Individualisierte Risikoabschätzung beim Einsatz zielgerichteter Tumortherapien in der pädiatrischen Hämatologie und Onkologie - GPOH
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Individualisierte Risikoabschätzung
beim Einsatz zielgerichteter Tumortherapien
in der pädiatrischen
Hämatologie und Onkologie
Leitfaden zur systematischen
Beschaffung, Aufbereitung und Verknüpfung
sicherheitsrelevanter Informationen
(Pharmakovigilanzleitfaden)
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 2

1. Einleitung ................................................................................ 5
2. Pharmakovigilanzleitfaden ........................................................ 5
2.1. Ziel des Pharmakovigilanzleitfadens .............................................................. 6
2.2. Individualisierte Risikoabschätzung .............................................................. 6
2.3. Standardisierung der Prozesse für die individualisierte Risikoabschätzung
anhand des Leitfadens ................................................................................... 7
2.4. Vertraulichkeit ............................................................................................... 9
2.5. Recherchestrategie und Informationsaufbereitung ....................................... 9
2.5.1. Allgemeine substanzspezifische Informationen ................................................... 10
2.5.1.1. Informationsquellen ............................................................................ 10
2.5.1.1.1.European Public Assessment Report ............................................ 10
2.5.1.1.2.Produktinformationen zugelassener Arzneimittel der FDA .............. 11
2.5.1.1.3.Produktinformationen des Zulassungsinhabers ............................. 16
2.5.1.1.4.Weitere Informationsquellen für Produktinformationen ................. 19
2.5.2. Sicherheitsrelevante Informationen beim Einsatz von zielgerichteten Tumortherapien
bei Kindern und Jugendlichen ........................................................................... 23
2.5.2.1. Meldungen aus Spontanmeldesystemen ................................................ 23
2.5.2.1.1.VigiAccess™ .............................................................................. 24
2.5.2.1.2.EMA ADR-Datenbank ................................................................. 25
2.5.2.1.3.BfArM und PEI ADR-Datenbank .................................................. 28
2.5.2.1.4. Aufbereitung der Rechercheergebnisse aus ADR-Datenbanken für
die individualisierte Risikoabschätzung........................................ 30
2.5.2.2. Individuelle Meldungen von Nebenwirkungen aus systematischen
Datensammlungen .............................................................................. 31
2.5.2.2.1. Pädiatrische, klinische Prüfungen ............................................... 31
2.5.2.2.2. INFORM Register ...................................................................... 31
2.5.2.2.3. Weitere systematischen Datensammlungen oder Patientenregister 32
2.5.2.3. Studienregister: Schwerpunkt pädiatrische Studien ................................ 32
2.5.2.4. Literaturrecherche ............................................................................... 35
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 3
2.5.3. Patientenspezifische Informationen ................................................................... 36
2.5.3.1. Individuelle, patientenspezifische Informationen .................................... 36
2.5.3.1.1. Demographische Daten ............................................................. 36
2.5.3.1.2. Klassifikation der onkologischen Erkrankung / Anamnese ............. 36
2.5.3.1.3. Arzneimittelanamnese (bisherige, allgemeine Chemotherapie) ...... 37
2.5.3.1.4. Toxizitäten und unerwünschte Ereignisse.................................... 37
2.5.3.1.5. Vorhergehende Behandlung mit zielgerichteten Tumortherapie(n) 38
2.5.3.1.6. Geplante Begleitmedikation im Kontext der zielgerichteten
Tumortherapie ......................................................................... 38
2.5.3.1.7. Pharmakogenomische Charakterisierung ..................................... 38
2.5.4. Individualisierte Risikoabschätzung.................................................................... 38
3. Fazit ....................................................................................... 39
4. Literaturverzeichnis ................................................................. 41
Annex I: Template zur strukturierten Informationsaufbereitung für die
individualisierte Risikoabschätzung gemäß Pharmakovigilanzleitfaden ... 43
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 4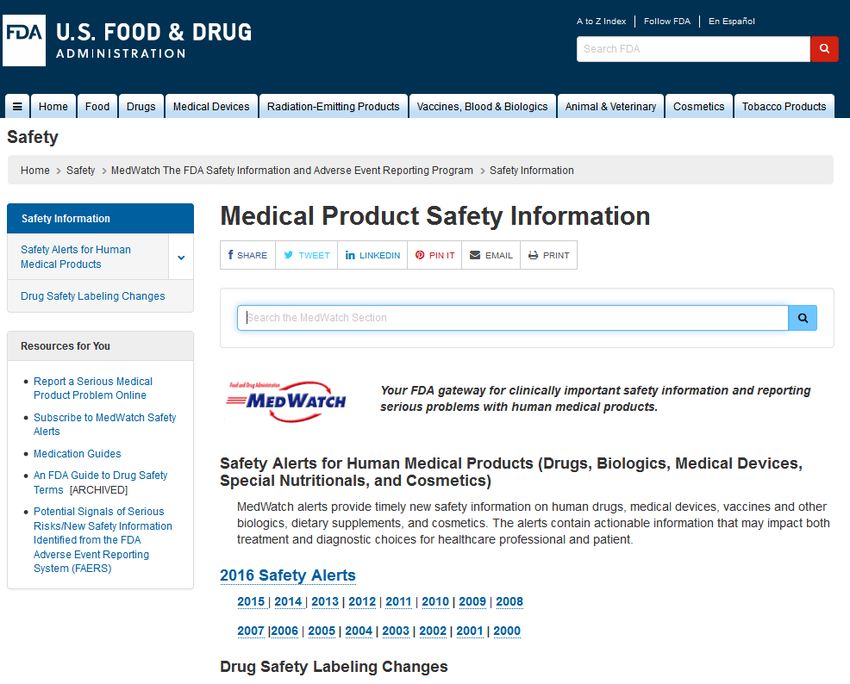
1. Einleitung In den vergangenen 15 Jahren wurden verstärkt gegen molekulare Strukturen der Tumorzellen gerichtete Arzneimittel für onkologische Indikationen zugelassen (Gravanis et al. 2014). Obwohl die Überlegenheit dieser Medikamente gegenüber den Standardtherapien bislang nur in spezifischen Situationen nachgewiesen wurde, ist die generelle Erwartungshaltung hinsichtlich einer besseren Wirksamkeit bei gleichzeitiger Reduktion unerwünschter Nebenwirkungen hoch (Pignatti et al. 2015). Die für Erwachsene zugelassenen, zielgerichteten Therapeutika werden zunehmend auch in der pädiatrischen Hämatologie und Onkologie „off-label“ eingesetzt, vor allem bei Patienten für die kein etabliertes Behandlungskonzept mehr zur Verfügung steht. Derzeit fehlen jedoch systematische Datensammlungen und Auswertungen zu Risiken und Nebenwirkungen des Einsatzes von zielgerichteten Tumortherapien bei Kindern. Für viele dieser pädiatrischen Indikationen sind die Erwartungen hypothetisch, da belastbare Daten zur Wirksamkeit bei diesem Patientenkollektiv derzeit nicht vorliegen (Dorris et al. 2016; Harris et al. 2016). Zudem fehlt für die Mehrheit der potentiell indizierten Substanzen nicht nur eine kindgerechte Arzneiform, sondern auch eine systematische Dosisfindung (Lu et al. 2016). Aufgrund dieser Wissenslücken ist derzeit für den individuellen, pädiatrischen Tumorpatienten und dessen behandelnden Arzt eine rationale, systematische Beurteilung der Risiken und Nebenwirkungen von zielgerichteten Tumortherapien nur ansatzweise möglich. Therapieentscheidungen folgen daher notgedrungen praktischen Kompromissen (Fried 2016) und sind – sowohl hinsichtlich der therapeutischen Chance als auch des potentiellen Risikos – eher weiche, probabilistische Annahmen. Mit zunehmenden Einsatz zielgerichteter Tumortherapien steigt der Bedarf für eine patientenzentrierte Nutzen-Risikobewertung, die die individuellen Bedürfnisse und Werte des Patienten bei der Therapieentscheidung berücksichtigt (Yu 2016). 2. Pharmakovigilanzleitfaden „Pharmakovigilanz ist die Gesamtheit der Maßnahmen zur Entdeckung, Erfassung, Bewertung und Vorbeugung von Nebenwirkungen sowie anderen arzneimittelbezogenen Problemen, die bei der Anwendung von Arzneimitteln auftreten“ (Aly 2015). Ziel der Pharmakovigilanz ist es, das arzneimittelassoziierte Risiko zu minimieren. Im Vergleich zu einigen europäischen Staaten und den USA stagniert die Pharmakovigilanzforschung in Deutschland unter anderem aufgrund der fehlenden Verknüpfung von relevanten Gesundheitsdatenbanken und limitierter nachhaltiger Forschungsförderung (Douros et al. 2016), obwohl die EU zwischenzeitlich den gesetzlichen Rahmen für eine proaktive Pharmakovigilanz geschaffen hat. Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 5
2.1. Ziel des Pharmakovigilanzleitfadens Aktuell sind Informationen und Daten zu Risiken und Nebenwirkungen zielgerichteter Tumortherapien in der pädiatrischen Hämatologie & Onkologie fragmentiert und lückenhaft. Um diese sicherheitsrelevanten Informationen und Daten für die patientenzentrierte Risikoabschätzung nutzbar zu machen, ist eine systematische Beschaffung, Aufbereitung und Vernetzung sicherheitsrelevanter Informationen und von Pharmakovigilanzdaten erforderlich. Die systematische Vorgehensweise wird anhand eines universell einsetzbaren Leitfaden (=Pharmakovigilanzleitfaden) beschrieben und i) führt die essentiellen Informationsquellen und wichtigsten Akteure auf, ii) definiert die verschiedenen Recherchestrategien für die eingesetzten Informationsquellen und iii) zeigt das systematische Vorgehen zur Vernetzung von substanz- und patientenbezogenen Daten mit dem Ziel der individualisierten Risikoabschätzung auf. Durch die Verknüpfung von substanz- und patientenspezifischen Daten wird die Grundlage für die individualisierte Risikoabschätzung geschaffen. Diese ist Voraussetzung für die rationale, patientenzentrierte Therapieentscheidung im Kontext des Einsatzes zielgerichteter Tumortherapien bei Kindern. Die mittels Pharmakovigilanzleitfaden erstellte individualisierte Risikoabschätzung beschränkt sich auf die Darstellung der Risiken zielgerichteter Tumortherapien; eine Darstellung des potenziellen Nutzens dieser Therapien ist nicht Gegenstand des Leitfadens. Darüber hinaus soll mit dem Leitfaden das Bewusstsein für Toxizitäten und Nebenwirkungen zielgerichteter Tumortherapien geschärft werden. Die Anwendbarkeit des Pharmakovigilanzleitfadens wurde anhand von drei verschiedenen Modellsubstanzen Dasatinib (Sprycel®), Trametinib (Mekinist®) und Palbociclib (Ibrance®) überprüft. Alle drei Substanzen sind bislang ausschließlich für Erwachsene zugelassen. Je nach aktuellem Stand des Entwicklungsprogramms der Substanz werden die Modellsubstanzen in unterschiedlichem Umfang bei Kindern eingesetzt. Die Auswahl der Modellsubstanz erfolgte anhand der Kriterien i) Zeitraum seit Erstzulassung, ii) fehlende pädiatrische Zulassung und iii) Einsatz im INFORM Register (Kap. 2.5.2.2.2). 2.2. Individualisierte Risikoabschätzung Der Pharmakovigilanzleitfaden dient der strukturierten Informationsaufbereitung für die individualisierte Risikoabschätzung. Dabei wird als generischer Prozess festgelegt, welche substanz- und patientenspezifischen Informationen zu erheben und zu verknüpfen sind, um eine patientenzentrierte Therapieentscheidung und eine individuelle Strategie zur Risikoüberwachung/-minimierung zu entwickeln. Im Zuge der individualisierten Risikoabschätzung werden nachfolgende Fragen patientenspezifisch abgeklärt: Besteht ein erhöhtes therapieassoziiertes Risiko Nebenwirkungen zu entwickeln? Welche Organklassen sind besonders betroffen? Welche klinischen Monitoringmaßnahmen werden eingeführt? Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 6
Welche Kombinationstherapien sind zu vermeiden?
Welche Begleitmedikation ist zu vermeiden?
Welche proaktiven Überwachungsmaßnahmen sind erforderlich?
Ist ein therapeutisches Drug monitoring (TDM) angezeigt?
Nutzung der individualisierten Risikoabschätzung
Die nach der Struktur des Leitfadens aufbereiteten Informationen bildet die Grundlage für
die klinisch-pharmakologische Einzelfallberatung. An dieser Einzelfallberatung beteiligen sich
idealerweise klinische und pharmakologische Experten. Die klinisch-pharmakologische
Einzelfallberatung bildet somit die Grundlage für die umfassende Patientenaufklärung im
Vorfeld der Therapieentscheidung.
Darüber hinaus kann der substanzspezifische Teil der individualisierten Risikoabschätzung
generell für die klinisch-pharmakologische Beratung von Ärzten zum Einsatz zielgerichteter
Tumortherapien bei Kindern und Jugendlichen eingesetzt (Star und Edwards 2014) oder für
die Planung pädiatrischer Studien verwendet werden.
2.3. Standardisierung der Prozesse für die individualisierte
Risikoabschätzung anhand des Leitfadens
Der Pharmakovigilanzleitfaden definiert das standardisierte Vorgehen der
Informationsbeschaffung, -aufbereitung und -verknüpfung für die individualisierte
Risikoabschätzung. Die Ergebnisse dieses Prozesses werden nach einem einheitlichen
Schema (Template zur strukturierten Informationsaufbereitung für die individualisierte
Risikoabschätzung gemäß Pharmakovigilanzleitfaden, Annex I) erfasst und dokumentiert.
Die individualisierte Risikoabschätzung gliedert sich in die nachfolgenden Hauptkapitel:
Substanzspezifisches Profil (Kapitel A)
Patientenspezifische Daten und Informationen (Kapitel B)
Individualisierte Risikoabschätzung: Verknüpfung substanz- und patientenspezifischer
Informationen (Kapitel C)
Tabelle 1 zeigt die relevanten Informationsquellen, die für die individualisierte
Risikoabschätzung herangezogen werden.
Tab. 1: Informationsquellen für die individualisierte Risikoabschätzung
Substanzspezifische Informationen Patientenspezifische Informationen
European Public Assessment Report (EPAR) (P) Patientenanamnese (C)
FDA Bewertungen und Zulassungsdokumente (P) Klassifikation der Tumorerkrankung (C)
Dossier zur Nutzenbewertung* (P) Arzneimittelanamnese (C)
Risikomanagementplan (C) Vorhergehende Tumortherapie (C)
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 7Substanzspezifische Informationen Patientenspezifische Informationen
Investigator’s Brochure (C) Begleitmedikation (C)
Development & Periodic Safety Update Reports (C) Bekannte Toxizitäten und Nebenwirkungen (C)
Pädiatrisches Prüfkonzept (C) Pharmakogenomische Daten, optional (C)
Studienregister (P, R)
Literaturrecherche (P)
Spontanmeldungen an Behörden / pharmazeutische
Unternehmen (P, R)
Nebenwirkungsmeldungen Studien und Register (C)
*Nutzenbewertung durch Gemeinsamen Bundesausschuss (G-BA): Einführung zum Stichtag 01.01.2011
C: vertrauliche Informationen; P: öffentlich verfügbare Informationen; R: teilweise veröffentlicht
Substanzspezifisches Profil (Kapitel A)
Für das substanzspezifische Profil werden die Informationsquellen (Tabelle 1) ausgewertet,
sicherheitsrelevante Informationen extrahiert und in drei Subkategorien erfasst.
- A.I: Zusammenfassung der Produktinformationen der Zulassungsbehörden und des
pharmazeutischen Unternehmens (Kap. 2.5.1).
- A.II: Sicherheitsrelevante Informationen beim Einsatz bei Kindern und Jugendlichen
(Kap. 2.5.2).
- A.III: Anlagen zu substanzspezifischen Detailauswertungen zum Nebenwirkungsprofil.
Um eine kontinuierliche Aktualisierung der Dokumente zu gewährleisten, wird der
substanzspezifische Teil für einzelne Arzneimittel in regelmäßigen Abständen (3 Monate)
oder bei substantiellen Änderungen der Zulassung / des Kenntnisstandes zum
Sicherheitsprofil aktualisiert. Zur Nachverfolgung des aktuellen Stands der
Informationsbeschaffung wird für jede Substanz das aktuelle Recherchedatum zu
substanzspezifischen Informationen mit Versionsnummern versehen und als Deckblatt
beigefügt (siehe Template, Annex 1).
Patientenspezifische Daten und Informationen (Kapitel B)
Im Gegensatz zum substanzspezifischen Teil variieren die patientenspezifischen
Informationen individuell für den einzelnen Patienten. Patientenspezifische Daten und
Information werden entweder vom behandelnden Arzt (nach Vorliegen des Einverständnisses
des Patienten) oder direkt vom Patienten anhand eines strukturierten Interviews oder eines
Fragebogens erfasst. Bei Vorliegen des Einverständnisses des Patienten können diese Daten
aufgrund der besonderen Strukturierung der pädiatrischen Onkologie & Hämatologie in
Deutschland teilweise auch von den entitätsspezifischen Studienzentralen der GPOH zur
Verfügung gestellt werden1.
1
http://www.gpoh.de; letzter Aufruf: 28.09.16
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 82.4. Vertraulichkeit
Aufgrund der unterschiedlichen Herkunft der Informationsquellen wird im Pharmakovigilanz-
leitfaden zwischen vertraulichen und nicht-vertraulichen Informationen differenziert.
Vertrauliche Informationen sind:
Informationen, die der Zulassungsinhaber vertraulich zur Verfügung stellt,
Detailinformationen aus dem Spontanmeldesystem (Narrative), die von den
Zulassungsbehörden für die einzelnen Substanzen angefordert werden,
Informationen und Daten aus laufenden, noch nicht veröffentlichten Studien,
Registern und sonstigen Projekten in Abstimmung mit den jeweiligen Projektleitern
und
alle patientenspezifischen Informationen.
In dem Template für die individualisierten Risikoabschätzung (Annex I) ist spezifisch
gekennzeichnet, welche Bestandteile ggf. vertraulich zu behandeln sind. Bei Weitergabe der
konkreten patienten- und substanzbezogenen Recherchen oder Aufbereitungen an Dritte
werden diese Textpassagen gemäß der Vertraulichkeitsabsprachen entsprechend
geschwärzt.
2.5. Recherchestrategie und Informationsaufbereitung
Die Informationen und Daten aus den verschiedenen Quellen (Tabelle 1) werden für die
individualisierte Risikoabschätzung einheitlich erfasst und aggregiert (Abb. 1). Die
Abbildung 1: Schematische Darstellung der strukturierten Aufbereitung und Aggregation der Daten und
Informationen für die individualisierte Risikoabschätzung gemäß Pharmakovigilanzleitfaden. Abkürzungen: EPAR
(European Public Assessment Report), SmPC (Summary of Product Characteristics, RMP (Risk Management
Plan), IB (Investigator’s Brochure), PIP (Paediatric Investigation Plan), DSUR / PSUR (Development / Periodic
Safety Update Report)
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 9Aufbereitung der Daten und Informationen erfolgt anhand der Struktur des Templates für die
individualisierte Risikoabschätzung (Annex I).
2.5.1. Allgemeine substanzspezifische Informationen
Die substanzspezifischen Informationen speisen sich aus verschiedenen
zulassungsrelevanten Dokumenten. Hierfür werden – je nach Stand des
Entwicklungsprogramms der Substanz – Dokumente der European Agency of Medicine
(EMA)2, der FDA und des Zulassungsinhabers herangezogen. Wichtigste Quellen sind hierbei
die arzneimittelspezifische Fachinformation (Summary of Product Characteristics, SmPC), der
European Public Assessment Report und Zulassungsdokumente der FDA.
2.5.1.1. Informationsquellen
2.5.1.1.1. European Public Assessment Report
Die Recherche erfolgt über die Webseite der EMA. Durch Eingabe des Wirkstoffnamens oder
des Arzneimittelnamens als Suchbegriff in der Kategorie „search for medicines (quick
search)“ wird der Zugriff auf den European Public Assessment Report (EPAR), weitere
zulassungsrelevante Dokumente (öffentlich
zugängliche Dokumente zum paediatric
investigation plan (PIP), orphan drug status)
und offene Verfahren (pending decisions)
ermöglicht (Abb. 2).
Der European Public Assessment Report
(Abb. 3) umfasst eine Kurzübersicht des
spezifischen Arzneimittels, Zugriff auf eine
allgemein verständliche Zusammenfassung
des EPAR und verweist im Einzelfall auf
weitere zulassungsrelevante Dokumente
(z. B. summary of risk management plan,
RMP). Detaillierte substanzspezifische
Informationen finden sich unter der Rubrik
Abbildung 2: Recherchestrategie und Zugriff auf
den EPAR und zulassungsrelevante Dokumente bei „Product Information“. Bestandteil der
der European Medicine Agency am Beispiel
Product Information ist die Summary of
Trametinib.
Product Characteristics (SmPC).
Dokumente der initialen Zulassung sowie nach Zulassung stehen in der Rubrik „Assessment
history“ als Download zur Verfügung. In dieser Rubrik findet sich mit dem Dokument
„Procedural steps taken and scientific information after the authorization” auch eine
2
www.ema.europa.eu; letzter Aufruf: 13.10.2016
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 10Gesamtübersicht über zulassungsassoziierte Verfahren und Entscheidungen der EMA im
Lebenszyklus des jeweiligen Arzneimittels.
Anhand des EPAR und der damit verknüpften Dokumente werden die Informationen zum
Zulassungsstatus, Indikationen, Wirkmechanismus, pharmakologischen Eigenschaften und
Interaktionen mit weiteren Informationsquellen abgeglichen und substanzspezifisch
dargestellt.
Abbildung 3: Übersicht über den Inhalt des European Public
Assessment Reports (EPAR) am Beispiel Trametinib. Rot markiert sind
die Rubriken „Product Information“ und „Assessment History“, die durch
Anklicken Zugriff auf weitere Dokumente ermöglichen.
2.5.1.1.2. Produktinformationen zugelassener Arzneimittel der FDA
Die Zulassung neuer Onkologika durch die FDA3 erfolgt um ca. 6 bis 12 Monate früher als in
der EU (Wörmann 2016). Insgesamt existieren ca. 1.600 von der FDA zugelassene,
verschreibungspflichtige Arzneimittel (Fang et al. 2016). Für Wirkstoffe, die in den USA
zugelassen und in Europa noch nicht zugelassen sind, bieten die Dokumente der FDA eine
3
http://www.fda.gov; letzter Aufruf: 25.10.2016
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 11wichtige Informationsquelle für den substanzspezifischen Teil der individualisierten
Risikoabschätzung (Template, Annex I).
Die Datenbank „Drugs@FDA“4 bietet die Möglichkeit anhand des Arzneimittel- oder
Wirkstoffnamens auf zulassungsrelevante Dokumente der FDA zuzugreifen. Vor allem für
bereits seit einem längeren Zeitraum zugelassene Arzneimittel bietet sich die Suche mit dem
Abbildung 4: Recherche in der FDA-Datenbank „Drugs@FDA“ am Beispiel Trametinib. Über die rot
markierten Begriffe ist ein Zugriff auf eine Vielzahl zulassungsrelevanter Dokumente möglich.
Arzneimittelnamen an, da hierdurch ein direkter Zugriff auf die initialen
Zulassungsdokumente möglich ist.
Eine Übersicht der zulassungsrelevanten Dokumente ist über den Link „Label Information“
oder alternativ über den Link „Approval History, Letters, Reviews, and Related Documents“
möglich (Abb. 4). Die Dokumente sind chronologisch sortiert. Die jeweilige(n)
Produktinformation(en) (Label), die Beurteilungen der Substanz für die initiale Zulassung
(substanzspezifisches Informationspaket) sowie weitere zulassungsrelevante Dokumente
(Korrespondenz, Labeländerungen etc.) stehen als Download zur Verfügung. Für die
individualisierte Risikoabschätzung werden die aktuellste Version der Produktinformation
sowie Teile des initialen, substanzspezifischen Informationspakets (Medical Review,
Pharmacology Review, Clinical Pharmacology Biopharmaceutics Review, Risk Assessment and
4
http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm; letzter Aufruf: 10.10.2016
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 12Risk Mitigation Review) ausgewertet. Dabei ist zu berücksichtigen, dass Art und Umfang der Reviews substanzspezifisch variieren können. Allgemeine Gliederung der FDA Produktinformationen Die Produktinformationen FDA zugelassener Arzneimittel enthalten jeweils eine halbseitige Zusammenfassung der wichtigsten Informationen zur Wirksamkeit und Sicherheit eines Arzneimittels („highlights of prescribing information“). Diese Informationen (Tab. 2) gewährleisten einen schnellen Überblick über die arzneimittelspezifischen Charakteristika und werden als substanzspezifische Informationsquelle für die individualisierte Risikoabschätzung verwendet. Bei nicht in Europa zugelassenen Arzneimitteln wird die vollständige Produktinformation mit insgesamt 17 Kapiteln (Fang et al. 2016) als Informationsquelle für die substanzspezifischen Informationen genutzt. Tab. 2: Zusammenfassung essentieller Informationen („Highlights of Prescribing Information“) zur Wirksamkeit und Sicherheit FDA zugelassener Arzneimittel Bezeichnung Kommentar Limitations Statement Hinweis auf Kurzfassung Product Names Name Arzneimittel und Wirkstoff, Formulierung und Gebrauch Initial U.S. Approval Datum der Erstzulassung durch FDA Boxed Warnings (optional) Optischer Warnhinweis auf besonders schwerwiegende Nebenwirkungen Recent Major Changes Kategorie, Monat, Jahr der substantiellen Änderung Indications and Usage Kurzfassung, Hinweis auf Kapitel 1 der vollständigen Produktinformation Dosage and Administration Kurzfassung, Hinweis auf Kapitel 2 der vollständigen Produktinformation Dosage Forms and Strengths Kurzfassung, Hinweis auf Kapitel 3 der vollständigen Produktinformation Contraindications Kurzfassung, Hinweis auf Kapitel 4 der vollständigen Produktinformation Warnings and Precautions Kurzfassung, Hinweis auf Kapitel 5 der vollständigen Produktinformation Adverse Reactions Kurzfassung, Hinweis auf Kapitel 6 der vollständigen Produktinformation Drug Interactions Kurzfassung, Hinweis auf Kapitel 7 der vollständigen Produktinformation Use in Specific Populations Kurzfassung, Hinweis auf Kapitel 8 der vollständigen Produktinformation Vor allem für Arzneimittel, die bislang nicht in Europa aber in den USA bereits zugelassen sind, bieten die FDA-Dokumente eine wichtige Informationsquelle zum Zulassungsstatus, Indikationen, Wirkmechanismus, pharmakologischen Eigenschaften, Interaktionen und Sicherheitsprofil. Darüber hinaus bieten der pharmakologische und klinische- pharmakologische Reviews eine sehr gute Übersicht über die pharmakologischen Eigenschaften und präklinische Daten, die substanzspezifisch (Template, Annex I) erfasst werden. Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 13
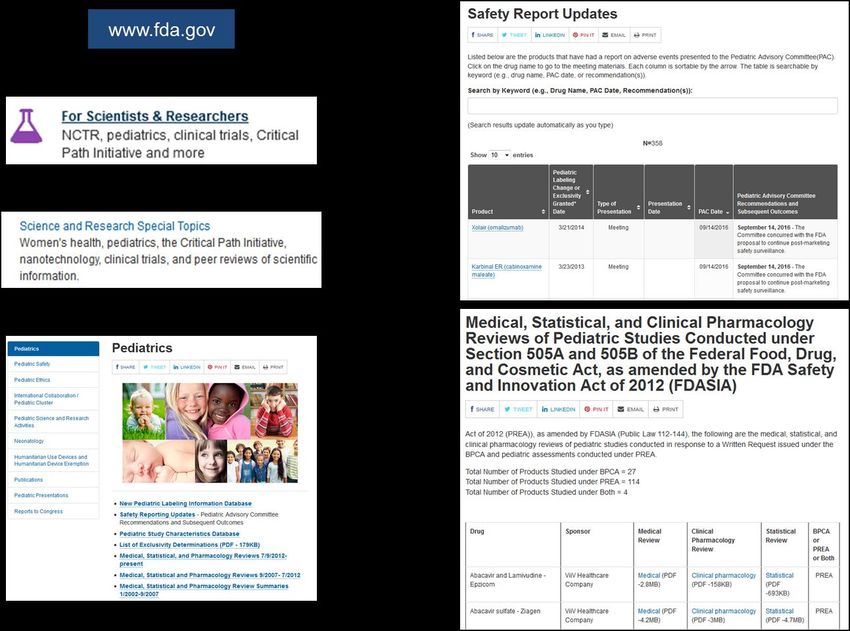
Dokumente der FDA zum Einsatz zielgerichteter Tumortherapien bei Kindern Der Approval Letter der initialen Zulassung enthält ein Kapitel zur pädiatrischen Beurteilung des jeweiligen Arzneimittels. Mit Hinweis auf die gesetzliche Grundlage (Pediatric Research Equity Act) ist definiert, ob der Zulassungsinhaber pädiatrische Studien zurückstellen kann, von diesen befreit ist oder pädiatrische Studien nicht anwendbar sind. Darüber hinaus veröffentlicht die FDA weitere Dokumente zum pädiatrischen Einsatz, die für die individualisierte Risikoabschätzung genutzt werden können. Die Recherchestrategie auf der Webseite der FDA ist in Abb. 5 dargestellt. Abbildung 5: Recherche von sicherheitsrelevanten Informationen für den pädiatrischen Gebrauch von Arzneimitteln bei der US-amerikanischen Zulassungsbehörde FDA. Die Safety Report Updates (Abb. 5) umfassen Arzneimittel, für die dem Pediatric Advisory Committee (PAC) ein Bericht über Nebenwirkungen vorgelegt wurde. Eine Detailsuche der Liste ist anhand des Arzneimittelnamens oder nach alphabetischer Listensortierung möglich. Die Unterlagen, die dem PAC vorgelegt wurden, sind durch Klicken des Arzneimittelnamens verfügbar. Die Liste der Medical, Statistical & Clinical Pharmacology Reviews (Abb. 5) ist unterteilt in verschiedene Zeiträume und alphabetisch sortiert. Sie umfasst, je nach Arzneimittel, verschiedene Reviews zu pädiatrischen Studien. Diese Dokumente stehen als Download zur Verfügung. Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 14
Für die individualisierte Risikoabschätzung ist zu überprüfen, ob für die jeweiligen Substanzen Safety Report Updates oder zusätzliche Reviews verfügbar sind. Dokumente der FDA vor Erstzulassung eines Arzneimittels Mit Zulassung eines Arzneimittels werden sicherheitsrelevante Dokumente durch die Zulassungsbehörden veröffentlicht (siehe oben, Kap. 2.5.1.1.1 und 2.5.1.1.2). Um auch Informationen zu Wirkstoffen, die sich im frühen klinischen Entwicklungsprogramm befinden und noch nicht zugelassen sind, für die individualisierte Risikoabschätzung nutzbar zu machen, kann auf wissenschaftliche Bewertungen der FDA zurückgegriffen werden. Hierdurch wird eine vorläufige Abschätzung des Sicherheitsprofils neuer, noch nicht zugelassener Substanzen, ermöglicht. Für nicht zugelassene Arzneimittel hat sich eine Freitextsuche auf der FDA Website als sinnvolle Recherchestrategie erwiesen. Hierzu wird der Wirkstoffname als Suchtext auf der Webseite der FDA verwendet (Abb. 6). Abbildung 6: Freitextsuche auf der Webseite der FDA für nicht zugelassene Wirkstoffe. Die rote Markierung zeigt das Textfeld für die Eingabe des Wirkstoffnamens. Am Beispiel des aktuell nicht von der FDA zugelassenen Wirkstoffs Tazemetostat (Stand: 25.10.2016) ist exemplarisch das Ergebnis der Freitextsuche dargestellt (Abb. 7). Als Download stehen hier Dokumente für die Bewertung des Wirkstoffs durch das Pediatric Oncology Subcommittee of the Oncologic Drugs Advisory Committee der FDA zur Verfügung. Je nach Substanz enthalten die FDA Dokumente Informationen zum Wirkmechanismus, zum Sicherheitsprofil und zum weiteren Entwicklungsprogramm. Diese können für den substanzspezifischen Teil der individualisierten Risikoabschätzung (Template, Annex I) verwendet werden. Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 15
Abbildung 7: Ergebnisse der Freitextsuche auf der Webseite der FDA am Beispiel Tazemetostat. Das Meeting
Information Package enthält wichtige substanzspezifische Informationen, die als Download verfügbar sind.
2.5.1.1.3. Produktinformationen des Zulassungsinhabers
Bei den substanzspezifischen Informationen, die der Zulassungsinhaber (Marketing
authorisation holder, MAH) für die individualisierte Risikoabschätzung zur Verfügung stellt,
handelt es sich in der Regel um vertrauliche Dokumente (Tabelle 1, Kap. 2.3). Ein Zugriff
oder eine Einsicht in substanzspezifische Dokumente hängt entsprechend von den
individuellen Verhandlungen mit dem MAH ab.
Prüferinformation (Investigator’s Brochure, IB), vertrauliche Information
Die Investigator’s Brochure (IB) enthält gemäß der Guideline for Good Clinical Practice5
neben den physikalischen, chemischen und pharmazeutischen Eigenschaften und
Darreichungsformen folgende, für die Risikoabschätzung relevante, Informationen:
5
http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002874.pdf;
S. 34-38, letzter Aufruf: 12.10.2016
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 16A. Nicht klinische Studien
Pharmakologie
Pharmakokinetik, Metabolismus beim Tier
Toxikologie
B. Effekte im menschlichen Organismus
Pharmakokinetik und Metabolismus am Mensch
Sicherheit und Wirksamkeit
Erfahrungen aus dem Markt
Darüber hinaus enthält die IB eine Zusammenfassung der Daten und Informationen für den
Prüfer in klinischen Prüfungen. Durch die regelmäßige Aktualisierung der IB sind
sicherheitsrelevante Informationen weitestgehend auf dem aktuellen Stand.
Pädiatrisches Prüfkonzept (Paediatric Investigation Plan, PIP), vertrauliche
Information
Gemäß der Guidance for Industry on pediatric study plans6 sind im PIP folgende
Informationen zu pädiatrischen Studien enthalten:
Generelle Strategie
Pädiatrische pharmakokinetische bzw. -dynamische Studien
Klinische Studien zur Wirksamkeit und Sicherheit
Zusammenfassung aller geplanten und/oder laufenden Studien
Details aller geplanten und/oder laufenden pädiatrischen Studien
Beschreibung anderer Studien
Modellierungs- und Simulationsstudien
Extrapolationsstudien
Bei den PIPs handelt es sich in aller Regel um vertrauliche Dokumente des
Zulassungsinhabers. Die EMA veröffentlicht lediglich die Entscheidungen des Paediatric
Committees der (PDCO) zum PIP, die jeweils über die arzneimittelassoziierten Webseiten des
EPAR (Kap. 2.5.1.1.1) einsehbar sind. Da der PIP den Konsens über eine verantwortbare
Entwicklungsstrategie bei Kindern darstellt, ergeben sich aus den publizierten
Entscheidungen des PDCO teilweise Rückschlüsse zum pädiatrischen Entwicklungsprogramm
und damit verbundenen sicherheitsrelevanten Aspekten, die für die individualisierte
Risikoabschätzung von Bedeutung sind. Eine umfassende Bewertung des Wirkstoffs für die
individualisierte Risikoabschätzung ist jedoch anhand der Entscheidungen des PDCO nicht
möglich.
6
http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm360507.pdf;
letzter Aufruf: 12.10.2016
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 17Development Safety Update Report (DSUR) / Periodic Safety Update Report
(PSUR), vertrauliche Information
DSUR
Der DSUR bietet einen Überblick über sicherheitsrelevante Informationen aus dem klinischen
Entwicklungsprogramm, die regelmäßig aktualisiert und mit dem bisherigen Wissensstand
abgeglichen werden. Ziel des DSUR ist es, sicherheitsrelevante Informationen aus klinischen
Studien zusammenzufassen, aufzubereiten und zu evaluieren. Aufgrund der Struktur von
DSUR und PSUR sind inhaltliche Überschneidungen in den Berichten möglich. Jeder Bericht
steht jedoch für sich allein, da die Berichte unterschiedliche Zielsetzungen verfolgen.
Patientenspezifische Informationen können aus den Line Listings im DSUR nicht extrahiert
werden, da es sich hierbei um aggregierte Daten handelt. Für die substanzspezifischen
Informationen der individualisierten Risikoabschätzung sind die Kapitel 3, 4 sowie 7-20
gemäß der in der ICH Guideline E2F beschriebenen DSUR-Struktur7 relevant.
PSUR
Der PSUR, dessen Inhalt und Format sich in der EU an der Guideline for the Periodic Benefit-
Risk Evaluation Report (PBRER)8 orientiert, dient der kontinuierlichen Evaluation des Nutzen-
Risiken-Verhältnisses eines Arzneimittels, auch wenn dieses bereits zugelassen ist.
Essentieller Bestandteil des PSUR ist dabei die regelmäßige Bewertung sicherheitsrelevanter
Informationen und Daten. Dabei kann die Berichtsfrequenz in Abhängigkeit vom Zeitpunkt
der Zulassung zwischen 6 Monaten und mehreren Jahren variieren. Patientenspezifische
Informationen können aus dem PSUR nicht extrahiert werden, da es sich hierbei um
aggregierte Daten handelt. Für die substanzspezifischen Informationen der individualisierten
Risikoabschätzung sind die Kapitel 3, 4, 6-12, 14-16 sowie 18-19 des PSUR relevant.
Risikomanagementplan (RMP), vertrauliche Information
Das Risikomanagement gemäß der Guideline on Good Pharmacovigilance Practice (GVP)9
umfasst die Aspekte i) Charakterisierung des Sicherheitsprofils eines Arzneimittels
einschließlich der Bewertung welche Informationen bekannt sind und welche fehlen, ii)
Planung von Pharmakovigilanz-Maßnahmen um arzneimittelassoziierte Risiken zu
charakterisieren und neue Risiken zu identifizieren sowie die Kenntnisse zum Sicherheitsprofil
auszubauen und iii) Planung und Implementierung von Risikominimierungsmaßnahmen und
Bewertung der Wirksamkeit dieser Maßnahmen. Die damit verbundenen Prozesse werden im
RMP, der kontinuierlich während des gesamten Lebenszyklus eines Arzneimittels aktualisiert
wird, dokumentiert.
7
http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/09/WC500097061.pdf;
letzter Aufruf: 12.10.2016
8
http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2012/12/WC-
500136402.pdf; letzter Aufruf: 12.10.2016
9
http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2016/02/WC-
500202424.pdf; letzter Aufruf: 12.10.2016
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 18Informationen für die individualisierte Risikoabschätzung finden sich im Part II “Safety
Specification” mit den Teilmodulen SI – SVIII, im Part III „Pharmacovigilance Plan“, im
Part V „Risk Minimisation Measures“ sowie im Part VI „Summary of Activities in the Risk
Management Plan by Medicinal Product“.
Seit dem 01.01.2016 werden Zusammenfassungen der RMP vom BfArM sukzessive über das
Portal für Arzneimittelinformationen des Bundes und der Länder (PharmNet.Bund)10
veröffentlicht. Eine weitere Informationsquelle für eine Zusammenfassung des RMP ist der
EPAR (Kap. 2.5.1.1.1) oder das Dossier zur Nutzenbewertung (siehe nachfolgendes Kap.
2.5.1.1.4).
Für die individualisierte Risikoabschätzung ist zu berücksichtigen, dass die Aussagekraft der
Zusammenfassungen der RMP begrenzt und nicht zwingend ausreichend ist. Diese Quelle ist
aber geeignet, Problemfelder für den frühen Einsatz bei Kindern zu identifizieren.
2.5.1.1.4. Weitere Informationsquellen für Produktinformationen
Arzneimitteldatenbanken
Für die umfassende Darstellung der substanzspezifischen Informationen für die
individualisierte Risikoabschätzung werden die Datenbanken DrugBank (Version 5.0) und
DRUGDEX® verwendet. Die Entscheidung für diese beiden Datenbanken erfolgte aufgrund
der lokalen Gegebenheiten. Prinzipiell kommen auch andere Arzneimitteldatenbanken für die
Informationsrecherche in Betracht.
DrugBank ist eine frei verfügbare Online-Datenbank11, die chemische, pharmakologische und
pharmazeutische Daten eines Arzneimittels mit Informationen der arzneimittelassoziierten
Zielstruktur (Sequenz, Struktur, Pathway) verknüpft. Die Datenbank enthält eine detaillierte
Übersicht der ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity)
Eigenschaften sowie der Arzneimittelinteraktionen. Schwerpunkt der Datenbank sind von der
FDA zugelassene Arzneimittel. Die Datenbank bietet über Verlinkungen Zugriff auf weitere,
externe Datenbanken und relevante Literatur.
DRUGDEX® ist eine gebührenpflichtige Datenbank mit über 2.300 Arzneistoffmonographien
mit ausführlichen Informationen zur Pharmakodynamik, Pharmakokinetik, Toxikologie und
Arzneimittelsicherheit. Die Datenbank bietet zu den einzelnen Rubriken sowohl
Kurzinformationen („schnelle Antworten“) als auch detaillierte Antworten. Basis der
Datenbankinformation bilden Dokumente der FDA und Literaturrecherchen. Dabei
unterliegen die Datenbankeinträge regelmäßigen Qualitätskontrollen (persönliche
Kommunikation mit dem Unternehmen). Die Datenbank ist nicht frei verfügbar, jährliche
Lizenzgebühren werden erhoben.
10
https://www.pharmnet-bund.de/static/de/index.html; letzter Aufruf: 25.10.2016
11
http://www.drugbank.ca/; letzter Aufruf: 12.10.2016
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 19Informationen aus den Arzneimitteldatenbanken werden für die individualisierte
Risikoabschätzung mit den Zulassungsdokumenten abgeglichen und für die
substanzspezifischen Informationen (Template, Annex I) verwendet.
Dossier zur Nutzenbewertung gemäß §35a SGB V, frei verfügbar
In Deutschland sind pharmazeutische Unternehmen seit dem 01.01.2011 verpflichtet, bereits
zur Markteinführung eines neuen Wirkstoffs bzw. bei der Zulassung neuer
Anwendungsgebiete, ein Dossier zum Nutzen des Arzneimittels gemäß §35a SGB V
vorzulegen. Die Nutzenbewertung erfolgt durch den Gemeinsamen Bundesausschuss (G-BA).
Die Recherche erfolgt über die Webseite des G-BA12 durch Eingabe des Wirkstoffnamens
(Abb. 8). Die Einzelmodule des Dossiers, Informationen zur zweckmäßigen
Vergleichstherapie, Dokumente zur Nutzenbewertung und Beschlüsse stehen über
Verlinkungen ebenfalls als Download zur Verfügung (Abb. 9). Für die individualisierte
Risikoabschätzung werden ausschließlich Informationen aus Modul 3 des Dossiers zur
Nutzenbewertung verwendet. Dabei finden sich die für die individualisierte Risikoabschätzung
relevanten Informationen im Kapitel 3.4 des Moduls 313 (zweckmäßige Vergleichstherapie,
Anzahl der Patienten mit therapeutisch bedeutsamem Zusatznutzen, Kosten der Therapie für
die GKV, Anforderungen an eine qualitätsgesicherte Anwendung). Dieses Kapitel enthält eine
Zusammenfassung des Risikomanagementplans und führt Vorschläge zur Risikominimierung
(Routine und zusätzliche Maßnahmen) auf. Dabei werden die Risiken in die Kategorien i)
wichtige identifizierte Risiken, ii) wichtige potenzielle Risiken und iii) wichtige fehlenden
Informationen aufgegliedert. Diese Unterlagen werden mit der Zusammenfassung des
Risikomanagementplans (RMP) abgeglichen und für den substanzspezifischen Teil der
individualisierten Risikoabschätzung genutzt.
Diese Quelle analysiert Studien zum Nutzen und hat vor allem sozialrechtliche Bedeutung.
Spezifische Fragen zu Kindern oder gar zum off-label Einsatz stehen naturgemäß nicht im
Vordergrund. Trotzdem können kumulierte Risikodaten dieses Dokumentes auch Hinweise
für den reflektierten Einsatz bei Kindern und Jugendlichen geben.
12
http://www.g-ba.de; letzter Aufruf: 15.10.2016
13
https://www.g-ba.de/downloads/17-98-3526/2013-04-18_Anl2_5_Modul3.pdf; letzter Aufruf: 24.10.2016
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 20Abbildung 8: Online-Recherche zu substanzspezifischen Dossiers der Nutzenbewertung
durch den G-BA.
Abbildung 9: Übersicht der Dokumente des Dossiers zur Nutzenbewertung durch den G-
BA am Beispiel Trametinib. Die verschiedenen Module (Modul 1-4) des Dossiers, die
Nutzenbewertung sowie Beschlüsse (rote Markierung) stehen ebenfalls über Verlinkungen
als Download zur Verfügung.
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 21Weitere Risikoinformationen zu zugelassenen Arzneimitteln
Rote-Hand-Briefe
In Deutschland sind pharmazeutische Unternehmen gesetzlich verpflichtet (§11 a, Absatz 2
AMG) therapierelevante Änderungen der Fachinformation den Fachkreisen in geeigneter
Form zugänglich zu
machen. Die Kommuni-
kation erfolgt in Form der
Rote-Hand-Briefe. Durch
diese wird gewährleistet,
dass die Fachkreise über
neue, unerkannte Arznei-
mittelrisiken und Maß-
nahmen zu deren
Vermeidung informiert
werden. Rote-Hand-Briefe
werden in Absprache mit
den Zulassungsbehörden
verbreitet und werden auf
den Webseiten der
Zulassungsbehörden ver-
öffentlicht.
Abbildung 10: Recherche zu Rote-Hand-Briefen am Beispiel Sprycel®
(Wirkstoff: Dasatinib). Durch Eingabe des Arzneimittelnamens werden die Die Recherche erfolgt
Rote-Hand-Briefe chronologisch gelistet und stehen als Download zur
Verfügung. Der Recherchepfad auf der BfArM-Webseite ist rot markiert.
über die Webseite des
BfArM14 durch Angabe
des Arzneimittelnamens oder des Wirkstoffnamens (Abb. 10). Dabei erwies sich die
Recherche mit dem Arzneimittelnamen als spezifischer gegenüber der Suche mit dem
Wirkstoffnamen. Bei Verwendung des Wirkstoffnamens werden auch solche Rote-Hand-
Briefe angezeigt, die nicht direkt mit dem Arzneimittel korrelieren.
Für biomedizinische Arzneimittel (monoklonale Antikörper, Immunglobuline, Sera,
Gerinnungsfaktoren) werden Rote-Hand-Briefe in Deutschland in Kooperation mit dem Paul-
Ehrlich-Institut (PEI)15 veröffentlicht. Entsprechend erfolgt die Recherche für diese Wirkstoffe
auf der Webseite des PEI nach dem für das BfArM beschriebenen Algorithmus.
FDA Safety Alerts
Die FDA kommuniziert Risikoinformationen online über das MedWatch System. Über das
Portal „Medical Product Safety Information“ können durch Eingabe des Arzneimittelnamens
oder des Wirkstoffs sicherheitsrelevante Informationen recherchiert werden (Abb. 11). Die
14
www.bfarm.de; letzter Aufruf: 25.10.2016
15
http://www.pei.de/DE/arzneimittelsicherheit-vigilanz/pharmakovigilanz/rote-hand-briefe/rote-hand-briefe-
node.html; letzter Aufruf: 09.11.2016
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 22Recherche mit dem Arzneimittelnamen ist spezifischer als mit dem Wirkstoffnamen. Neben
den „Drug safety communications“ enthält das Rechercheergebnis auch eine Übersicht aller
„Safety Labeling Changes (SLC)“. Die SLCs listen die einzelnen Änderungen der
Produktinformation auf.
Die Rote-Hand-Briefe und die Drug safety communications werden substanzspezifisch für die
individualisierte Risikoabschätzung genutzt (Template, Annex I).
Abbildung 11: FDA Internetportal MedWatch zur Recherche von sicherheitsrelevanten Informationen für
Arzneimittel. Die rote Markierung zeigt den Recherchepfad auf der FDA-Webseite.
2.5.2. Sicherheitsrelevante Informationen beim Einsatz von zielgerichteten
Tumortherapien bei Kindern und Jugendlichen
2.5.2.1. Meldungen aus Spontanmeldesystemen
Da klinische Prüfungen bei Kindern besonderen Rahmenbedingungen unterliegen (Hartford
et al. 2006), bietet sich die Nutzung bereits existierender Datenquellen – wie den
Spontanmeldesystemen – für die Verbesserung der Arzneimittelsicherheit bei Kindern an
(Osokogu et al. 2015). Dabei können Daten zum Nebenwirkungsprofil, die beim Einsatz im
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 23medizinischen Versorgungsalltag gewonnen werden, trotz der beschränkten Übertragbarkeit
auf Kinder, für die Risikoabschätzung für Kinder genutzt werden. Durch Spontanmeldungen
können zudem sicherheitsrelevante Informationen zum off-label-Einsatz generiert werden
(Stammschulte et al. 2010).
Für die individualisierte Risikoabschätzung werden Daten der drei Spontanmeldesysteme
VigiAccess™ (WHO),
EudraVigilance (EU) und
BfArM ADR-Datenbank (DE) bzw. PEI ADR-Datenbank (DE)
genutzt. Da für die Verwendung von Spontanmeldungen aus dem FDA System16 spezifische
technische und statistische Werkzeuge für die Datenverarbeitung erforderlich sind, wird auf
die Auswertung der Spontanmeldungen der FDA verzichtet.
2.5.2.1.1. VigiAccess™
Die Datenbank VigiAccess™ beruht auf VigiBase®, der größten globalen ADR-Datenbank, die
seit 1968 betrieben wird (Kroger et al. 2015). Dabei erfolgen die Meldungen an VigiBase® in
regelmäßigen Abständen (quartalsweise Aktualisierung) durch nationale Zentren von über
110 Ländern. Die Datenbank17
enthält über 14 Millionen
Einzelberichte (Stand: Sept.
2016) und umfasst über
100.000 verschiedene
Arzneimittel. Seit 2015 ist eine
frei zugängliche Recherche zu
Verdachtsfällen von uner-
wünschten Arzneimittelwirk-
ungen über die Datenbank
VigiAccess™ möglich18. Die
Recherche erfolgt durch Angabe
des Wirkstoff- oder des
Arzneimittelnamens (Abb. 12).
Abbildung 12: Recherche zu ADRs in der Datenbank VigiAccess™ der Die ADRs werden nach Anzahl,
WHO am Beispiel Palbociclib (Ibrance®). geografischer Verteilung, Alters-
gruppen und Geschlecht sowie
nach Meldejahr angezeigt und sind nach der MedDRA- Nomenklatur klassifiziert und online
16
http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects
/ucm082193.htm; letzter Aufruf: 24.10.2016
17
http://www.who-umc.org/DynPage.aspx?id=98082&mn1=7347&mn2=7252&mn3=7322&mn4=7326; letzter
Aufruf: 24.10.2016
18
http://www.who-umc.org/DynPage.aspx?id=132936&mn1=7347&mn2=7252&mn3=7254&mn4=7753; letzter
Aufruf: 24.10.2016
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 24abrufbar (Abb. 13). Ein Export der VigiAccess-Daten ist nicht möglich. Für die
individualisierte Risikoabschätzung werden die Daten gemäß Kap. 2.5.2.1.4 manuell
aufbereitet und verwertet.
Abbildung 13: Klassifizierung der ADRs nach der MedDRA-Nomenklatur
von der SOC (Systemorganklasse) bis zum Preferred Term am Beispiel
Palbociclib und Herzerkrankungen.
2.5.2.1.2. EMA ADR-Datenbank
Die europäische ADR-Datenbank19 speist sich aus den verschiedenen Komponenten des
EudraVigilance-Systems20 der EMA. Dabei werden sowohl Verdachtsfälle von
Nebenwirkungen aus dem Spontanmeldesystem als auch aus klinischen Prüfungen im
EudraVigilance-System erfasst. Daten aus dem EudraVigilance-System werden in der
europäischen ADR-Datenbank veröffentlicht. Eine Aktualisierung der Daten findet jeweils zu
Beginn des Folgemonats für den vorhergehenden Monat statt.
Die ADR-Datenbank umfasste (Stand: 31.12.2015) insgesamt 9.530.295 Adverse Reaction
Reports, entsprechend 6.212.064 individuelle Patientenberichte (=Individual Case Safety
Reports, ICSR) (Jahresbericht 2015, European Medicines Agency).
19
http://www.adrreports.eu/; letzter Aufruf: 24.10.2016
20
http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/q_and_a/q_and_a_detail_000166.jsp; letzter
Aufruf: 24.10.2016
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 25Von besonderem Interesse für die individualisierte Risikoabschätzung sind ADRs bei Kindern
und Jugendlichen (Altersgruppe ≤ 18 Jahre). Ein Zugriff auf die Narrative von pädiatrischen
ADRs ist nicht möglich. Für Forschungszwecke kann ein separater Antrag zur Bereitstellung
von erweiterten Datensätzen gestellt werden. Da die ADR-Narrative auch in erweiterten
Datensätzen für wissenschaftliche Zwecke nicht zur Verfügung stehen, werden für die
individualisierte Risikoabschätzung nur die öffentlich zugänglichen Daten aus der ADR-
Datenbank ausgewertet.
Im Gegensatz zu der Recherche in den Datenbank VigiAccess (Kap. 2.5.2.1.1) liefert die
Suche mit dem Wirkstoffnamen und dem Arzneimittelnamen in der EMA ADR-Datenbank
unterschiedliche Ergebnisse. Dabei zeigt sich, dass die Recherche mit dem Wirkstoffnamen
deutlich mehr ADRs umfasst. Im Zuge der individualisierten Risikoabschätzung wird daher
die Recherche in der ADR-Datenbank der EMA ausschließlich mit dem Wirkstoffnamen
durchgeführt, eine Einschränkung der Recherche ist nicht möglich.
Die Rechercheergebnisse (Abb. 14) werden online in folgenden vier verschiedenen Rubriken
dargestellt:
- Number of Individual cases: Einfache Zählung der individuellen Fälle kategorisiert nach
Geschlecht / nach Altersgruppen / nach geografischer Region (EEA/Non-EEA)
- Number of Individual Cases By Reaction Groups: Graphische Darstellung der Anzahl
der ADRs kategorisiert nach SOCs in Abhängigkeit von der Altersgruppe / Geschlecht /
Berichterstatter / geografischer Region
- Number of Individual Cases for a selected Reaction Group: Graphische Darstellung der
Anzahl der ADRs für einzelne SOCs (Auswahlfunktion) in Abhängigkeit von Alter und
Geschlecht / Berichterstatter / geografischer Region
- Number of Individual Cases for a selected Reaction: Graphische Darstellung der Anzahl
einzelner ADRs (Auswahl über SOC) in Abhängigkeit von Alter und Geschlecht /
Berichterstatter / Outcome
Ein Download der korrelierenden Tabellen ist nur für die Rubrik Number of Individual Cases
möglich. Für die weiteren Rubriken können die korrelierenden Daten durch Anklicken der
Balken und der Bestätigung „Switch to table“ eingesehen werden. Ein Download dieser
Tabellen ist nicht möglich. Für die individualisierte Risikoabschätzung werden die Daten
gemäß Kap. 2.5.2.1.4 manuell aufbereitet und verwertet (Kap. A.II.1 Template , Annex I).
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 26Abbildung 14: Rechercheergebnisse in der EMA ADR-Datenbank am Beispiel Trametinib. Rot markiert sind die verschiedenen Rubriken zur Darstellung der ADRs bzw. ICSRs. Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 27
2.5.2.1.3. BfArM und PEI ADR-Datenbank
BfArM ADR-Datenbank
Um auch Daten aus dem nationalen Spontanmeldesystem für die individualisierte
Risikoabschätzung nutzen zu können, wird die Recherche
auf die ADR-Datenbank des BfArM ausgeweitet. Diese
Datenbank umfasst seit 1995 gemeldete Verdachtsfälle von
unerwünschten Arzneimittelwirkungen (suspected adverse
drug reactions, ADRs). Die in der ADR-Datenbank erfassten
Verdachtsfälle wurden nicht im Rahmen von klinischen
Prüfungen gemeldet. Nicht in die Datenbank aufgenommen
sind zudem Verdachtsfälle, wenn der Hinweis auf eine
Literaturquelle vorhanden ist und der Ersteingang des
Verdachtsfalles vor dem 1.1.2005 liegt. Eine Aktualisierung
der Datenbank findet jeweils zum Monatsbeginn statt.
Zeitverzögerungen bei der Aktualisierung der Verdachtsfälle
können auftreten, sofern diese zeitnah zum Monatswechsel
gemeldet wurden21.
Im Jahr 2015 wurden in Deutschland insgesamt 27.334
Berichte zu Verdachtsfällen von Arzneimittelneben-
wirkungen erfasst (Kayser und Paeschke N 2016), dies
entspricht 57.111 gemeldeten ADRs (Bundesinstitut für
Arzneimittel und Medizinprodukte 2016, 2016). Analog zur
EMA ADR-Datenbank (Kap. 2.5.2.1.2) führt die Suche mit
dem Wirkstoffnamen in der BfArM-Datenbank zu deutlich
mehr ADR-Verdachtsfällen. Entsprechend erfolgt die
Recherche ausschließlich mit dem Wirkstoffnamen, ohne
weitere Einschränkung hinsichtlich Nebenwirkung,
Anwendung, Alter, Geschlecht oder Meldejahr. Die
Rechercheergebnisse sind in verschiedenen Tabellen
zusammengefasst (Abb. 15). Da für die Verteilung der
Nebenwirkungen pro SMQ (standardisierte
Zusammenstellung von Nebenwirkungsbegriffen) bei der
Recherche eine vorhergehende Einschränkung der
Nebenwirkungen erforderlich ist, enthält die
individualisierte Risikoabschätzung ausschließlich einfache
Abbildung 15: Rechercheergebnisse ADR-Zählungen und Verteilungen der ADR pro SOC.
in der BfArM ADR-Datenbank am
Beispiel Trametinib. Die Verteilung der Eine übersichtliche Zusammenfassung der tabellarischen
Nebenwirkungen pro SMQ werden Ausgabe der Rechercheergebnisse ist nicht vorhanden, da
nicht für die individualisierte
Risikoabschätzung genutzt.
21
http://nebenwirkung.bfarm.de/apex/f?p=100:1:0:::::; letzter Aufruf: 10.10.2016
Leitfaden zur individualisierten Risikoabschätzung zielgerichteter Tumortherapien, V.1.3 28Sie können auch lesen

















































