptionen verfügbar (HPP, XLH,…) Vermeidung nicht hilfreicher / potentiell schädlicher Therapien (BP / GC bei HPP) Ermöglicht zielgerichtete Zusatzdiagnostik Ermöglicht adäquate genetische Beratung, auch prädiktiv Optimale sozialmedizinische Versorgung (Langzeit-Physiotherapie, GdB, Selbsthilfegruppen,…) Vermeidung Stigmatisierung durch Fehldiagnose bzw. fehlende Diagnose
Sie können auch lesen

















































