Häufigkeit der HLA-Null-Allele in der Deutschen Stammzellspenderdatei (DSSD) Ulm - oparu
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

Institut für Klinische Transfusionsmedizin und Immungenetik Ulm (IKT)
Geschäftsführer: Prof. Dr. med. Hubert Schrezenmeier
Häufigkeit der HLA-Null-Allele in der
Deutschen Stammzellspenderdatei (DSSD)
Ulm
Dissertation
zur Erlangung des Doktorgrades der Medizin der
medizinischen Fakultät
der
Universität Ulm
vorgelegt von
Michael Begović
geboren in Gaildorf
2017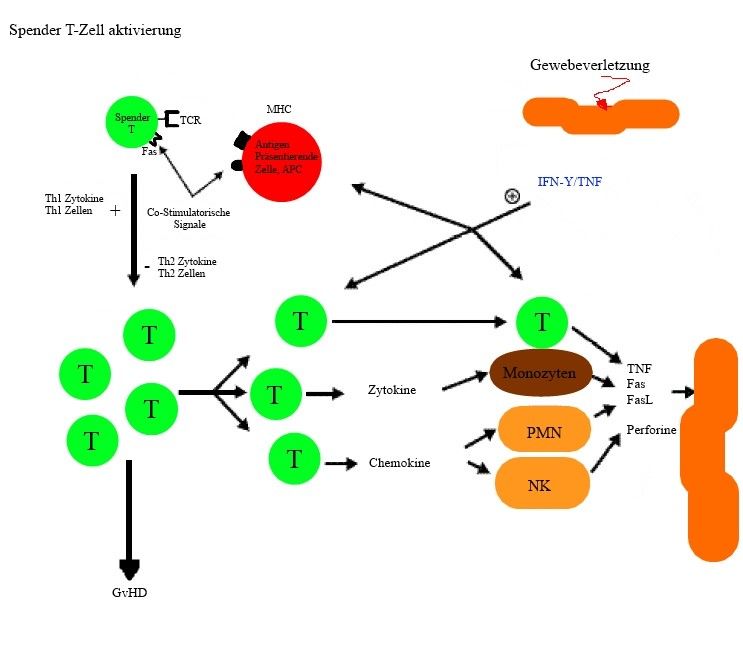
Amtierender Dekan: Prof. Dr. Thomas Wirth 1. Berichterstatter: Prof. Dr. Hubert Schrezenmeier 2. Berichterstatter: Prof. Dr. Hayrettin Tumani Tag der Promotion: 15.06.2018
Meiner Frau Yvonne und unseren Kindern Nicole und Tristan gewidmet
Inhaltsverzeichnis
Inhaltsverzeichnis
Abkürzungsverzeichnis IV
1. Einleitung 1
1.1 Der Haupthistokompalitätskomplex, MHC
(Major Histocompatibility Complex) 1
1.1.1 Bedeutung der MHC-Region 1
1.2 Genetik, allelischer Polymorphismus und Nomenklatur der HLA-
Komplexe 2
1.2.1 Genetik 2
1.2.2 Allelischer Polymorphismus 5
1.2.3 Nomenklatur 5
1.3 HLA-A-Moleküle 6
1.3.1 HLA-Klasse I-Moleküle 6
1.3.2 HLA-Klasse II Moleküle 8
1.4 HLA Null Allele 8
1.5 Allogene Erkennung der HLA-Antigene 10
1.6 GvHD (Graft-versus-Host-Disease) 10
1.7 Serologische Typisierung 14
1.8 Molekulargenetische Typisierung 15
1.9 Ziel der Arbeit 17
2. Material und Methoden 18
2.1 Material 18
2.1.1 Geräte 18
2.2 verwendete Chemikalien 20
2.3 verwendete Datenverzeichnisse 23
2.4 Methoden 24
2.4.1 Patientenkollektiv 24
IInhaltsverzeichnis
2.4.2 Polymerase Chain Reaction 24
2.4.3 Agarose-Gelelektrophorese (Nachweis von PCR Produkten) 32
2.4.4 HLA-Typisierung mit der Luminex LiquiChip LABType SSO 35
2.4.5 Vorbereitung für die Luminex-Analyse 35
2.4.6 Die einzelnen Schritte der Luminex-Analyse 35
2.4.7 Vorbereitung der Liqui Chip Workstation 39
2.4.8 Auswertung . 41
2.4.9 Freigabe der Ergebnisse 41
2.4.10 Dokumentation 42
2.4.11 Verwendete Statistik 42
3. Ergebnisse 43
3.1 Diskrepanzen nach der Molekulargenetischen Typisierung 43
3.2 Vermutliche oder vermutete HLA-Null Allele 45
3.3 Low Expression Allele 48
3.4 Erläuterung zur Bestätigung der HLA-Null Allele 48
3.5 Erläuterung der Allele mit atypischen Expressionsmuster 50
3.6 Erläuterung zur Bestätigung der Low-Expression Allele 51
4. Diskussion 52
4.1 Bedeutung der HLA-Null Allele in der Stammzell-Transplantation und
Deren Typisierung 52
4.2 Molekulargenetische vs. Serologische Typisierung 52
4.3 Diskussion der HLA-Null Allele Ergebnisse 53
4.4 Diskussion der Allele mit atypischen Expressionsmuster 56
4.5 Diskussion der Low-Expression Allel Ergebnisse 58
5. Zusammenfassung 60
6. Literaturverzeichnis 62
Danksagung 84
IIInhaltsverzeichnis
Lebenslauf 85
IIIAbkürzungsverzeichnis
Abkürzungsverzeichnis
% Prozent
° Grad
°C Grad Celsius
µl Mikroliter
A Adenin
AEG Allgemeine Elektricitäts-Gesellschaft
al andere
Amp Ampere
APC American Power Conversion
ATC Anatomisch-therapeutisch-chemischen
Bf Bright Field
B-Lymphozyten Bursa fabricii-Lymphozyt
bp Basenpaar
C Component
C2 Component 2
C4 Component 4
C4A Component 4 aktivated
C4b Component 4 binding
CAG CytosinAdeninGuanin,
CD4 Cluster of Differentiation 4
CGC CytosinGuaninCytosin
cGvHD Chronic Graft Versus Host Disease
CHIP Chromatin-Immunpräzipitation
CTG CytosinThyminGuanin
dATP Desoxyadenosintriphosphat
dCTP Desoxycytidintriphosphat
dGTP Desoxyguanosintriphosphat
dl Deziliter
DMIX Gemisch aus dNTP-Mix-Puffer, deoxyriboNucleoside TriPhosphates
DNA Deoxyribonucleicacid
DNS Desoxyribonukleinsäure
IVAbkürzungsverzeichnis
dNTP Desoxyribonukleosidtriphosphate
DSSD Deutsche Stammzellspenderdatei
dTTP Desoxythymidintriphosphat
EPS Electrophoresis Power Supply
et und
FasL Fas ligand
FI Fluor
G Guanin
GAC Guanin Adenin Cytosin
GAG Guanin Adenin Guanin
GAT Guanin Adenin Thymin
GCC Guanin Cytosin Cytosin
GGC Guanin Guanin Guanin
GvH Graft-versus-Host
GvHD Graft-versus-Host Disease
HI-Virus Humane Immundefizienz-Virus
HLA Humanes Leukozyten Antigen
HSP-70 Hitzeschockprotein 70
IBMTR International Bone Marrow Transplant Registry
IFN Interferone
IMGT international ImMunoGeneTics
IS Instrumentation
kDa Kilodalton
L Low
LAB Laboratory
LE Low Electroendosmosis
LiChrosolv Liquid Chromatography
Liqui Liquidation
LT Lymphotoxin
MACS Magnetic Cell Separation
mg Milligramm
MHC Major Histocompatibility Complex
ml Milliliter
mRNA messenger RNA
VAbkürzungsverzeichnis
n Anzahl
N Null
NK Natürliche Killerzelle
NMDP National Marrow Donor Program
PBS Phosphate-buffered saline
PC Personal Computer
PCR polymerase chain reaction
PE Phycoerythrin
PMN Polymorphkernige neutrophile Granulozyten
PTC Peltier Thermal Cycler
PTC Positive Temperature Coefficient
SAPE Streptavidin Phycoerythrin
SBT Sequence Based Typing
SSO Sequence-specific oligonucleotide
SSOP Sequence-Specific Oligonucleotide Probe
SSP Single Specific Primer
T Thymin
TAC Thymin Adinin Cytosin
TAG Thymin Adenin Guanin
Taq Thermus aquaticus
TCR T cell receptor
Th Thymus-Helfer-Zelle
T-Helferzellen Thymus-Helferzellen
T-Lymphozyten Thymus-Lymphozyten
TNF Tumornekrosefaktor
U/µl Units/Mikroliter
UV Ultraviolett
UV-Licht Ultraviolettes-Licht
Var Variation
Vs Versus
VWR Van Waters und Rogers
WHO World Health Organization
α Alpha
β Beta
VIEinleitung
1. Einleitung
1.1 Der Haupthistokompalitätskomplex, MHC [ Major
Histocompatibility Complex]
1.1.1 Bedeutung der MHC-Region
MHC-Moleküle finden sich in allen Wirbeltieren. Neben Kontrolle der Immunantwort
gegenüber von Transplantaten, hat der Haupthistokompatibilitätskomplex eine große
Bedeutung in der Einleitung der spezifischen immunologischen Antwort als
antigenbindende Struktur [43,17,15].
Bei jeder Spezies wird der Begriff MHC durch einen separaten, für diese Art spezifischen
Namen bezeichnet. Das menschliche MHC-System wird, als HLA (Human Leukocyte
Antigen)-System bezeichnet [46]. Es werden zwei Klassen von MHC-Molekülen
unterschieden: MHC-Klasse I und MHC-Klasse II [45,19,26]. Werden in den MHC-Klasse
I Molekülen Proteine gebunden, die nicht vom eigenen Körper stammen, dann werden
diese von CD8+ T-Lymphozyten erkannt. Nach diesem stimulierenden Kontakt auf
zellulärer Ebene, werden aus CD8+ Zellen, zytotoxische T-Lymphozyten, welche den Tod
deren Zielzelle bewirken [29,37,28,41,42]. MHC Klasse II-Moleküle, die ein Antigen
präsentieren, werden von CD4+T-Zellen, sog. T-Helferzellen, erkannt und diese wiederum
veranlassen die Bildung von Zytokinen, welche andere Zellen aktivieren, wodurch es dann
zu einer Potenzierung der Immunantwort kommt [40, 28,20,32].
HLA-Moleküle stellen Oberflächenglykoproteine dar, deren Hauptfunktion ist, die
Präsentation von Antigenpeptidfragmenten z.B. durch Makrophagen, dendritische Zellen
und B-Lymphozyten an T-Lymphozyten zu gewährleisten, wodurch die Immunantwort
eingeleitet wird [56 ,39,18,38,15,11,2].
1Einleitung
1.2 Genetik, allelischer Polymorphismus und Nomenklatur der
HLA- Komplexe
1.2.1 Genetik
Das HLA-System wird in der Nähe des Zentromers auf dem kurzen Arm des menschlichen
Chromosoms 6 (6p21.1-6p21.3) genetisch kodiert [49,58,16,77].
Siehe Abbildung 1.
Abb.1: Struktur des HLA-Systems auf Chromosom 6. Abbildung selber angefertigt. Dargestellt ist hier der
Bereich im Chromosom 6, in den die HLA-Gene codiert werden. Die Region 6p21.31 zeigt vergößert die
Gen-Klassen I-III. Die Gene der HLA Klasse I (gelb) liegen nahe des Telomers auf dem kurzen Arm, Gene
der Klasse II (blau) befinden sich nahe des Zentromers und die Gene der Klasse III (grün) befinden sich
zwischen den Klasse I und II. Abkürzungen: class, englisch für Klasse, p = petit für kurz, q = queue, für lang.
Bedeutung: Telomer = griechisch für Endteil. Centromer = griechisch für Mittelteil.
In Abbildung 1 wird das Chromosom 6 dargestellt, in dessen Bereich von 6p21.1 –
6p21.31 sich ein Genabschnitt mit ca 4.000 Kilobasenpaaren befindet [5,9].
In diesem Abschnitt sind die drei verschiedenen HLA-Regionen [Klasse I, II und III] zu
finden [77,78,86].
Die Klasse II-Region liegt zentromerwärts, während der Klasse I-Bereich in Richtung
Telomer orientiert ist.
Dazwischen liegt die Klasse III-Region, die weder zu Klasse II noch zu Klasse I gezählt
werden kann, und auch keine eigentlichen HLA-Merkmale kodiert [50].
2Einleitung
Abb.2: Humanes MHC (englisch für Major Histocompatibility Complex), Haupthistokompatibilitätskomplex.
Der Komplex ist in drei Regionen eingeteilt: HLA (Humanes Leukozyten Antigen) class (englisch für Klasse) I
(gelb), II (hellblau) und III (grün). Jede dieser Regionen enthält zahlreiche Gene. Hier sind die wichtigen,
exprimierten Gene dargestellt.
Bei der HLA-Klasse I Genregion unterscheidet man Genorte, die für HLA-Klasse Ia Moleküle, die sog.
klassischen HLA-Klasse I Antigene A (hellblau), B (hellblau), C (hellblau), kodieren von weiteren HLA-Klasse
Ib Genorten, zu denen HLA-E (hellblau), F (hellblau), G (hellblau), MICA (hellblau)/MICB (hellblau) gehören.
Innerhalb der HLA-Klasse II Genregion werden drei Subregionen unterschieden, die für verschiedene HLA-
Klasse II Antigene kodieren. Die HLA-DR-Subregion enthält ein monomorphes Gen für A-Kette (DRA, rot )
und neun Gene für B-Ketten (DRB1-DRB9), wobei es sich bei DRB2 (rot), DRB6-DRB9 um Pseudogene
handelt. Die verschiedenen B Kettengene sind vermutlich durch Genduplikationen entstanden. Die HLA-DQ
Subregion weist jeweils zwei potentiell funktionelle Genorte für A- und B-Ketten (DQA1, DQA2, DQB1,
DQB2, rot) auf. Nur die Loci DQA1 und DQB1 kodieren entsprechende Proteinketten. Die HLA-DP Subregion
enthält zwei funktionelle Loci für jeweils eine A- und B-Kette (DPA1, DPB1) von HLA-Klasse II Antigenen
sowie zwei Pseudogene für eine A- und B-Kette (DPA2, DPB2). Zwischen der HLA-Klasse I und HLA-Klasse II
Region befindet sich eine sog. HLA-Klasse III Region von ca. 1000kb, in der einzelne HLA-Klasse I Gene sowie
eine Vielzahl von anderen Genloci (z.B. Gene für Komplementfaktoren, Tumornekrosefaktor,
Hitzeschockproteine usw.) zu finden sind, die nur z.T. Funktionen im Immunsystem besitzen, in den
strukturellen Eigenschaften sich von HLA-Molekülen deutlich unterscheiden und daher nicht zum
eigentlichen HLA-System gerechnet werden. Abkürzungen: HLA = humanes Leukozyten Antigen, MICA =
Haupthistokompatibilitätskomplex Klasse I ähnliche Kette A (engl.: major histocompatibility complex class I-
realted chain A), MICAB = Haupthistokompatibilitätskomplex Klasse I ähnliche Kette B (engl.: major
histocompatibility complex class I-realted chain B), Antigene = -A,-B, -C, -D, -E, -F, -G, HFE (hellblau) =
(englisch für High Iron Fe) hereditäre-Hämochromatose-Protein, TAPBP = Tapasin TAP Bindentes Protein,
DPB2 = DP beta 1, DPB2 = DP beta 2, DPA1 = DP alpha 1, DOA = DO alpha, DMA = DM alpha, DMB = DM
beta, PSMB9 = Proteasome Subunit beta 9, LMP2 = Large Multifunctional Proteasome 2, PSMB8 =
Proteasome Subunit beta 8, LMP1 = Large Multifunctional Proteasome 1, DOB = DO beta, DQB1 = DQ beta
2, DOA1 = DO alpha 1, DRB1 = DR beta 1, DRB2 = DR beta 2, DRB3 = DR beta 3, DRA = DR alpha, C21B =
Cytochrom P 450 Subfamilie XXI, BF = Komplementfaktor B, C2 = Komplementkomponente 2, C21B =
Cytochrome P450 Klasse 21 Unterklasse A Teil 2, C4A und C4B = Komplementkomponenten 4A und 4B, BF =
Komplement Faktor B, HFE = Hereditäre-Hämochromatose-Protein, HSP = Hitzeschockprotein, LMP =
Multifunktionelle Protease, LTA und LTB = Lymphotoxine A und B, P450 = Cytochrom P-450, PSMB8 und 9 =
Proteasom Untereinheit Beta Typ 8 und 9, TAP1 und TAP2 = Antigenpeptid-Transporter assoziiert mit der
Antigenverarbeitung 1 und 2, TNF-α = Tumor Nekrose Faktor alpha, HSPA1A, HSPA1B und HSPA1L =
Hitzeschockproteine Typ 1A,1B und 1L. (Quelle: www.uni-
ulm.de/~wflegel/STUD/FOLIEN/Mytilineos2008HLA.pdf, mit freundlicher Genemigung von PD Dr.
Mytilineos)
3Einleitung
Der HLA-Komplex wird, wie oben bereits erwähnt, in drei Regionen eingegliedert.
HLA-Klasse I-Moleküle bestehen aus einer α- und eine ß-Kette. Die α-Kette der HLA-
Klasse I-Moleküle wird in einem Gen kodiert. Die leichte Kette des HLA-Klasse I-
Moleküls, die ß-Kette, auch ß-Microglobulin genannt, wird nicht im Chromosom kodiert
und dient lediglich zur Stabilisierung des HLA-Klasse I Moleküls. Die wichtigsten HLA-
Klasse I-Moleküle sind HLA-A, -B, -C –E und –G [92]. Darüber hinaus existieren
sogenannte Pseudogene denen bisher keine Genprodukte nachgewiesen werden konnten
[95].
Zu diesen Pseudogenen zählen, HLA-H, -J, -K, -L, -N, -S, -X und –Z [44,52,47,57].
HLA-Klasse I-Gene sind von kodierenden Abschnitten, sogenannten Exons, aufgebaut
[65]. Vor und hinter einem Exon, steht ein nicht-kodierender Abschnitt, welcher als Intron
bezeichnet wird. Somit besteht ein HLA-Klasse I-Gen aus sieben Exons und acht Introns.
Die wichtigsten HLA-Klasse II-Moleküle sind HLA-DR, -DQ und –DP [54]. Diese
bestehen jeweils aus zwei Ketten,αundβ.Die α-Ketten werden von A-Genen kodiert (z.B.
DRA1, DQA1, DPA1),währenddieβ-Ketten in B-Genen kodiert (DRB1, DQB1, DPB1)
werden [51,62]. A-Gene bestehten aus jeweils vier Exons und vier Introns, die B-Gene aus
fünf Exons und fünf Introns.
Die HLA-Klasse III-Region befindet sich zwischen der HLA-Klasse I- und -Klasse II-
Region und enthält die Gene für die Komplementfaktoren C2, C4, Bf, C4A und C4B
[55,59], die Tumornekrosefaktoren TNF-α, -β und LT, das Hitzeschockprotein HSP-70
und die Steroid-21-Hydroxylase [63,73,68,70,66,53,60].
4Einleitung
Abb.3: Anordnung der Exons und Introns am Beispiel des Muskelproteins Tropomyosin in einem
eukaryotischen und prokaryotischen Genes. Blau, grün, orange, violet, rot und gelb steht für Exone. Grau
steht für Introne. Abbildung selbst erstellt.
Abkürzungen: DNA = Deoxyribonucleic acid, englisch für Desoxyribonukleinsäure, E = Exone, I =
Introns
1.2.2 Allelischer Polymorphismus
Das HLA-System ist das polymorphste System im humanen Genom. Durch
Punktmutationen die durch äußere Einflüsse oder spontan auftreten können, führt dies
dazu, dass sich ein Gen in einer Vielzahl von Formen sich präsentiert [1,7,12]. Diese
Punktmutationen, welche zu neuen Allelen eines Gens führen, werden als Polymorphismus
bezeichnet [61,64]. Der HLA-Polymorphismus ist das wichtigste genetische Hindernis bei
einer Organ-[Gewebe-] oder Blutstammzelltransplantation. HLA-Klasse-I- und HLA-
Klasse-II-Moleküle weisen einerseits eine ausgeprägte Homologie ihrer DNA- und
Proteinsequenzen auf, andererseits lässt sich innerhalb der genetischen Regionen eine
ausgeprägte allelische Variabilität finden. Dieser ausgeprägte Polymorphismus ist vor
allem in den Exonen 2 und 3, bei HLA-Klasse I, und in Exon 2 bei HLA-Klasse II zu
finden [14,17,79,81,93]. Die Konzentration der Variabilität auf die Region der
Antigenbindungsstelle legt nahe, dass die allelische Variabilität die Grundlage für die
effektive Bindung strukturell verschiedener Antigenfragmente ist [46].
1.2.3 Nomenklatur
In Anbetracht des starken Polymorphismus innerhalb des HLA-Systems ergab sich früh die
Notwendigkeit, einen Weg zur systematischen Klassifizierung und Benennung von HLA-
Gruppen zu entwickeln. Die entsprechende HLA-Terminologie wurde durch das WHO
(World Health Organisation) Nomenklatur Komitee entwickelt und gepflegt. Bisher sind
16.251 Allele bekannt.
HLA-A * 24 : 02 : 01 : 02 L
Abb. 4: Nomenklatur des HLA-Systems. Abbildung selber erstellt.
Der Buchstabe A bezeichnet den Genort. Der Stern zeigt, dass es sich um eine molekulargenetische und nicht
um eine serologische Testung handelte. Das Hauptantigen wird durch die ersten zwei Zahlen (=erstes Feld)
angegeben (hier durch die Zahl 24). Die Zahl im zweiten Feld steht für die Allel-Spezifität dieses Antigens.
Im dritten Feld werden stille Mutationen (= Mutationen, die bei der Übersetzung des Gens zu Proteine keine
5Einleitung
Auswirkung haben, da sie nicht zu einer eigenständigen Proteinvariante führen) beschrieben. Das vierte Feld
zeigt Polymorphismen, die außerhalb der kodierenden Region dieses Gens vorliegen (Introns, regulatorische
Genabschnitte). Der Buchstabe am Ende, hier: L (steht für Low Expression, niedrig exprimiert)
charakterisiert den Expressionsstatus des jeweils durch den Allel-Namen angegebene HLA-Allels. Die
einzelnen Felder werden durch ``:``- Zeichen voneinander getrennt [84,122,126].
1.3 HLA-A-Moleküle
1.3.1 HLA-Klasse I-Moleküle
HLA-Klasse I-Antigene (HLA-A, B, C) bestehen aus einer schweren Kette mit 44 kDa die
nichtkovalentanβ-2–Mikroglobulin angelagert ist. Letzteres wird auf dem Chromosom 15
kodiert [3,4,67]. Der extrazelluläre Anteil der α–Kette besteht aus drei Domänen, von
denen die äußeren beiden durch eine Aminosäuresequenz determinierten alloantigenen
Determinanten tragen. Die beiden äußeren Domänen bilden eine Rinne, an die die Peptide
binden, welche von MHC–Molekülen präsentiert werden, siehe Abb.5 [8,13,80].
HLA-Klasse I-Antigene finden sich auf der Oberfläche von allen kernhaltigen Zellen,
Thrombozyten und Spermatozoen.
6Einleitung
Abb.5: Struktur eines HLA-Klasse I Moleküls. Gebildet wird die Peptid-Bindungsstellte durch die links und
rechts miteinander verbundenen α1- und α2-Domäne (rot). An die α3-Domäne (rot) hängt eine
Transmembranregion, welche mit dem Zytoplasmamembran (blau) verbunden ist, und mit dem
zytoplasmatischen Schwanz mit dem Zellinneren (grau) verbunden ist. Das β2 Mikroglobulin (violett), die
leichte Kette des Klasse I Molekül, ist nicht-kovalent an das HLA-A-Molekül gebunden ist.
Abkürzungen:α1=alpha1,α2=alpha2,α3=alpha3,β2m = Beta2Mikroglobulin, TM = Transmembran.
(Quelle: www.uni-ulm.de/~wflegel/STUD/FOLIEN/Mytilineos2008HLA.pdf, mit freundlicher Genemigung
von PD Dr. Mytilineos)
7Einleitung
1.3.2 HLA-Klasse II Moleküle
Die HLA-Klasse II-Moleküle, werden als Heterodimere [87] bezeichnet, da Sie aus einer
schweren α-Kette mit ca. 34 kDa Molekulargewicht, und einer leichteren β-Kette mit
einem Molekulargewicht von ca. 27 kDa bestehen (siehe Abb. 6). Die HLA-Klasse II-
Antigene sind nicht auf allen Zellen des Organismus, wie die HLA-Klasse I-Moleküle, zu
finden [6,10,14,17]. Man findet Sie auf B-Lymphozyten, Antigen-präsentierenden Zellen,
Makrophagen, Monozyten, aktivierten T-Zellen, aktivierte Epithelzellen, Endothelzellen
und B-Lymphozyten [25,36].
Abb.6: Struktur eines HLA-Klasse II Moleküls. Hier wird die Pepdit-Bindungsstelle durch die α1-(gelb) und
β2-Domäne (orange) gebildet. Diese Domänen sind jeweils mit ihrer entsprechender α2-(gelb) bzw. β2-
Domäne (orange) verbunden. Diese sind mit einer Transmembranregion mit der Zytoplasmamembran
verbunden, welche durch die Plasmammembran (blau) führt und mit ihrem zytoplasmatischem Schwanz im
Zellinneren (grau) verbunden sind.
Abkürzungen:α1=alpha1,α2=alpha2,β1=beta1,β2=beta2,TM=Transmembran. (Quelle: www.uni-
ulm.de/~wflegel/STUD/FOLIEN/Mytilineos2008HLA.pdf, mit freundlicher Genemigung von PD Dr.
Mytilineos).
8Einleitung
1.4 HLA Null Allele
HLA-Null-Allele sind durch die fehlende Expression des korrespondierenden HLA-
MerkmalsaufderZelloberflächecharakterisiert.DieUrsacheliegtmeistensin„nonsense“-
Mutationen, die durch ein sog. Stopp-Codon zu einem frühzeitigen Abbruch in der
Proteinsynthese führen. Solche werden häufig durch eine Punkt-Mutation verursacht oder
durch Mutationen in der Promoter Region, die zu einer Veränderung in der Gentranslation
führen können [91,151,154,157,158]. Solche Mutationen können aber auch für den
Austausch von Genabschnitten, Löschen oder Einfügen von Nukleotiden [83,153], sowie
Rekombinationen [91] verantwortlich sein. Bei nicht Erkennen eines Null-Alles kann es
dann zu einer unerwünschten Abstoßungsreaktion kommen [71,82,91].
Über 190 HLA Null-Allele der Klasse I und Klasse II wurden bisher identifiziert [84]. Die
Häufigkeit der HLA-Null-Allele wird bisher mit 0,003-0,07% angegeben [89,90]. Der
Nachweis der HLA-Null-Allele mit modernen molekulargenetischen Methoden ist
schwierig, da routinemäßig nur ein Teil des Gens untersucht wird (meist Exon 2 und 3)
[156], jedoch die Mutationen, welche zur nicht-Expression eines HLA-Merkmals führen,
an beliebiger Stelle der Exone oder der Promoter Region eines HLA-Gens vorkommen
können [69,76]. Der Nachweis der Null-Allele kann mit kombinierter Anwendung eines
serologischen HLA-Typisierungsverfahrens (Lymphozytotoxizitätstest, der auf eine
Antigen-Antikörper-Reaktion und somit auf die Expression des Moleküls beruht) und
molekulargenetischen Typisierung der HLA-Merkmale erfolgen [72,74,91]. Dabei kann
eine Diskrepanz zwischen diesen zwei Typisierungsverfahren auf das Vorliegen eines
Null-Allels hinweisen [75,94,96]. Ein endgültiger Nachweis kann lediglich nur durch die
Sequenzierung des entsprechenden Genabschnittes erfolgen.
Bei Vorliegen eines Null-Allels kann es zu Fehltypisierungen kommen, was folglich zu
einer Inkompatibilität zwischen Spender und Empfänger führen kann [71]. Inwieweit bei
Verdacht auf ein Null-Allel dieses weiter differenziert werden sollte, wird aktuell debattiert
[148,159]. Von der NMDP “Policy for Confirmatory Typing Requirements“ wurde
empfohlen, bei keiner Eindeutigkeit des Typisierungsergebnisses zumindest die in der
Tabelle 1 aufgezeigten Null-Allele auszuschließen, wenn der in der Tabelle dazugehörige
haplotypische Zusammenhang besteht [152].
9Einleitung
Tabelle 1: Die zwei häufigsten Null-Allele bei den HLA-A und HLA-B-Allelen [150]. Dargestellt sind die
Null Allele, welche bei einem HLA-Typisierungsergebnis ausgeschlossen werden müssten, da diese zu einer
Inkombatibilität von Spender und Empfänger führen könnten. Links sind die HLA-Null-Allele aufgelistet.
Mitte das Allel, das differentialdiagnostisch am ehesten in Frage kommt und die rechte Spalte gibt den
Haplotypen an, der am häufigsten mit dem entrsprechenden Null-Allel assoziiert ist.
Abkürzungen: N = Null-Allel, A-,B-,Cw,J = Antigene, DRB1 = DR beta
Null Allel Alternativ Gemeinsames Assoziierte Allele im
Allel Haplotyp
A*24:09 N A*24:02:01:01 B*40 oder B*27
B*51:11 N B*51:01:01 A*02:01 und DRB1*04:02
und Cw*15BJ
1.5 Allogene Erkennung der HLA-Antigene
Eine Transplantation zwischen genetisch differenten Individuen der gleichen Spezies wird
als allogene Transplantation bezeichnet [97]
Bei der Unterscheidung zwischen „Selbst“und„Nicht-Selbst“habenHLAAntigeneeine
sehr wichtige Bedeutung. Im Fall der Transplantation, die einen nicht physiologischen
Vorgang darstellt, werden die HLA-Merkmale von allogenen Transplantaten als Fremd
erkannt und die entsprechenden Zellen abgestoßen [98,99,110]. Die auf den Spenderzellen
getragenen HLA-Merkmale können im Organismus des Empfängers zelluläre und
humorale Abwehrmechanismen [Graft-versus-Host Reaktion] auslösen. Dies ist der Grund
weshalb bei einer Transplantation eine hohe Übereinstimmung der HLA Merkmale
zwischen dem Spender und dem Empfänger angestrebt wird [100].
1.6 GvHD [Graft-versus-Host-Disease]
Die Graft-versus-Host-Disease (dt: die Graft-versus-Host-Krankheit) kann in zwei
Reaktionen unterteilt werden [101,117]. Die akute und die chronische GvHD. Sie kann bei
einer allogenen Knochenmark- bzw. Stammzelltransplantation als immunologische
Reaktion auftreten. Die T-Lymphozyten, welche sich im Transplantat des Spenders
befinden, reagieren gegen den Empfänger [104,105]. Diese Reaktion, die meist mit
Hautveränderungen, bis zu Manifestationen an Organen [Haut, Leber, Augen, Lunge,
Gastrointestinal Trakt] einhergeht, kann letztendlich zum Tode des Patienten führen
[102,111,112,103]. Bei der akuten GvHD können die Symptome auch nach drei Monaten
10Einleitung
nach erfolgter Transplantation auftreten. Nach diesem Zeitraum geht die akute in die
chronische GvHD über. Die akute GvHD zeigt sich klinisch unterschiedlich ausgeprägt
(siehe Tabelle 2). Die Stadieneinteilung, welche 1974 nach Glücksberg deklariert wurde,
zeigt die Ausprägungen an Haut, Leber, Gastrointestinal Trakt sowie den klinischen
Zustand des Patienten.
Tabelle 2: Einteilung nach Stadium der akuten GvHD gemäß Glücksberg 1974. Auflistung der GvHD
Stadien und der dazu gehörigen Veränderungen der Haut, Leber und im Magen-Darm Trakt. Der klinische
Zustand (ganz rechts) zeigt in Abhängigkeit des Stadiums, die aufgelisteten Symptome des Patienten.
Abkürzungen: < = kleiner als, > = größer als, / = pro, % = Prozent, mg/dl = Miligramm pro Deziliter, ml =
Milliliter, - = bis
Stadium Haut Leber Gastrointestinal Klinischer
makulopapulöses Bilirubinwert Trakt Zustand
Exanthem Diarrhö
0 kein Ausschlag < 2 mg/dl keine Diarrhö normal
oder < 500
ml/Tag
1 < 25 % der 2 – 3 mg/dl > 500 ml/Tag normal
Körperoberfläche
2 25 – 50% der 3,01 – 6 mg/dl > 1000 ml/Tag leichte
Körperoberfläche Einschränkung
3 generalisierte 6,01 – 15 mgdl > 1500 ml/Tag mäßige
Erythrodermie Einschränkung
4 generalisierte > 15 mg/dl starke schwere
Erythrodermie abdominale Einschränkung
Schmerzen mit
oder ohne Ileus
Eine weitere Einteilung zeigt die überarbeitete Version (siehe Tabelle 3, in Anlehnung der
Glücksberg-Einteilung, von der Consensus Conference von 1995 [107,108]. Diese zeigt
ebenfalls eine Gradeinteilung der Schwere der GvHD jedoch ohne den klinischen Zustand
des Patienten.
Tabelle 3: Graduierung der akuten GvHD nach der Consensus Conference 1995, in Anlehnung an die
Glücksberg Einteilung (siehe Tabelle 2). Die Schwere der Erkrankung wird hier von Grad I bis Grad IV
unterteilt, wobei das Stadium aus der oben gezeigten Glücksberg-Einteilung mit eingebracht wird. Z.B.: Grad
II beudetet: Hautveränderung zeigt bei Stadium 3 generalisierte Erythrodermie, Stadium 1 der Leber mit
erhöhten Billirubinwerte 2-3 mg/dl und Stadium 1 des Gastrointestinaltrakt mit Diarrhöen vom mehr als 500
ml pro Tag.
Abkürzungen: GvHD = Graft-versus-Host-Disease (dt: Graft-versus-Host-Krankheit)
Grad Haut Leber Darm
I Stadium 1 oder 2 keine Beteiligung keine Beteiligung
II Stadium 3 Stadium 1 Stadium 1
III Stadium 1 – 3 Stadium 2 oder 3 Stadium 2 oder 4
IV Stadium 4 Stadium 4 Stadium 4
11Einleitung
Eine weitere Einteilung wurde 1997 von der International Bone Marrow Transplant
Registry (IBMTR-Index) eingeführt [106,109,113]. Diese Einteilung wurde entwickelt, da
es immer wieder bei Patienten, welche an einer bestimmten Schweregrad der gleichen
GvHD litten, jedoch andere Organverteilungsmuster aufzeigten. Auch diese Einteilung
beinhaltet nicht den klinischen Zustand des Patienten.
Tabelle 4: Einteilung der Stadien der IBMTR Severity Index, mit Anlehnung an die Glücksberg Einteilung
(siehe auch Tabelle 2).
Index Haut [maximales Leber [maximales Darm [maximales
Stadium] Stadium] Stadium]
A 1 0 0
B 2 1–2 1–2
C 3 3 3
D 4 4 4
Die akute GvHD kommt in ca. 50% der Blutstammzell- bzw. Knochenmark-
Transplantierten auf. Die seltene chronische GvHD zeigt sich in ca. 30 % der Fälle. Auch
bei der chronischen GvHD gab es Einteilungen die jedoch 2005 von Filipovich modifiziert
wurden. Letztere korreliert die Anzahl der betroffenen Organe mit dem aktuellen
Schweregrad. Die Einteilung erfolgte in Score, wobei Score 0 keine Symptome, Score 1
milde Symptome, Score 2 mäßig, starke Symptome und Score 3 ausgeprägte, starke
Symptome bezeichnet [114,115,116] siehe Tabelle 5.
Tabelle 5: Score Einteilung der chronischen GvHD. Diese Tabelle zeigt die Zuordnung der GvHD in Indices
abhänging von der Organspezifischen, symptomorientierte Stadiumeinteilung nach Glücksberg (siehe dafür
Tabelle 2).
Abkürzungen: GvHD = Graft-versus-Host-Reaktion (dt: Graft-versus-Host-Krankheit)
Anzahl der milde cGvHD mäßig starke Ausgeprägte starke
Organe cGvHD cGvHD
1 Organ Score 1 Score 2 Score 3
2 Organe Score 1 Score 2 Score 3
3 Organe Score 1 Score 3
Lungenbefall Score 1 Score 2
12Einleitung
Abb.7: Schematische Darstellung einer Graft-versus-Host-Reaktion. Abbildung selbst erstellt. Die
Pathophysiologie der GvHD beruht auf drei aufeinander folgenden Phasen. Die 1. Phase beginnt mit der
Gewebeverletzung (host tissue, orange) welche zur Freisetzung von Warnsignalen, sogennante
proinflamatorische Zytokine, IFN-β (blau) und TNF (blau) führt. Die 2. Phase führt zur Donor-T-Zell (grüne
Kreise) Aktivierung, sowie zur Proliferation und Differenzierung. Die Aktivierung beginnt wenn ein Donor-
T-Zellrezeptor an einen MHC/Peptid-Komplex der an einem APC-Empfänger (roter Kreis) sitzt, bindet. In
Anweseneheit von sogennanter Co-Stimulatorischer Signale kommt es zur weiteren Aktivierung von T-
Tellen. Aktivierte Donor-T-Zellen unterliegen einer weiteren Prolifertaion und Differenzierung und beginnen
Zytokine zu produzieren. Die 3. Phase ist die Zelluläre und inflamatorische Phase. Durch die Aktivierung
von T-Zellen kommt es zur Entstehung von Effektorzellen welche direkt oder über die Produktion von
entzündlichen Zytokinen (Monozyten hellbraun) Organe schädigen.
Abkürzungen: GvHD = Graft-versus-Host-Reaktion, IFN-γ= Interferon gamma, TNF =
Tumornekrosefaktor, APC = Antigen-Präsentierende Zelle, MHC = Major-Histokompatibilitäts-Komplex, T
= T-Zelle, TCR = T-Zell-Rezeptor, Fas = Fas-Rezeptor, Th1 = T-Helfer-Zelle 1, Th2 = T-Helfer-Zelle 2, NK
(hellbraun) = Natürliche Killer-Zelle, PMN (hellbraun) = Polymorphkernige Neutrophile Granulozyten,
FasL = Fas-Rezeptor Ligand
In Abbildung 7 sieht man die Phasen der Graft-versus-Host-Reaktion. Man erkennt schon
zu Beginn, nach T-Lymphozyten Aktivierung, dass bereits Zytokine ausgeschüttet werden.
Diese setzen weitere Reaktionen in Gang. Dies wiederum führt zu weiteren Reaktionen,
die als Ergebnis Schäden an der Haut, Leber, Gastrointestinal Trakt, usw. mit sich ziehen.
13Einleitung
Nach dieser Schädigung an zuvor erwähnten Organen, werden erneut Zytokine
ausgeschüttet und somit der zerstörende Kreislauf fortgesetzt [34,118,125,127].
Für eine erfolgreiche Transplantation und ein möglich langes Überleben des Patienten ist
die Übereinstimmung der HLA-Merkmale zwischen Spender und Empfänger äußerst
wichtig und entscheidend. Der häufigste Grund für eine Transplantatabstoßung oder eine
GvHD wird auf eine HLA-Inkompatibilität zurückgeführt. Je nach Organ das transplaniert
wurde, ist der Einfluss der Spender-Empfänger Kompatibilität unterschiedlich.
1.7 Serologische Typisierung
Das serologische HLA-Typisierungsverfahren, das in HLA-Laboratorien angewendet wird,
geht auf den Lymphozytotoxizitätstest von Paul Terasaki zurück. HLA Klasse I – und/oder
Klasse II Antigene, lassen sich mit spezifischen Antikörpern (Alloantikörper) nachweisen.
[119].
Bei der HLA-Klasse I-Typisierung werden normalerweise unseparierte periphere
Blutlymphozyten eingesetzt, die aus ca. 80% T-Lymphozyten und ca. 20% B-
Lymphozyten bestehen [122].
Jedoch exprimieren T-Lymphozyten keine Klasse II Moleküle, deshalb wird die HLA-
Klasse II-Testung mit angereicherten B-Lymphozyten-Präparationen durchgeführt.
Zur Isolierung von B-Lymphozyten kommen zwei Methoden zur Anwendung. Zur
Gewinnung kann die MACS-Methode verwendet werden, welche die Zellen mit
monoklonalen Antikörpern, die auf Magnetperlen konjugiert sind, bindet. Die gängigste
Methode ist die Verwendung von Nylon-Watte [120,121]. Die Nylon-Watte wird in eine
Säule gefüllt und die Zellsuspension hinzugegeben. Hierbei binden die B-Lymphozyten
beim Durchwandern an die Wolle. Die T-Lymphozyten passieren ohne anzuheften.
Anschließend werden die B-Lymphozyten von der Nylon-Wolle herausgepresst.
Bei der Inkubation der Testzellen mit spezifischen HLA-Alloantiseren binden diese an die
Lymphozyten und durch Hinzugabe von Komplement werden letztere lysiert.
Durch Zugabe eines speziellen Farbstoffes, der in die oben genannten lysierten Zellen
eindringt, kann die Bindung von Antikörpern an ein HLA-Merkmal indirekt sichtbar
gemacht werden. Die lysierten Lymphozyten nehmen den Farbstoff auf (=sog. positive
14Einleitung
Reaktionen), während der Nachweis einer negativen Reaktion darauf beruht, dass die
vitalen Zellen keine Färbung aufnehmen. Führt man dieses Verfahren mit Antiseren gegen
jedes HLA-Merkmal durch, kann jedes der vorliegenden Gewebemerkmale nachgewiesen
(=typisiert) werden [123].
1.8 Molekulargenetische Typisierung
Nachteile des serologischen Typisierungsverfahrens sind eine niedrige Auflösung, und die
starke Abhängigkeit der Testung von der Vitalität der zu untersuchenden Zellen. Daraus
resultiert eine höhere Fehlerrate. Durch die molekulargenetische Verfahren wird eine
höhere Auflösung erzielt. Sowohl die Zuverlässigkeit als auch die Genauigkeit dieser
Verfahren sind der serologischen Typisierung überlegen [124,128].
Die Polymerase-Ketten-Reaktion, die erstmalig 1985 beschrieben wurde [129], dient als
Grundlage der molekulargenetischen Typisierungsverfahren.
Aufgrund der stetig steigenden Zahl von identifizierten HLA-Antigenen wurde inzwischen
die serologische Typisierung, vor allem für den Nachweis von HLA-Klasse II Antigenen,
durch die molekulargenetische Verfahren weitgehend abgelöst.
Die am häufigsten angewandten molekularen Typisierungsverfahren in der klinischen
HLA-Diagnostik sind die “Sequence-Specific Oligonucleotide Probe Analyse“, kurz PCR-
SSOP [130,131], die “Sequence-Specific Priming-Analyse“, kurz PCR-SSP [134,138,140]
und die “Sequenze-Based Typing-Analyse“ kurz PCR-SBT [132,138,140]. Ganz aktuell
kam hinzu das Verfahren„NextGenerationSequencing“(NGS),dasvermutlichdieHLA-
Diagnostik in den nächsten Jahren entscheidend prägen wird [201,202].
Bei der PCR-SSOP [130,135,136] werden einzelne Allele oder Allel-Gruppen nach einer
genortspezifischen Vervielfältigung mittels Hybridisierung mit sequenzspezifischen
Gensonden differenziert. Die Hybridisierung kann in diesem Falle nur ablaufen, wenn eine
vollständige Komplementarität zwischen dem Oligonukleotid-Primer und der
amplifizierten Test-DNA besteht. Sollten Oligonukleotide nicht komplementär sein,
können diese nicht binden.
Die PCR-SSP ist die heute am stärksten verbreiterte Variante der molekulargenetischen
HLA-Typisierung. Diese Methode beruht darauf, dass sequenzspezifische Primer während
der Amplifikation einzelne Allele erkennen [137,139]. Bei der PCR-SSP werden
15Einleitung
verschiedene Primerpaare eingesetzt, welche sequenzspezifisch sein müssen. Hier kommt
es nur zur Amplifizierung, wenn jeweils beide Primer eines Ansatzes an ihrer genauen
Zielsequenz binden. Pro Typisierung werden somit viele verschiedene
Primerpaarkombinationen für alle möglichen Allelgruppen bzw. Allele eingesetzt. Im
Anschluss wird mittels Gelelektrophorese das PCR-Produkt, bei passender
Basenkombination, von definierter Länge, welche im Gel als Bande zu erkennen ist,
nachgewiesen. Sollte keine Amplifikation stattgefunden haben, fehlt die Bande. Zur
Kontrolle wird zu jedem PCR-Primerpaar immer ein Kontroll-PCR-Primerpaar beigefügt,
dessen PCR-Produkte sich bei der Gelelektrophorese immer sichtbar darstellen lässt.
Geeignet ist die PCR-SSP vor allem bei einem geringen Probendurchsatz [141,142,147].
Die Methode mit der höchsten Auflösung ist die sogenannte PCR-SBT (Sequenz-based
Typing). Bei diesem Verfahren wird die zu untersuchende polymorphe HLA-Region (wie
z.B. HLA-DRB1) zunächst mittels PCR Locus-spezifisch bzw. nach Möglichkeit Allel-
oder Gruppen-spezifisch amplifiziert und anschließend mittels Sanger-Sequenzierung
analysiert [143,144,146]. Im Vergleich zu den zuvor erwähnten Typisierungsverfahren ist
die SBT zwar die aufwändigste zugleich jedoch die zuverlässigste Methode um HLA-
Genotypen zu charakterisieren [145].
Für die PCR wird als Ausgangsprodukt genomische DNS, welche primär aufgereinigt und
isoliert wurde. Anschließend wird die DNA mittels Polymerasekettenreaktion amplifiziert.
Die dafür benutzen spezifischen Primer, sind mit Biotin markiert. Die Amplifikate
(biotinylierte DNA-Kopien) werden im weiteren Schritt eingesetzt.
Bei der Hybridisierungsreaktion erfolgt die Charakterisierung der amplifizierten
Gensegmente mit sequenzspezifischen, immobilisierten Oligonukleotiden. Die
biotinylierten Amplifikate werden denaturiert und während der Hybridisierung mit den
sequenzspezifischen Gensonden (Oligonukleotide) gebunden.
16Einleitung
1.9 Ziel der Arbeit
Das Ziel dieser Arbeit war, die Ermittlung der Häufigkeit sowie die Analyse von HLA-A-
und HLA-B-Null Allelen in einer normalen, gesunden, lokalen Kohorte [89,90].
Auch sollten möglicherweise neu entdeckte, Null-Allele charakterisiert werden. Diese
Arbeit sollte darüber hinaus als Grundlage dienen, um die Strategie zum
Ausschluss/Erkennen von Null-Allelen in der HLA-Diagnostik besser zu planen bzw. die
Möglichkeit Konsequenzen eines Nicht-Nachweises von Null-Allelen genauer einschätzen
zu können.
17Material und Methoden
2. Material und Methoden
2.1 Material
2.1.1 Geräte
Thermocycler Gene Amp 9700 Firma Applied Biosystems
PCR Systems
Thermocycler PTC-200 Firma MJ Research
Sequenzier Gerät 3100, 3730 Firma Applied Biosystems
Spektralphotometer Firma Beckmann
Mikrowelle AEG Mikromat AEG
8- oder 12- Kanal Pipette 5-50µl Firma Socorex, Firma Eppendorf
und Spitzen T.I.P.S.
Standard 2-200µl
Multipette Firma Eppendorf
Pipetten und Filterspitzen Firma Eppendorf und Firma Biozym
10µl, 100µl, 1000µl
Analysen-Waage Sartorius L610D Firma Sartorius
Vortexer VWR International
Optical 96-Well Reaktion Platte Firma Applied Biosystems mit Barcode
Optical Verschlüsse Firma Applied Biosystems
(Otical Caps)
18Material und Methoden
Reaktionsgefäße Firma Eppendorf
Sequenziergerät 3730 Firma Applied Biosystems
Thermocycler 9700 Firma Applied Biosystems
Zentrifuge 5415 D Firma Eppendorf
neoLab-Regenzglasmixer Vortex Firma neoLab Laborbedarf
Laborzentrifuge SIGMA 2-6 Firma Sigma Laborzentrifugen GmbH
Luminex Liqui Chip Firma Qiagen 4072 Hieden
Biozym Electronik Firma Biozym Scientific GmbH
Pipettor 50-1200
Ultraschall-Reinigungsgerät Firma VWR International
USC 100
Schüttelgerät REAX top Firma Heidolph
Gel-Elektrophoresekammer Firma Pharmacia Biotech
Spannungsversorger Firma Pharmacia Biotech
Pharmacia Biotech EPS 300
UV Transilluminator Firma MWG Biotech
Video copy Processor Firma Mitsubishi
zur Fotodokumentation
Klebefolie für 96 Well Platten Firma Nunc
Sequence Pilot-HLA SBT Firma JSI Medical Systems
19Material und Methoden
Allele Identification Software
2.2 Verwendete Chemikalien
LABType® SSO A Locus Typing Test Firma BMT
Bestandteile:
SSO Bead Mix 400µl
Primer Set 400 µl
Primer Set D-mix 2 x 690 µl
Hybridization Buffer 3,4 ml
Denaturation Solution 2,25 ml
(Natriumhydroxid)
Wash Buffer 55 ml
SAPE Buffer 4,95 ml
Neutralization Buffer 2,5 ml
LABType® SSO B Locus Typing Test Firma BMT
Bestandteile:
SSO Bead Mix 400µl
Primer Set 400 µl
Primer Set D-mix 2 x 690 µl
Hybridization Buffer 3,4 ml
Denaturation Solution 2,25 ml
(Natriumhydroxid)
Wash Buffer 55 ml
SAPE Buffer 4,95 ml
Neutralization Buffer 2,5 ml
20Material und Methoden
Denaturierungslösung ( Natriumhydroxid) ONE LAMBDA,INC.
Neutralisierungspuffer ( Essigsäure) ONE LAMBDA,INC.
Dinatriumethylenediamintetraacetatdihydrat ONE LAMBDA,INC.
SAPE-Puffer Firma BMT
Stamm-SAPE-Lösung Firma BMT
D-Mischung Firma BMT
HLA Locus-spezifische Primermischung Firma BMT
LABType® SSO Bead-Mischung Firma BMT
Taq DNA Polymerase Firma Roche
SeaKem LE Agarose Firma Biozym
Ethidiumbromid Firma Eurobio
Olerup SSP DNA Size Marker Firma GenoVision
Loading Dye Firma Fermentas
Marker9 Firma Fermentas
LiquiChip™SystemFluid Firma Qiagen
PE-Conjugated Streptavidin (100xSAPE) Firma BMT
LiquiChip Calibration Bead Kit Firma Qiagen
21Material und Methoden
LiquiChip Control Bead Kit Firma Qiagen
Amplifikationsmischung
Bestandteile:
Primer 4,0 µl
DMIX 13,8 µl
Taq (5U/µl) 0,2 µl
(Isolierte DNA-Proben: DNA 2,0 µl)
SEAKEM Le Agarose Firma Cambrex
Ethidiumbromid Firma Innotrain
Ladepuffer LoadingDyeSol Firma MB Fermentas
Boratpuffer Firma Fermentas
Längenmarker Phix 174-DNA Firma MB Fermentas
LE Agarose Firma Biozym
Loading Buffer 1:3 Firma MB Fermantes
QIAmp Kit Firma Qiagen
Aqua ad iniectabilia Firma Braun
LiChrosolv Wasser Firma Merk
PBS Firma C.C.pro
22Material und Methoden
Ethanol reinst Firma Merk
SeaKem LE Agarose Firma Biozym
Olerup SSP DNA Size Marker Firma GenoVision
2.3 verwendete Datenverzeichnisse
Immunogenetische Datenbank (IMGT/HLA) des Anthony Nolan Research Institute
http://www.ebi.ac.uk/imgt/hla/align.html
The HLA dictionary 2004: A summary of HLA-A, -B, -C, -DRB 1/3/4/5 and –DQB1
alleles and thei association with serologically defined HLA-A, -B, -C, -DR and –DQ
antigens (Schreuder G.M.Th. et al. 2004)
http://www.ncbi.nlm.nih.gov/pubmed/15686589
Exon identities and ambiguous typing combinations des Anthony Nolan Research Institute,
Stand Januar 2005
http://www.ebi.ac.uk/imgt/hla/pdf/ambiguity_v280.pdf
23Material und Methoden
2.4 Methoden
2.4.1 Patientenkollektiv
Die Proben, die wir für die Analyse benötigten, stammten von Spendern, die sich in der
Deutschen Stammzellspenderdatei Süd (DSSD Süd) freiwillig registriert hatten. Die
Spender gaben ihr Einverständnis dazu, dass Ihre HLA-Merkmale getestet werden.
Die Anzahl der Proben, die in die Studie aufgenommen wurden, betrug 10.690. Diese
wurden zunächst serologisch durch Mitarbeiter des Instituts für Transfusionsmedizin in
Ulm typisiert und in die Knochenmarkspenderdatei aufgenommen.
Unter den initial typisierten Spendern wurden diejenigen herausgesucht, die primär
serologisch als HLA-A- bzw. HLA-B-homozygot eingestuft wurden. (Homozygot
bedeutet, dass das Genom einer Zelle zwei identische Allele enthält, Heterozygot bedeutet,
dass das Genom zwei unterschiedliche Allele aufweist). Diese Proben wurden
anschließend molekulargenetisch nachtypisiert.
Die molekulargenetische Typisierung erfolgte mit Hilfe des Luminex-Verfahrens.
Zeigte sich im Endresultat eine Diskrepanz zwischen der serologischen und der
molekulargenetischen Typisierung, deutete dies auf ein Null-Allel hin, wobei zu beachten
war, dass niedrig exprimierte Antigene zu einer sogenannten vorgetäuschten Homozygotie
führen können, ebenso wie neue, bisher unbekannte HLA-Gruppen, gegen die keine
Alloantiseren existieren sowie Null-Allele. Das Projekt wurde von der Ethikkommission
der Universität Ulm positiv beurteilt (Antrag-Nr. 286/07).
2.4.2 Polymerase Ketten Reaktion
Für die Typisierung benötigte DNA musste zunächst mittels PCR vervielfältigt werden.
Zuvor musste ein LABType PCR-Protokoll erstellt werden.
Dieses Protokoll besteht aus zwei Seiten, siehe Abbildung 8 und 9.
Hierbei musste zunächst dokumentiert werden, welcher Genort (HLA-A bzw. HLA-B)
amplifiziert werden sollte. Anschließend musste berechnet werden, welche Menge und mit
welcher Zusammensetzung ein PCR-MIX benötigt wird.
Auf der weiteren Seite ist schematisch eine mit 96-Kavitäten (= Well-Platte) abgebildet,
auf welcher die Nummern der zu testenden DNA Proben notiert sind. Jede einzelne Zahl
(von 1-96) steht für eine Vertiefung in einer 96-Well-Platte.
24Material und Methoden
Abb.8: PCR-Protokoll, Vorderseite. Auf diesem Protokoll werden die eingesetzten Volumina aller
verwendeten Chemikalien und Primergemische pro Ansatz aufgezeichnet.
25Material und Methoden
Abkürzungen: BH = Baden-Württemberg-Hessen, FB = Formblatt, PCR = Polymerase-Ketten-Reaktion,
HLA = Humanes Leukozyten Antigen, IKT = Immunogenetik, UL = Ulm, A = Antigen A, B = Antigen B,
Cw = Antigen Cw, DRB1 = DR beta 1 Antigen, DQB1 = DQ beta 1, MIX = Mixtur, Hyp.-Puffer =
Hybridisierungspuffer, DMIX = Gemisch aus dNTP-Mix-Puffer, deoxyriboNucleoside TriPhosphates, Taq =
Thermus aquaticus, U = Units, µl = Mikroliter, SAPE = Streptavidin Phycoerythrin, FN-UL-L-
528B_LABType PCR-Protokoll = selbstgenerieter Dateiname (Quelle: Scan eines PCR-Protokols, mit
freundlicher Genehmigung von PD Dr. Mytilineos).
26Material und Methoden
Abb.9: PCR-Protokoll, Rückseite. Auf diesem Protokoll wird in jeder „Box“ (= 1 – 96) die in der
entsprechenden Kovitität der PCR-Platte eingesetzte DNA-Id-Nummer dokumentiert.
27Material und Methoden
Abkürzungen: BH = Baden-Württemberg-Hessen, FB = Formblatt, PCR = Polymerase-Ketten-Reaktion,
HLA = Humanes Leukozyten Antigen, IKT = Immunogenetik, UL = Ulm, FN-UL-L-528B_LABType PCR-
Protokoll = selbstgenerieter Dateiname. (Quelle: Scan eines PCR-Protokols, mit freundlicher Genehmigung
von PD Dr. Mytilineos).
Die PCR dient, um eine Vervielfältigung der DNA-Region zu ermöglichen, die
anschließend untersucht werden soll. Aus einem einzelnen Molekül ausgehend gewinnt
man nach der PCR die millionenfache Menge, die einem dann erlaubt, die vervielfältigte
Region der DNA weiter zu analysieren, z.B. mittels Sequenzanalyse oder Hybridisierung
[174,178,176].
Für die PCR benötigt man pro Vertiefung der 96-Well-Platte:
2,0μlDNA
4,0 µl Primer
13,8 µl DMIX-Lösung
0,2 µl Taq-Polymerase, welche eine Gesamt Menge von 20 µl ergibt.
Was geschieht in einer Polymerase Ketten-Reaktion (PCR):
28Material und Methoden
Abb.10: Schematische Darstellung eines PCR-Zyklus. Abbildung selber erstellt. Nr.1 1zeigt die zu
vervielfältigende DNA (schwarzer Strang) (rot) welche als Template (englisch für Matrize, Muster)
verwendet wird. Nr. 2 zeigt die Denaturierung bei 94 °C in zwei einzelne Stränge. Nr. 3 zeigt wie die
zugesetzten Primer (Vorwärts- und des Rückwärtsprimer, grüner und oranger Punkt) bei 60 °C an deren
Komplementären Position auf jeden Einzelstrang anlagern. Nr. 4 zeigt die Bildung der neuen DNA-
Einzelstränge durch Verlängerung der Primersequenzen in ihre jeweilige Richtung. Nr. 5 zeigt die nun sich
neugebildete DNA, und der Zyklus läuft nun von vorne erneut.
Abkürzungen: PCR = Polymerase Ketten-Reaktion, Nr = Nummer, DNA = Desoxynukleinsäure
Die PCR beruht auf sich zyklisch wiederholenden drei Schritten:
1. Denaturierung: Aufspalten des DNA-Doppelstranges in Einzelstränge
2. Anlagerung (Annealing) der Primer-Oligonukleotide
3. Polymerisation (Neubildung) einer doppelsträngigen DNA mit Hilfe der
Primer, von denen sie ausgeht, und des Enzyms Polymerase sowie der vier
Desoxynukleotide (dATP, dTTP, dCTP, dGTP) [180,175,177]
29Material und Methoden
Ad 1:
bei der Denaturierung wird durch das Einwirken einer hohen Temperatur, hier 94°C, der
DNA-Doppelstrang in zwei Einzelstränge getrennt.
Erst nach dieser Trennung, können die Primer an die passenden DNA-Abschnitte binden.
Ad 2:
das Annealing, zuDeutsch,„dieAnlagerung“,bezeichnetdenSchritt,währenddessensich
die Primer an den homologen DNA-Abschnitt anlagert. Dies geschieht bei einer
Temperatur von 54°C.
Primer sollten im Überschuss vorhanden sein, damit sie die Anlagerung des DNA-
Gegenstranges kompetitiv verdrängen.
Ad 3:
AlsLetzteswirddasEnzym„DNA-Polymerase“eingesetzt.
Dabei handelt es sich um ein Enzym, das die Neubildung der neuen DNA-Stränge
ermöglicht. Hierbei wird ein DNA-Abschnitt als Ablese-Matrize benutzt und
komplementär dazu, von Primer ausgehend der neue DNA-Abschnitt synthetisiert.
Bevor die 2 µl DNA in die Vertiefung der 96-Well-Platte hinzugefügt wird, werden in
einem separaten Eppendorf-Gefäß, das DMIX (dNTP-Mix-Puffer, DeoxyriboNucleoside
TriPhosphates), die Primer und die Taq-Polymerase zusammen gefügt. Diese wird dann als
PCR-MIX bezeichnet.
Pro Vertiefung der 96-Well-Platte, werden (wie oben erwähnt) 13,8 µl der DMIX, 4,0 µl
des Primers und 0,2 µl der Taq-Polymerase benötigt, was eine Gesamt Menge von 20 µl
ausmacht. Bei bekannter DNA-Menge, kann das Gesamt-Volumen des PCR-MIX
berechnet, und dieser in einem separaten Eppendorf-Gefäß zusammengefügt werden.
30Material und Methoden
Beispiel-Rechnung:
Wird die komplette 96-Well-Platte verwendet, rechnet man die Menge für 95 Proben (die
96. Probe ist die Negativ-Kontrolle) also der PCR-MIX ohne DNA.
Bei der Mischung der PCR-MIX, sollte man folgende Reihenfolge beachten.
Zuerst werden das DMIX und Primer zusammengefügt. Die beiden Reagenzien werden
zuvor gut per Vortex vermischt (Reagenzglaßschüttler). Die Taq-Polymerase wird zum
Schluss hinzugefügt, da sonst die Gefahr bestünde, dass der PCR-MIX anfängt mit sich zu
reagieren (= Gefahr der Bildung von Primerdimeren).
Nachdem in jeder Vertiefung der 96-Well-Platte jeweils 2 µl DNA hinzu pipettiert wurden,
fügt man 18 µl der PCR-MIX Lösung in jede der Vertiefungen hinzu, so dass sich ein
Gesamt-Volumen von 20 µl pro Well ergibt.
Anschließend verschließt man die 96-Well-Platte luftdicht mit Hilfe einer Thermo-Stabilen
selbstklebenden Folie und legt sie in den Thermocycler.
Das PCR-Programm kann gestartet werden.
31Material und Methoden
PCR-Programm:
Tabelle 6: Die gesamte zyklische Amplifikationsreaktion mit einer Gesamtmenge von 20 µl pro Vertiefung
der 96-Well-Platte dauert etwa 90 Minuten. In der abgebildeten Tabelle, wurden zum Verständnis, die
einzelnen Schritte dargestellt.
Abkürzungen: °C=GradCelsius;∞=unendlich.
Temperatur Inkubationszeit Anzahl der
in Grad in Minuten Zyklen
Schritt 1 96°C 03:00 1
Schritt 2 95°C 00:20 5
60°C 00:20
72°C 00:20
Schritt 3 96°C 00:10 30
60°C 00:15
72°C 00:20
Schritt 4 72°C 10:00 1
Schritt 5 4°C ∞ 1
2.4.3 Agarose-Gelelektrophorese (Nachweis von PCR Produkten)
Die Gel-Elektrophorese die im Anschluss der PCR-Amplifikation erfolgt, dient zum semi-
quantitativen visualisierenden Nachweis der PCR-Amplifikate [179,182]. Die Gel-
Elektrophorese beruht auf dem Prinzip, dass Amplifikate aufgrund ihrer Größe und
elektrischen Ladung in einem elektrischen Feld unterschiedlich schnell laufen und dadurch
voneinander getrennt werden [186,185]. Aufgrund deren Molekülgröße laufen größere
Moleküle langsamer, die Kleineren schneller und weiter durch das Gel. Für die
Elektrophorese wird ein 0,2% iges Agarosegel hergestellt. Hierfür werden exakt 300 ml
Boratpuffer in einem entsprechend großen Glasgefäß eingefüllt und zusätzlich 6 g Agarose
hinzugefügt. Diese Suspension wird in der Mikrowelle bei 600 Watt so lange aufgekocht
bis eine klare, reine, homogene Lösung entsteht. Die Lösung wird nun auf 60°C abgekühlt.
Bei der erreichten Temperatur von 60° werden 10 Tropfen Ethidiumbromid dazu gegeben.
Anschließend wird die abgekühlte Agaroselösung in eine abgedichtete Gelwanne, in
welcher Kämme mit Zähnen vorhanden sind, eingefüllt. Diese Prozedur sollte blasenfrei
32Material und Methoden
erfolgen. Die Gelwanne wird während der Polymerisation in einer luftdicht
abgeschlossenen Ablufthaube für mindestens 30 Minuten gestellt. Ist die Agaroselösung
ausgehärtet, wird Boratpuffer in die Gelwanne eingefüllt bis das Agarose-Gel komplett
durch den Puffer umspült ist und die Kämme werden entfernt. Durch die Zähne der
Kämme entstehen Geltaschen mit einem Volumeninhalt von 15 µl.
Bevor die DNA in die Geltaschen einpipettiert wird, werden von jeder amplifizierten DNA
4 µl in eine neue 96-Well-Platte mit jeweils 2 µl Loading Farbstoff pipettiert. Die restliche
Menge des DNA-Amplifikats, die sich noch in der 96-Well-Platte befindet, wird Luftdicht
mit einer sterilen selbstklebenden Folie verklebt und zur Lagerung im Kühlschrank
deponiert. Diese gesamte Menge von nun 6 µl wird vorsichtig in die Geltaschen pipettiert.
In die vorletzte Geltasche kommt eine Negativ Probe, also Loading Farbstoff ohne DNA.
Um feststellen zu können, wie groß die jeweiligen Moleküle sind, wird der Gel-
Elektrophorese ein bestimmter Längen-Marker mit bekannten Basenpaaren
(Molekulargewichtsstandart 50-1000bp) in eine separate Geltasche gegeben. Anhand
dieses Markers kann abgelesen werden, wie groß das jeweilige Amplifikat ist. Nachdem
alle Proben in den Geltaschen gefüllt sind, wird die Gel Wanne für 15 Minuten unter
Spannung von 180 Volt und ca. 140 mA gesetzt.
Am Ende des Vorganges wird die Spannung ausgeschaltet. Zum eigenen Schutz werden
nun Handschuhe angezogen, und mit einer Pinzette und einem Skalpell das Gel-Stück, in
dem die Proben sich befinden, separat ausgeschnitten. Dieses Gel-Stück wird auf den UV-
Transilluminator zur Begutachtung und Fotodokumentation gelegt [184,181]. Vor
Einschaltung des UV-Lichtes werden die Augen zum Schutze mit einer speziellen Brille
bedeckt. Unter UV-Licht bei einer Wellenlänge von 254 nm und dem zuvor eingefügtem
Ethiumbromid zum Einfärben der Nukleinsäuren [183], werden die Banden, welche die
DNA beim Durchlaufen unter angelegter Spannung hinterlässt, sichtbar gemacht. Die UV
Bestrahlung sollte jedoch so kurz wie möglich gehalten werden, da ultraviolettes Licht die
Bildung von Pyrimidin-Dimeren begünstigen kann, was zur Folge haben könnte, das
Mutationen oder Strangbrüche entstehen könnten. Anschließend wird zur Dokumentation
mit einer integrierten Fotographie Einheit ein Bild (siehe Abbildung 11) des Gel-Stückes
gemacht.
33Sie können auch lesen

















































