Morphologische Analyse dendritischer Spines und synaptischer Kontakte in ProSAP/Shank-Mausmutanten im dorsolateralen Striatum - OPARU
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Universität Ulm
Institut für Anatomie und Zellbiologie
Direktor: Prof. Dr. Tobias M. Böckers
Morphologische Analyse dendritischer Spines und
synaptischer Kontakte in ProSAP/Shank-Mausmutanten
im dorsolateralen Striatum
Dissertation
zur
Erlangung des Doktorgrades der Medizin
der Medizinischen Fakultät der Universität Ulm
vorgelegt von
Sarah-Catharina Kroegel
geboren in Freiburg i. Br.
2019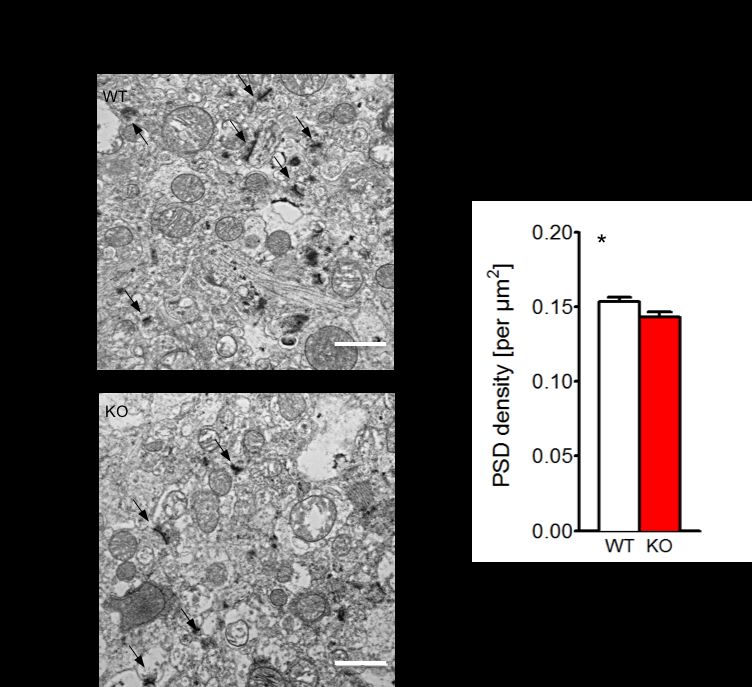
Amtierender Dekan: Prof. Dr. Thomas Wirth 1. Berichterstatter: Prof. Dr. Tobias Böckers 2. Berichterstatter: Prof. Dr. Bernd Knöll Tag der Promotion: 15.04.2021
Meinen geliebten Eltern
INHALTSVERZEICHNIS
Inhaltsverzeichnis
1 EINLEITUNG............................................................................................................... 1
1.1 AUTISMUS-SPEKTRUM-STÖRUNGEN ............................................................................ 1
1.1.1 ÜBERBLICK ............................................................................................................. 1
1.1.2 SYNDROMALER AUTISMUS ....................................................................................... 4
1.2 DIE SYNAPSE ............................................................................................................. 7
1.2.1 ÜBERSICHT ............................................................................................................. 7
1.2.2 MORPHOLOGIE DENDRITISCHER SPINES.................................................................... 9
1.2.3 DIE POSTSYNAPTISCHE DICHTE .............................................................................. 11
1.3 DIE PROSAP-/SHANK-PROTEINFAMILIE .................................................................. 12
1.4 PROSAP/SHANK-MAUSMODELLE ............................................................................. 15
1.4.1 SHANK1 ................................................................................................................ 16
1.4.2 SHANK2 ................................................................................................................ 16
1.4.3 SHANK3 ................................................................................................................ 19
1.4.4 ZUSAMMENFASSUNG DER MORPHOLOGISCHEN ERKENNTNISSE ................................ 22
1.5 BASALGANGLIEN UND STRIATUM .............................................................................. 26
1.6 ZIELSETZUNG DIESER ARBEIT ................................................................................... 27
2 MATERIAL UND METHODEN .................................................................................. 29
2.1 2.1 MATERIAL .......................................................................................................... 29
2.1.1 CHEMIKALIEN ........................................................................................................ 29
2.1.2 TIERE ................................................................................................................... 29
2.1.3 GERÄTE ................................................................................................................ 30
2.2 METHODEN .............................................................................................................. 30
2.2.1 GOLGI-COX-FÄRBUNG ........................................................................................... 30
2.2.2 LOKALISATION DER NEURONE IM STRIATUM ............................................................ 31
2.2.3 ANALYSE DER SPINES............................................................................................ 33
2.2.4 ELEKTRONENMIKROSKOPIE .................................................................................... 35
2.2.5 PSD-ANALYSE ...................................................................................................... 36
2.2.6 STATISTISCHE AUSWERTUNG ................................................................................. 38
3 ERGEBNISSE ........................................................................................................... 39
3.1 MORPHOLOGISCHE ANALYSE DER SPINES IM DLS ..................................................... 39
3.1.1 PROSAP1/SHANK2-/- ............................................................................................. 39
3.1.2 PROSAP2/SHANK3-/- ............................................................................................. 41
3.2 MORPHOLOGISCHE ANALYSE DER PSDS IM DLS ....................................................... 43
3.2.1 GESCHLECHTSSPEZIFISCHE ANALYSE DER PSD...................................................... 43
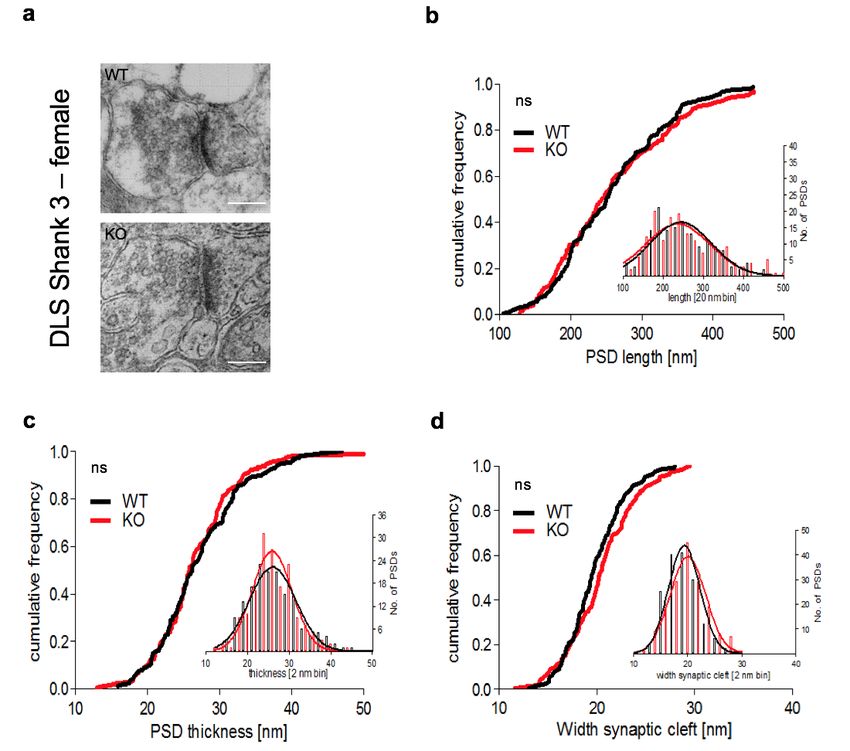
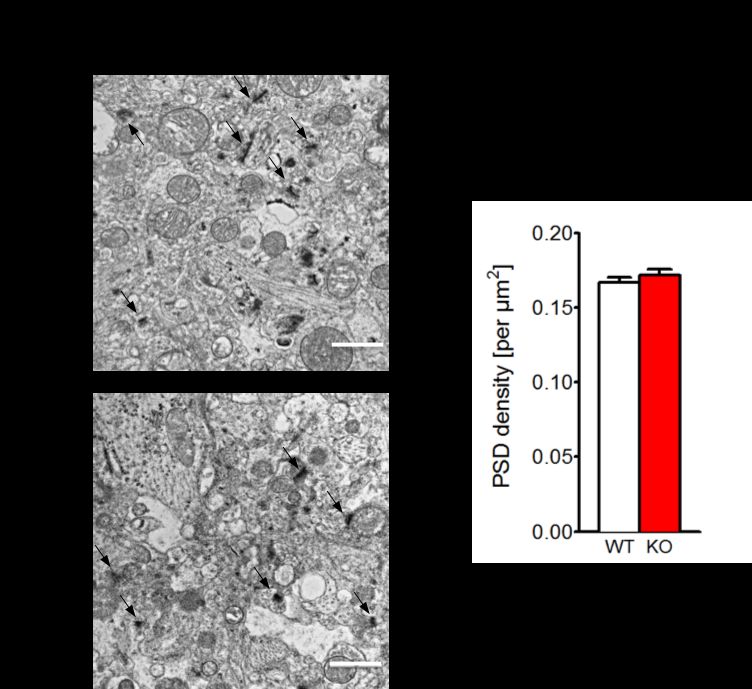
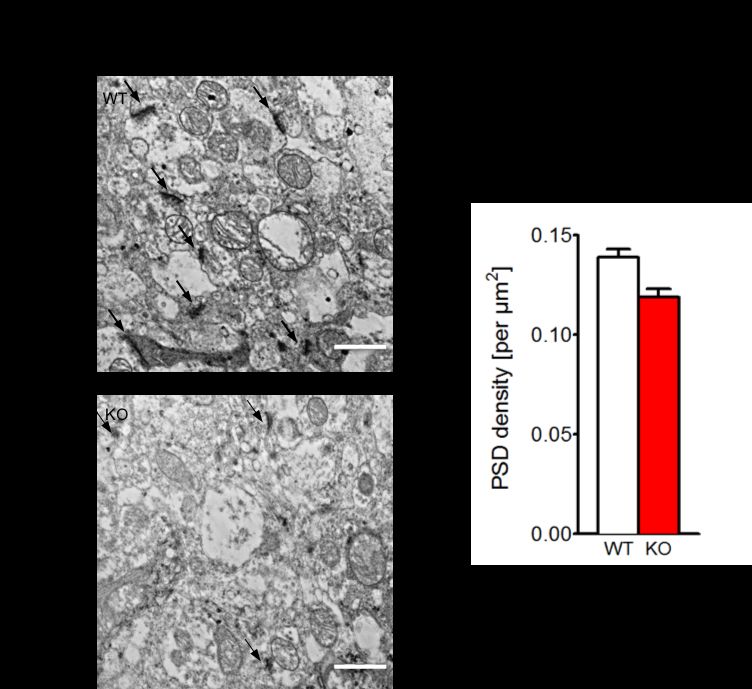
3.2.2 GESCHLECHTSÜBERGREIFENDE ANALYSE DER PSD................................................ 49
3.3 ZUSAMMENFASSUNG DER ERGEBNISSE ..................................................................... 52
4 DISKUSSION ............................................................................................................ 54
4.1 DISKUSSION DER SPINE-ANALYSE IM STRIATUM ........................................................ 54
4.1.1 SPINEDICHTE ........................................................................................................ 54
4.1.2 SPINELÄNGE ......................................................................................................... 56
4.1.3 SPINEBREITE......................................................................................................... 57
4.1.4 SHANK2 VERSUS SHANK3 ...................................................................................... 57
I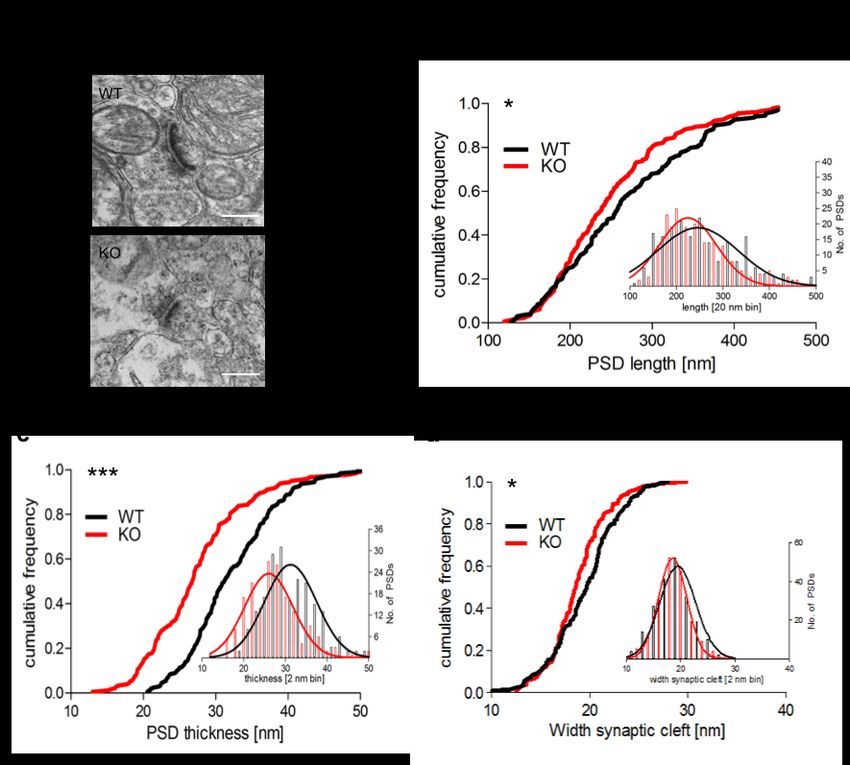
INHALTSVERZEICHNIS
4.1.5 ZUSAMMENFASSUNG ............................................................................................. 58
4.2 DISKUSSION DER PSD-ANALYSE IM STRIATUM .......................................................... 59
4.2.1 SYNAPSENDICHTE ................................................................................................. 59
4.2.2 PSD-LÄNGE.......................................................................................................... 61
4.2.3 PSD-BREITE ......................................................................................................... 61
4.2.4 SYNAPTISCHER SPALT ........................................................................................... 62
4.2.5 MALE VS. FEMALE ................................................................................................. 62
4.2.6 ZUSAMMENFASSUNG ............................................................................................. 64
5 ZUSAMMENFASSUNG ............................................................................................ 66
6 LITERATURVERZEICHNIS ...................................................................................... 68
7 ABBILDUNGSVERZEICHNIS ................................................................................... 79
8 TABELLENVERZEICHNIS........................................................................................ 81
9 DANKSAGUNG ........................................................................................................ 82
10 LEBENSLAUF ........................................................................................................ 83
II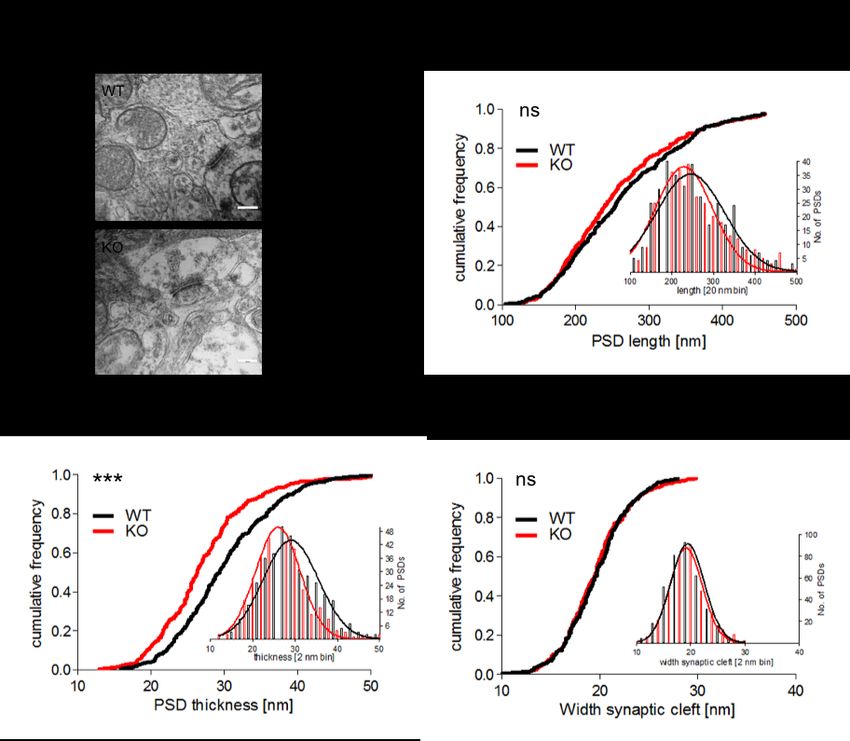
ABKÜRZUNGSVERZEICHNIS
Abkürzungsverzeichnis
Å Ångström (1 Å = 0,1 nm)
Abp1 Actin-binding-protein1
ACR Acrosin
AMPA(R) α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
(receptor)
ANK Ankyrin
ASD(s) Autism spectrum disorder(s)
CA1 Cornu Ammonis 1
CA3 Cornu Ammonis 3
CAM(s) Cell adhesion molecule(s)
CDC Centers for Disease Control and Prevention
Cortactin Cortical-actin-binding protein
Cre/loxP Rekombinationssystem
DKO Double Knock-out
DLS Dorsolaterales Striatum
DMS Dorsomediales Striatum
DS Dorsales Striatum
DSM Diagnostic and Statistical Manual of Mental Disorders
EM Elektronenmikroskopie
FMR1 Fragile-X-mental-retardation-protein-1
FMRP Fragile-X-mental-retardation-protein
FXS Fragiles-X-Syndrom
GA Glutaraldehyd
GKAP Guanylate-kinase-associated protein
GluA1 AMPAR Untereinheit A1
GluA2 AMPAR Untereinheit A2
GluA3 AMPAR Untereinheit A3
GluR Glutamat-Rezeptor
GRIP Glutamate receptor-interacting protein
Hbs Homer binding site
IP3-R Inositol-Triphosphat-Rezeptor
IQ Intelligenzquotient
III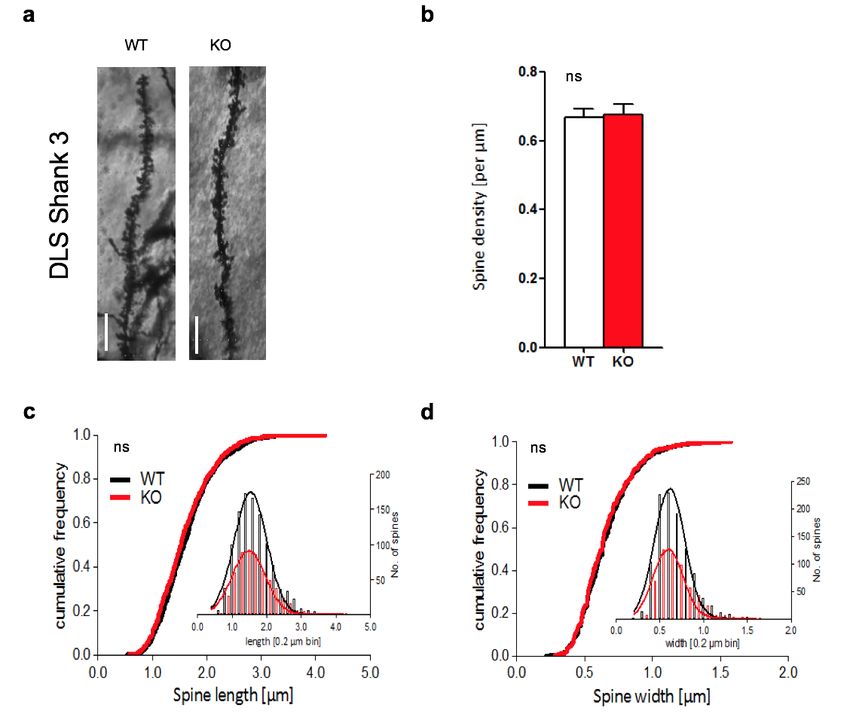
ABKÜRZUNGSVERZEICHNIS
kb Kilobasen
KI Knock-in
KO Knock-out
LTP Langzeit-Potenzierung
MAGUK Membrane-associated guanylate kinase
MECP2 Methyl-CpG-binding protein 2
mGluRs metabotrope Glutamatrezeptoren
mRNA Messenger-Ribonukleinsäure
MSN(s) Medium spiny neuron(s)
NF1 Neurofibromatose 1
NLGN Neuroligin
nm Nanometer
NMDA(R) N-Methyl-D-aspartic acid (receptor)
NUS National University of Singapore
NRXN Neurexin
PBS Phosphatgepufferte Salzlösung
PDD-NOS Pervasive developmental disorder not otherwise
specified
PDZ PSD-95, DLG und ZO-1
PFA Paraformaldehyd
PMS Phelan-McDermid-Syndrom
ProSAP Proline rich synapse associated protein
PSD(s) Postsynaptische Dichte(n)
PSD-95 Postsynaptic density protein 95
RABL2B Rab-Like Protein 2B
RNA Ribonukleinsäure
SAM Sterile alpha motif
SH3 Src homology3-Domäne
UBE3A Ubiquitin-protein ligase E3A
VS Ventrales Striatum
WT Wildtyp
ZNS Zentrales Nervensystem
μm Mikrometer
IV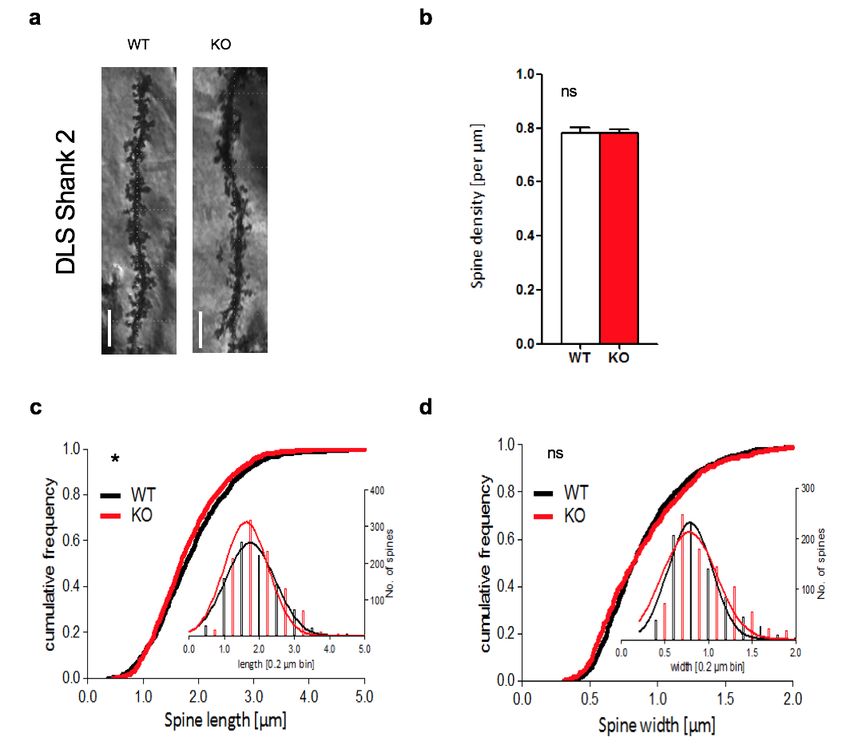
EINLEITUNG
1 EINLEITUNG
1.1 Autismus-Spektrum-Störungen
1.1.1 Überblick
Autismus ist eine angeborene neuropsychiatrische Entwicklungsstörung mit einer
starken genetischen Verankerung. Der Begriff bezeichnet ein klinisches Syndrom,
welches sich aus Defiziten in sozialer Interaktion und Kommunikation sowie
eingeschränkten Interessen und repetitiven Verhaltensweisen zusammensetzt.
Autismus weist eine hohe ätiologische Heterogenität auf. Dabei ist jeder
„empfindliche“ Genlokus nur für einen Bruchteil der Erkrankungen verantwortlich
(44). In vielen Fällen haben eine frühe angewandte Verhaltensanalyse und eine
intensive Verhaltensinterventionstherapie eine positive Wirkung. Autismus bleibt
jedoch ein Krankheitsbild, das in der Regel keine Remission zeigt. Aus diesem
Grund ist die Entwicklung gezielter Therapien basierend auf pathophysiologisch und
ätiologisch definierten Subtypen des Autismus gegenwärtig von großem Interesse
und Gegenstand intensiver Forschung (44).
Im Jahre 1943 wurde Autismus erstmals als ein einheitliches Krankheitsbild
beschrieben: Der austroamerikanische Jugendpsychiater Leo Kanner
veröffentlichte damals in seinem Artikel „Autistic disturbances of Affective
Contact“ elf Fallvorstellungen. Kanner deklarierte hierzu Folgendes: „We must, then
assume that these children have come into the world with innate inability to form the
usual, biologically provided affective contact with people, just as other children come
into the world with innate physical or intellectual handicaps“ (69). Er beobachtete,
dass die betroffenen Kinder von Geburt an eine ausgeprägte Tendenz zum
Alleinsein zeigten, ohne auf Reize der Außenwelt zu reagieren. Sie waren in der
Lage, eine zweckorientierte Beziehung zu Objekten aufzubauen, die ihr Alleinsein
nicht gefährdeten. Gegenüber Personen jedoch waren sie von Beginn an entweder
unruhig und angespannt oder gleichgültig. Kanner bemerkte: „All of the children’s
activities and utterances are governed rigidly and consistently by the powerful desire
for aloneness and sameness“ (69).
In den frühen 1980er Jahren wurde Autismus schließlich als eine
1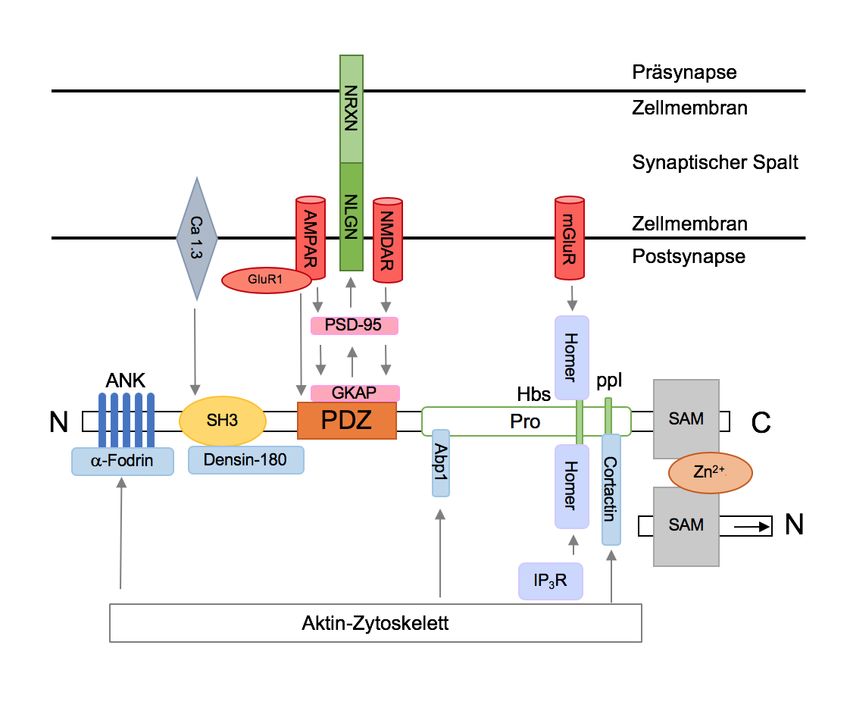
EINLEITUNG
Entwicklungsstörung klassifiziert, differentialdiagnostisch von einer Psychose im
Sinne einer kindlichen Schizophrenie abgegrenzt und als Krankheit mit
biomedizinischer Ätiologie anerkannt. Bis dato war man von pathologischen
Familienverhältnissen und einer schlechten Erziehung als Ursache ausgegangen
(25, 42). Studien konnten jedoch schließlich nachweisen, dass das Leiden hereditär
und mit einer Reihe anderer genetischer Syndrome assoziiert war (41, 104, 129).
Jede Form des Autismus kann einer von zwei großen Gruppen zugeteilt werden,
wobei sich zusammenfassend der Begriff der Autismus-Spektrum-Störungen
etabliert hat (ASD, autism spectrum disorder): Man unterscheidet den syndromalen
vom nicht-syndromalen Autismus. Im ersten Fall geht die autistische Symptomatik
mit einer bereits bekannten monogenetischen Störung, wie beispielsweise dem
Fragilen-X-Syndrom (FXS), einher. Beim nicht-syndromalen Autismus steht das
Krankheitsbild für sich, ohne eine erkennbare Komorbidität. Hierzu zählen unter
anderem der frühkindliche Autismus (auch Kanner-Syndrom) und das Asperger-
Syndrom sowie der sogenannte atypische Autismus. Letzterer beschreibt sämtliche
nicht weiter klassifizierbaren Formen der Erkrankung und wird in den USA im dort
gebräuchlichen psychiatrischen Diagnosehandbuch DSM-IV „tiefgreifende
Entwicklungsstörung, nicht anders bezeichnet“ (PDD-NOS, pervasive
developmental disorder not otherwise specified) genannt (44).
Um die Diagnose einer ASD korrekt zu stellen, müssen bestimmte klinische
Kriterien erfüllt werden, die das Auftreten schwerer neuropsychiatrischer
Dysfunktionen vor dem Alter von drei Jahren beschreiben. Dazu zählen i. Defizite
in Sprache und Kommunikation, ii. Defizite in sozialer Interaktion und iii. das
Vorhandensein repetitiver Verhaltensweisen und restriktiver Interessen und
Aktivitäten. Diese Symptome müssen dabei eine deutliche Beeinträchtigung des
Betroffenen auf sozialer Ebene bewirken und dürfen nicht durch eine vermeintliche
geistige Behinderung anderer Ätiologie zu erklären sein (5). Der Intelligenzquotient
(IQ) variiert in Realität in den meisten Fällen stark. In 40-60 % gehen die
autistischen Symptome mit einer schweren intellektuellen Störung einher; der IQ
kann jedoch auch im normalen Bereich liegen (97).
Neben den Defiziten in den oben genannten Kerngebieten können in einem Großteil
der mit Autismus diagnostizierten Kinder weitere klinische Auffälligkeiten
beobachtet werden: Über 90 % der Betroffenen leiden an einer pathologischen
2EINLEITUNG
sensorischen Perzeption, sodass einige Experten fordern, die beeinträchtige
Sinneswahrnehmung als ein Diagnosekriterium zu etablieren (134). Auch werden
häufig motorische Defizite in Form von Hypotonie oder Apraxie beschrieben.
Darüber hinaus können Schlafstörungen und gastrointestinale Symptome auftreten
(44, 87). Weiterhin ist es von großer Bedeutung für die Patienten, eine fragliche
Epilepsie als eigenständiges Krankheitsbild von entsprechenden Anfällen im
Rahmen der ASD abzugrenzen (44). Autismus und Frühgeburtlichkeit sowie
physische Fehlbildungen weisen ebenfalls eine nicht unerhebliche Korrelation auf
(82).
Die Prävalenz von Autismus lag 2013 weltweit bei 62 von 10 000 Kindern, das heißt,
eines von 160 Kindern leidet an einer ASD. Die amerikanische Gesundheitsbehörde
Centers for Disease Control and Prevention (CDC) veröffentlichte 2016 noch
dramatischere Zahlen und berichtete von einem betroffenen Kind unter 68 Kindern
in den USA (29). Damit nimmt die Zahl der Erkrankten seit den ersten
epidemiologischen Studien Ende des 20. Jahrhunderts kontinuierlich zu (89, 97).
Betrachtet man die Prävalenz der Erkrankung hinsichtlich der
Geschlechterverteilung, leiden Jungen bis zu fünf Mal häufiger an Autismus als
Mädchen (22).
Die Diagnose nach DSM-IV beruht allein auf der Beobachtung von Verhalten und
Kognition der Betroffenen – Kriterien, welche sich nur schwer absolut und objektiv
bewerten lassen. In Wirklichkeit gehen physiologische Zustände aber fließend in
Pathologien über. Umso erstrebenswerter ist also die ätiologische Identifikation
verschiedener autistischer Syndrome und damit ein Verständnis der Erkrankung auf
genetischer Ebene. In den letzten Jahren wurden hier erhebliche Fortschritte
gemacht: So ist mittlerweile bekannt, dass de novo Mutationen und solche, die ihm
Rahmen eines genetischen Syndroms vererbt werden, zusammengenommen für
etwa 10-20 % der ASDs verantwortlich sind; jede einzelne hingegen lediglich die
Ursache für ein Hundertstel der Fälle darstellt (1, 44). Weiterhin konnte in
Familienstudien gezeigt werden, dass das relative Risiko eines Kindes, an Autismus
zu erkranken, um das 25fache erhöht ist, wenn die Erkrankung bereits bei einem
Geschwisterkind diagnostiziert wurde (67). Zwillingsstudien ergaben zudem für
monozygote Zwillinge deutlich erhöhte Konkordanzraten (70-90 %) im Vergleich zu
zweieiigen Zwillingen (0-10 %) (7, 120). Das relative Risiko bei eineiigen Zwillingen
3EINLEITUNG
bezugnehmend zum Risiko in der Normalbevölkerung beläuft sich auf 153,0, bei
zweieiigen Zwillingen auf 8,2 und bei Geschwistern auf 10,3 (89, 109). Die
epidemiologischen Studien bestätigen somit die starke genetische Komponente in
der Pathogenese von ASDs.
Autismus-Spektrum-Störungen werden zu den sogenannten Synaptopathien
gezählt. Darunter versteht man Erkrankungen des ZNS, die auf eine Fehlfunktion
der Synapsen zurückzuführen sind. So haben sich in einem Großteil der
Betroffenen Mutationen in Genen gezeigt, welche mit Struktur und Funktion von
Synapsen assoziiert werden. Die häufigsten Einzelgen-Mutationen in ASDs als
Formen des syndromalen Autismus stellen unter anderem das Fragile-X-Syndrom
(FXS, FMR1-Gen), die Neurofibromatose (NF1-Gen), das Angelman-Syndrom
(UBE3A-Gen), das Rett-Syndrom (MECP2-Gen) und das Phelan-McDermid-
Syndrom (SHANK3) dar (11). Mutationen in Neuroliginen (NLGN) sowie Neurexinen
(NRXN) können nicht-syndromale ASDs hervorrufen (64). Obwohl diese Auflistung
die Vielzahl der mit Autismus-assoziierten Gene nicht annähernd ausschöpft,
kommt man nicht umhin festzustellen, dass all diese Mutationen in Genen auftreten,
welche kritische Regulatoren synaptischer Funktion darstellen (141).
1.1.2 Syndromaler Autismus
Im Folgenden soll auf drei ausgewählte Krankheitsbilder eingegangen werden, um
Gemeinsamkeiten und Unterschiede der Syndrome und deren zugrundeliegende
synaptische Pathophysiologie herauszuarbeiten.
Phelan-McDermid-Syndrom
Als eigenständiges Krankheitsbild fand das Phelan-McDermid-Syndrom (PMS,
auch 22q13.3-Deletionssyndrom oder Mikrodeletion 22q13.3) erstmalig im Jahre
1985 Erwähnung – damals assoziiert mit einem auffälligen Phänotyp, geistiger
Behinderung und fehlender Sprachentwicklung. Weitere Fallberichte folgten, die
von ähnlichen Entwicklungsstörungen berichteten und diese einer Deletion im
langen Arm des Chromosoms 22 zuschrieben (32, 133). Schließlich gelang es
Anderlid et al., die kritische Region auf etwa 100 Kilobasen (kb) im Bereich 22q13
einzugrenzen (2): 1999 wurde das Chromosom 22 als zweitkleinstes Chromosom
4EINLEITUNG
des menschlichen Genoms vollständig sequenziert, sodass drei Kandidatgene
identifiziert werden konnten – Acrosin (ACR), PROSAP2/SHANK3 und RABL2B
(38). Während sich ACR als Proteinase im Akrosom reifer Spermien befindet und
das Eindringen des Spermiums in die Eizelle ermöglicht, spielt RABL2B als G-
Protein im Transport zellulärer Vesikel eine entscheidende Rolle (40, 73).
PROSAP2/SHANK3 codiert für ein Protein in der postsynaptischen Dichte, das in
vielen Bereichen des Gehirns exprimiert wird (19). Mit den Veröffentlichungen von
Bonaglia et al. 2001 sowie Wilson et al. 2003 konnte ein Wegfall von SHANK3
maßgeblich PMS-typischen Symptomen zugeordnet werden (20, 135). In 80-85 %
der Fälle manifestiert sich dieser Verlust als Haploinsuffizienz aufgrund einer de
novo Mutation paternalen Ursprungs (95).
0,5 % aller ASDs sind auf das 22q13-Deletionssyndrom zurückzuführen, wobei
davon auszugehen ist, dass die Prävalenz aufgrund unzureichender Diagnostik
unterschätzt wird (76). Das PMS lässt sich durch typische Symptome
charakterisieren: Betroffene zeigen üblicherweise eine moderate bis
schwerwiegende Entwicklungsstörung mit postnataler Hypotonie, geistiger
Retardierung sowie verzögerter bis fehlender Sprachentwicklung und einer
beeinträchtigten sozialen Interaktion. Auch sind repetitive Verhaltensweisen zu
beobachten – stereotype Bewegungen oder Laute, ungewöhnliche sensorische
Empfindungen und Schlafstörungen (95). Hinzu kommen milde kraniofaziale
Dysmorphien (32). Bei ausgeprägter klinischer Heterogenität muss
differentialdiagnostisch an ähnliche syndromale Krankheitsbilder gedacht werden
wie beispielsweise die Folgenden.
Fragiles-X-Syndrom
Das Fragile-X-Syndrom (FXS) ist mit einer Prävalenz von 1:5 000 unter Männern
und etwa halb so viel betroffener Frauen die häufigste Ursache angeborener
geistiger Retardierung und wird außerdem als eine der Hauptursachen von
Autismus angesehen (30, 130). Bis zu drei Prozent aller mit ASD diagnostizierten
Patienten leiden molekulargenetisch am FXS, welches determiniert ist durch eine
Mutation im X-chromosomalen FMR1-Gen. Dieser Fehler wiederum bedingt eine
geringere Expression des Fragile X mental retardation protein (FMRP), welches als
RNA-bindendes Protein die Translation von mRNA und damit die Proteinsynthese
in Dendriten reguliert (12). Neben moderater bis schwerer geistiger Retardierung
5EINLEITUNG
zeigen Betroffene autistische Züge wie stereotype Verhaltensweisen und eine
verzögerte sprachliche Entwicklung. Charakteristisch für das FXS sind zudem ein
erhöhtes epileptogenes Potential, ein Aufmerksamkeitsdefizit, Hyperaktivität,
abnormales Wachstum, typische kraniofaziale Auffälligkeiten sowie ein
Makroorchidismus (70).
Morphologische Studien zeigten, dass ein fehlerhaftes FMR1-Gen auch ein
phänotypisches Korrelat findet: In Fmr1-KO-Mäusen ließen sich im Kortex abnormal
geformte Spines im Sinne von dünneren und längeren Spines nachweisen (3, 63).
Rett-Syndrom
Das Rett-Syndrom wurde erstmals 1966 vom österreichischen Neuropädiater
Andreas Rett beschrieben, indem er Beobachtungen von 22 Patienten
zusammenfasste (103). Klinische Akzeptanz erhielt es jedoch erst 1983, als
Hagberg und Kollegen eine Serie von 35 Fallstudien aus verschiedenen Ländern
veröffentlichten (54, 55, 80). Heutzutage liegt die Prävalenz bei etwa einer von
1 000 Frauen. Als monogene postnatale Entwicklungsstörung beeinträchtigt das
Rett-Syndrom die Gehirnentwicklung während der frühen Kindheit: Patienten
entwickeln sich in den ersten sechs bis 18 Monaten vermeintlich normal,
altersgerechte Meilensteine werden erreicht – so beispielsweise die Fähigkeit, zu
laufen oder einzelne Worte zu sprechen. Meist im Verlauf des zweiten Lebensjahres
treten jedoch die ersten Symptome der Erkrankung auf. Charakteristisch ist hier ein
typisch autistischer Phänotyp, einhergehend mit einer schweren geistigen
Retardierung, stereotypen Handbewegungen und eine allgemeine motorische
Schwäche. Darüber hinaus sind eine erhöhte Krampfneigung, Defizite im Sprechen
und Lernen sowie abnorme Atemmuster zu beobachten (27, 141). In nahezu allen
Fällen liegt dem Rett-Syndrom eine de novo Mutation im X-chromosomal-fixierten
MECP2-Gen (methyl-CpG-binding-protein 2) zugrunde. Dieser nicht vererbte,
sondern sporadisch entstehende Defekt führt zum Verlust des entsprechenden
Genprodukts und damit eines wichtigen Proteins zur Aufrechterhaltung der
synaptischen Homöostase (8). Ähnlich dem FXS zeigt auch dieses Syndrom
morphologische Auffälligkeiten: So ist die Spine-Dichte und das Maß des dendritic
branching in Mecp2-KO-Mäusen geringer als in der WT-Vergleichskohorte. Auch
zeigt sich eine kortikale Atrophie (9, 71). In einer Umkehrstudie von Bird und
6EINLEITUNG
Kollegen konnte interessanterweise gezeigt werden, dass die Aktivierung eines
zuvor inaktiven Mecp2-Gens in entsprechend vom Rett-Syndrom betroffenen
Mäusen eine Normalisierung einiger Symptome bewirkte (36, 53).
Die hier angeführten Krankheitsbilder zeigen zum einen Autismus-typische
Phänotypen und weisen zum anderen eine ähnliche Pathogenese auf: Durch
Mutationen in den genannten Genen kommt es zum Verlust von Genprodukten, die
eine bedeutende Rolle in Aufbau und Funktion von Synapsen spielen. Dies führt zu
einem Ungleichgewicht in der synaptischen Homöostase. Somit scheint die
Synapse eine Schlüsselrolle in der Pathogenese von Autismus einzunehmen (60).
1.2 Die Synapse
1.2.1 Übersicht
Führende Aufgabe von Nervenzellen ist es, mit anderen Nervenzellen zu
kommunizieren. Obwohl einige Neurone elektrische Signale über Gap junctions
senden, findet die meiste interzelluläre Kommunikation im zentralen Nervensystem
(ZNS) mittels chemischer Synapsen statt (56). Der Begriff „Synapse“ wurde 1897
von Charles Scott Sherrington etabliert. Er setzt sich zusammen aus den
griechischen Wortbausteinen „syn“ für „zusammen“ und „haptein“ für „greifen,
fassen, tasten“ (118).
Klassischerweise sitzt die Synapse am Ende eines Axons und verbindet dieses mit
einem anliegenden Dendriten eines anderen Neurons. Auch Gewebe nicht
neuronalen Ursprungs kann von Synapsen angesteuert werden. So beispielsweise
an der motorischen Endplatte, wo das Signal auf eine Muskelzelle übergeht und
dort eine entsprechende motorische Reaktion hervorruft.
In aller Kürze lässt sich die Übertragung chemischer Signale wie folgt
zusammenfassen: Eintreffende Aktionspotentiale bewirken eine Depolarisation im
präsynaptischen Endknöpfchen. Dadurch öffnen sich spannungssensitive Calcium-
Kanäle, so dass Calcium in die Zelle einströmen kann. Erhöhte intrazelluläre
Calciumkonzentrationen setzen in Vesikeln gespeicherte Neurotransmitter via
Exozytose in den synaptischen Spalt frei. Hier diffundieren die Botenstoffe durch
7EINLEITUNG
den Interzellulärraum, um an entsprechenden Rezeptoren der postsynaptischen
Membran anzudocken und sie zu aktivieren. Ionenkanäle öffnen oder schließen sich
und verändern damit die elektrische Aktivität der Postsynapse. Zuletzt dissoziieren
die Transmitter von den Rezeptoren und werden mittels Diffusion, Re-Uptake oder
enzymatischer Inaktivierung aus dem synaptischen Spalt eliminiert. Innerhalb der
Nervenzelle werden die Vesikel durch axonalen Transport oder lokal via
Endozytose wiederhergestellt, sodass das nächste Aktionspotential nach
demselben Prinzip weitergeleitet werden kann (23, 56, 125).
Lange Zeit waren die strukturellen Grundlagen der Informationsübertragung
zwischen Neuronen unklar, bis Neuroanatomen im späten 19. Jahrhundert mittels
damals fortschrittlicher Silberfärbungen axonale Boutons identifizierten, die
Dendriten oder Spines anderer Dendriten zu berühren schienen. Die Idee einer
protoplasmatischen Kontinuität zwischen präsynaptischem und postsynaptischem
Element geriet ins Wanken (52, 117). Doch erst mit Einführung der
Elektronenmikroskopie (EM) wurde diese Theorie endgültig revidiert. Es war nun
möglich, den synaptischen Spalt eindeutig zu visualisieren (35, 90): So brachten die
ersten elektronenmikroskopischen Studien vor über 60 Jahren wesentliche
Erkenntnisse über den Aufbau einer Synapse. Gray prägte den Begriff des
„Synaptolemma“ als ein gegenüber liegendes Membranpaar, dass sich
zusammensetzt aus i. dem präsynaptischen Element als Fortführung der Membran
des Telodendrons, welches den letzten Abschnitt eines Axons oder Dendriten
determiniert, und ii. dem postsynaptischen Apparat als Spezialisierung der
Plasmamembran des gegenüberliegenden Dendriten. „The base of each ending is
closely opposed to the surface of the dendrite, following faithfully for a short distance
the small indentations and undulations of its surface, but clearly separated from the
dendrite by a space approximately 200 Å wide. At no point do the limiting
membranes of the ending and dendrite fuse” (90). Damit war der synaptische Spalt
erstmals beschrieben.
In der Präsynapse fallen elektronenmikroskopisch insbesondere die Vielzahl an
Vesikeln und zahlreiche Mitochondrien auf, welche notwendig sind, um den hohen
Energiebedarf des synaptischen Apparats zu decken (90). Das postsynaptische
Element zeigt dagegen innerhalb der Membran verdichtete Bereiche
beziehungsweise dickere Abschnitte. Diese Region wird als postsynaptische Dichte
(PSD) bezeichnet und soll im Rahmen dieser Arbeit eine essentielle Rolle spielen
8EINLEITUNG
(50). Anders als die Bezeichnung suggeriert, ist der synaptische Spalt keineswegs
ein etwa 20nm messender „leerer“ Raum zwischen präsynaptischem Axon und
postsynaptischem Gegenstück, sondern vielmehr gefüllt mit elektronendichtem
Material (79, 142). Dieses dient dazu, die Synapse zu stabilisieren und spielt
darüber hinaus auch im reibungslosen Ablauf der Erregungsübertragung eine
entscheidende Rolle (58).
Grays Erkenntnisse führten noch weiter: Er differenzierte zwischen zwei
verschiedenen Typen von Synapsen – Typ 1 oder asymmetrischen Synapsen, die
sich später als exzitatorisch beziehungsweise glutamaterg herausstellen sollten,
und Typ 2 oder symmetrische, später inhibitorische Synapsen. Mengenmäßig
führend sind eindeutig die Typ 1-Synapsen, die an Dendriten und deren Spines
sitzen und die Unmengen axo-dendritischer Verbindungen herstellen. 88 % von
ihnen zeigen über nahezu die gesamte Strecke (70-100 %) der postsynaptischen
Membran die bereits oben beschriebene Verdichtung, während die präsynaptische
Membran als sehr zart imponiert. Auffällig ist, dass prä- und postsynaptisches
Element innerhalb der verdickten Bereiche einen größeren Abstand zueinander
einnehmen können und der synaptische Spalt bis zu 30 nm messen kann. Gray
bezeichnet diesen als intermediäres Band. Die zarten Bereiche der Membranen
sind dagegen nur durch einen 20 nm messenden interzellulären Raum getrennt.
Typ 2-Synapsen finden sich insbesondere auf den Nervenzellköpern selbst sowie
auf Dendriten und sind so für die axo-somatischen Kontakte zuständig. Sie zeigen
die postsynaptische Verdickung lediglich über maximal 40 % der Membran und
auch das intermediäre Band erscheint verschwommener (31, 51). Van der Los
prägte 1963 den Begriff der subsynaptischen Organelle als Merkmal der axo-
dendritischen und somit Typ 1-Synapsen und beschrieb damit die verdichteten
Areale der PSD (127).
1.2.2 Morphologie dendritischer Spines
Dendritische Spines finden sich als postsynaptische Elemente auf dem Großteil der
exzitatorischen Synapsen im Gehirn, wo sie Input von glutamatergen Axonen
erhalten. Erstmals Erwähnung fanden diese Fortsätze bei Ramon und Cajal im
späten 19. Jahrhundert, die sie als „Dornen oder Stacheln“ beschrieben und ihnen
bereits eine Rolle in der Konnektivität von Neuronen zugestanden (13, 101). Die
9EINLEITUNG
Fähigkeit des Dendriten, in Abhängigkeit der synaptischen Aktivität neue Spines
auszubilden und vorhandene Spines umzuwandeln oder abzustoßen, legt nahe,
dass dendritische Spines eine zentrale Funktion hinsichtlich synaptischer Plastizität
einnehmen. Damit stellen Spines ein zelluläres Korrelat für Lernen und Gedächtnis
dar (138). In der Konsequenz ist es nicht überraschend, dass viele
neuropsychiatrische Erkrankungen, die mit einer hohen Rate an mentaler
Retardierung einhergehen, eine atypische Spine-Ausprägung und -Morphologie
aufweisen. Diese Pathologie wird als „spine dysgenesis“ bezeichnet (92, 98).
Dendriten entstehen im sich entwickelnden Gehirn zunächst gänzlich ohne Spines
oder Synapsen. Während der Synaptogenese formen sich äußerst flexible, finger-
ähnliche Fortsätze, Filopodien genannt, und bilden erste, sich formende Synapsen
mit nahegelegenen Axonen. Schließlich werden diese durch Spines ersetzt (140).
Die Dichte dendritischer Spines ist abhängig vom Entwicklungsstadium des Gehirns:
Nach der Spinogenese folgt die Elimination von nahezu 50 % aller Synapsen und
Spines (96).
Dendritische Spines setzen sich typischerweise zusammen aus einem Hals, der aus
dem entsprechenden Dendriten wächst und einem Kopf, auf welchem sich
exzitatorische Synapsen bilden. Die Morphologie kann hierbei variieren – sowohl in
der Gesamtlänge der Spines als auch in der Kopfform oder der Länge des Halses.
Man unterscheidet hauptsächlich drei morphologische Typen: i. sogenannte „stubby
spines” ohne einen sichtbaren Hals, ii. „mushroom spines” mit einem großen Kopf
und dünnem Hals, und iii. „thin spines” mit einem kleinen Kopf und einem langen,
dünnen Hals (93, 96).
Relevant werden diese Erkenntnisse aber vor allem durch Studien, die zeigen
konnten, dass die Morphologie von Spines im Rahmen von synaptischer
Transmission, Synapsenbildung sowie Spine-Stabilität eine Rolle spielt (59, 83).
Auch zeigt sich eine positive Korrelation zwischen der Kopfgröße, der AMPA- und
NMDA-Rezeptoren und der synaptischen Stärke (77, 78, 123). Diese Korrelationen
weisen deutlich darauf hin, dass die Morphologie der Spines abhängt von der
Funktion ihrer Synapsen. Es liegt also nahe, dass eine gestörte Funktion dieser
Synapsen – zum Beispiel bedingt durch ASDs – mit einer atypischen Form
einhergeht.
10EINLEITUNG
1.2.3 Die postsynaptische Dichte
Die postsynaptische Dichte (PSD) ist an der Spitze dendritischer Spines lokalisiert.
Sie wurde erstmals elektronenmikroskopisch wie folgt beschrieben: „a fuzzy
electron-dense thickening of the postsynaptic membrane that is apposed to the
presynaptic active zone“ (50). Damit stellt die PSD eine Spezialisierung der
postsynaptischen Membran dar und rechtfertigt maßgeblich den Begriff der
Synapsen vom asymmetrischen Typ (116, 119). So sind PSDs zwar typischerweise
scheibenähnlich konfiguriert, können aber auch erhebliche Unregelmäßigkeiten
aufweisen (26). Die PSD beherbergt ein komplexes Netzwerk aus zahlreichen
Proteinen, welche verantwortlich sind für die transsynaptischen Signalwege sowie
für die strukturelle Organisation der Spines. Man kann diese Proteine in folgende
Gruppen einteilen: i. Zelladhäsions-Moleküle, ii. Gerüstproteine, sogenannte
Scaffold Proteine, iii. membran-gebundene Rezeptoren und G-Proteine, iv.
zytoskelettale Proteine und v. Signalproteine (15, 116). Morphologisch betrachtet,
lässt sich die PSD in drei Schichten einteilen: Die äußerste Schicht direkt an der
postsynaptischen Membran setzt sich aus Zelladhäsionsmolekülen (cell adhesion
molecules, CAMs) und Transmembranproteinen wie den Neuroliginen sowie
Glutamatrezeptoren wie AMPA- und NMDA-Rezeptoren zusammen. Hier findet die
Interaktion und molekulare Verbindung mit der Präsynapse statt. Darunter folgen
Proteine, welche mittels zahlreicher Bindungsdomänen als Adaptermoleküle
fungieren und für die Protein-Protein-Interaktion innerhalb der Postsynapse
verantwortlich sind. Hervorzuheben sind an dieser Stelle die Familie der GKAPs
(Guanylate-kinase-associated proteins) sowie das PSD-95 (Postsynaptic density
protein 95), aber auch Homer und GRIP (Glutamate receptor-interacting protein).
Den Abschluss bildet die dritte Schicht, welche sich zusammensetzt aus GKAP-
Adapterproteinen und der ProSAP/SHANK-Proteinfamilie. Letztere dienen der
Verankerung mit den Aktin-Filamenten des Zytoskeletts und bilden damit das
stabilisierende Gerüst der Synapse (Abbildung 2) (47, 107, 114, 128).
Der komplexe Aufbau der PSD und die große Anzahl wie auch Vielfalt an
Rezeptoren und Strukturmolekülen lassen vermuten, dass eine Störung in Form
und Funktion der PSD Auswirkungen auf die Funktion der Synapse und damit der
Interaktion zwischen Neuronen hat.
11EINLEITUNG
1.3 Die ProSAP-/SHANK-Proteinfamilie
Die ProSAP-/SHANK-Proteinfamilie findet sich charakteristischerweise in der
postsynaptischen Dichte exzitatorischer Synapsen. Sie umfasst drei Moleküle:
ProSAP1/SHANK2, ProSAP2/SHANK3 und SHANK1 (im Folgenden auch
bezeichnet als SHANK2, SHANK3 und SHANK1). Die Begriffe „ProSAP“ und
„SHANK“ sind als Akronym zu verstehen und heben einzelne Domänen der
Moleküle hervor. So steht ProSAP für „proline-rich synapse associated protein“ und
SHANK für „SH3-domain and ankyrin repeat containing protein“ (19, 115).
ProSAP/SHANK-Moleküle sind an der Schnittstelle von Rezeptoren der
postsynaptischen Membran und den zytoskelettalen Elementen der Postsynapse
positioniert und rekrutieren, ordnen und vernetzen diese, damit ein funktionsfähiges
Gefüge entsteht. Sie gelten dabei als „master scaffold proteins“ (am ehesten:
Haupt-Gerüstproteine), die das komplexe Netzwerk postsynaptischer Moleküle
organisieren und stabilisieren (76, 128). Alle drei Gene gleichen sich in 63-87 % der
Aminosäuren und setzen sich aus den folgenden Bestandteilen zusammen: N-
terminal reihen sich zunächst fünf Ankyrin-Wiederholungen (ANK) aneinander,
gefolgt von der Src homology3-Domäne (SH3). Es schließt sich die sogenannte
PDZ-Domäne an, welche sich aus PSD-95, DLG und ZO-1 zusammensetzt. Diesem
geht ein Prolin-reicher Abschnitt nach, welcher die sogenannte Homer binding site
(Hbs) und ppI-Domäne beinhaltet; am C-terminalen Ende findet sich abschließend
eine SAM-Domäne (sterile alpha motif) (Abbildung 1). Durch alternatives Splicing
entstehen dabei die zum Verständnis dieser Arbeit wichtigen verschiedenen
Isoformen der Proteine (Abbildung 3) (19, 128).
Abbildung 1: Schematische Darstellung des ProSAP/SHANK-Proteins
N (N-terminal), ANK (Ankyrin repeats), SH3 (Scr homology 3), PDZ (PSD-95/DLG-/ZO1), Pro
(Prolin-reiche Domäne), Hbs (Homer binding site), SAM (sterile alpha motif), C (C-terminal); eigene
Darstellung in Anlehnung an Jiang und Ehlers (2013).
12EINLEITUNG
Über diese Domänen kann ProSAP/SHANK zahlreiche Protein-Protein-
Interaktionen gewährleisten. Unter anderem binden beispielsweise GKAPs
(Guanylate-kinase-associated protein) über ihren C-Terminus an die PDZ-Domäne
von ProSAP/SHANK, während sie N-terminal mit MAGUKs (Membrane-
associated guanylate kinase) wie beispielsweise PSD-95 in Kontakt treten (19).
Diese wiederum sind Bindungspartner von transmembranären NMDA- und AMPA-
Rezeptoren (NMDAR, AMPAR). AMPAR können zudem über ihre GluR1-Unterheit
(Glutamat-Rezeptor 1) auch direkt an die PDZ-Domäne anknüpfen (128). Über den
Prolin-reichen Abschnitt – genauer die Homer binding site (Hbs) – werden Homer-
Proteine gebunden. In Richtung der PSD dockt Homer 1a an die Hbs an und
gruppiert metabotrope Glutamatrezeptoren (mGluRs) mGluR1a und mGluR5 an der
postsynaptischen Membran, während Homer 1b/c in Richtung des
zytoplasmatischen Raums Inositol-Trisphosphat-Rezeptoren (IP3-R) bindet. Diese
spielen eine wichtige Rolle im Spine-Wachstum (107, 108). Über diese Verknüpfung
kann Einfluss auf den mGluR-abhängigen Calcium-Influx in die Zelle genommen
werden (62). Hier kommt die SH3-Domäne ins Spiel, welche mit dem
spannungsgesteuerten L-Typ-Calciumkanal 1.3 vernetzt ist und auch darüber den
Calciumeinstrom steuern kann (88, 139). Über alpha-Fodrin können die N-terminal
gelegenen Ankyrin-Domänen von ProSAP/SHANK an das F-Aktin dendritischer
Spines anknüpfen, was ebenfalls Einfluss auf die intrazelluläre Calciumhomöostase
hat (16). Weiterhin kann Aktin mit Hilfe von Cortactin (Cortical-actin-binding protein)
und Abp1 (Actin-binding-protein1) gebunden werden. Beide Proteine docken dabei
an den Prolin-reichen Abschnitt des SHANK-Moleküls an. Im Zusammenspiel
tragen all diese Proteine maßgeblich zur Spinemorphologie bei (17, 37, 99). Densin-
180 bindet an die SH3-Domäne und antagonisiert übermäßiges dendritic branching
zugunsten der Entwicklung funktionaler Spines und Synapsen (100). Am C-
Terminus findet sich abschließend die SAM-Domäne, welche mit Hilfe von Zink für
die Anordnung von SHANK2 und SHANK3 in der Postsynapse selbst ist (46).
Weiterhin können SHANK-Proteine direkt und indirekt über PSD-95 an die ebenfalls
postsynaptisch liegenden Neuroligine binden, welche wiederum über ihren
transmembranären Abschnitt mit Neurexinen der Präsynapse interagieren können.
Dadurch tragen sie maßgeblich zur Entstehung von Synapsen und intakter
Kommunikation zwischen Prä- und Postsynapse bei (Abbildung 2) (121, 122).
13EINLEITUNG
Abbildung 2: Schematische Darstellung der Protein-Protein-Interaktionen des SHANK-
Proteins
N (N-terminal), ANK (Ankyrin repeats), SH3 (Scr homology 3), PDZ (PSD-95/DLG-/ZO1), Pro
(Prolin-reiche Domäne), Hbs (Homer binding site), SAM (sterile alpha motif), C (C-terminal), Ca 1.3
(spannungsabhängiger Ca2+-Kanal 1.3), AMPAR (alpha-amino-3-hydroxy-5-methyl-4-isoxazole
propionic acid-Rezeptor), GluR1 (Untereinheit des AMPAR), NMDAR (N-methyl-D-aspartat-
Rezeptor), NLGN (Neuroligin), NRXN (Neurexin), mGluR (metabotroper Glutamatrezeptor), PSD-
95 (Postsynaptisches Dichteprotein-95), GKAP (Guanylate-kinase-associated protein ), IP3R
(Inositoltriphosphat-Rezeptor), Zn2+ (Zink-Ion), Abp1 (Actin binding protein 1); eigene vereinfachte
Darstellung in Anlehnung an Jiang und Ehlers (2013).
Dieser komplexe Aufbau der PSD macht deutlich, welche essentielle Rolle die
ProSAP/SHANK-Proteine in der Vernetzung membranständiger Rezeptoren mit
dem Zytoskelett sowie mit zytoplasmatischen Molekülen spielen. Sie ermöglichen
damit einerseits ein gesundes Spinewachstum und eine funktionelle
Spinemorphologie. Andererseits sind sie beteiligt an der Steuerung intrazellulärer
Calciumlevel und damit synaptischer Aktivität.
14EINLEITUNG
Shank-Proteine zeigen eine sehr heterogene Verteilung im Gehirn (Tabelle 1):
Zusammenfassend kann festgehalten werden, dass Shank2 überwiegend im Cortex,
im Hippocampus und in der Molekularschicht des Kleinhirns exprimiert wird,
während Shank3 vor allem im Striatum und in der Körnerzellschicht des
Cerebellums zu finden ist (91, 111).
Tabelle 1: Expression der Shank-Proteine im Gehirn
Cortex Hippocampus Striatum Mesencephalon Cerebellum
Shank1 +++ ++ + ++ +/++
+++ (Molekularschicht)
Shank2/ProSAP1 ++ +/++ + +
Ø (Körnerzellschicht)
Ø (Molekularschicht)
Shank3/ProSAP2 + + ++ sehr niedrig
++ (Körnerzellschicht)
Neben ihrer Expression im Gehirn findet man Shank-Proteine – mit Ausnahme von
Shank1 – auch in nicht-neuronalem Gewebe. Während Shank2 in Thymus, Leber,
Niere, Pankreas und Keimzellen im Hoden vorkommt, findet man Shank3 in nahezu
jedem Gewebe (76, 128).
Shank-Proteine sind bereits am ersten Tag nach der Geburt in dendritischen Spines
von Nagetiergehirnen nachzuweisen. Sie vermehren sich im weiteren Verlauf, bis
sie zwischen der dritten und vierten Woche ihren Höhepunkt erreichen (17, 18, 124).
So bleibt anzunehmen, dass ProSAP/Shank gerade in der frühen Phase der
Synaptogenese und Spineentwicklung essentiell sind, finden diese Prozesse doch
verstärkt im frühen Lebensabschnitt statt (66, 106).
1.4 ProSAP/Shank-Mausmodelle
Wie im vorangegangenen Kapitel beschrieben, nimmt die Shank-Proteinfamilie eine
zentrale Rolle in Struktur und Funktion der Synapse ein. Aus diesem Grund liegt
nahe, dass sich bereits einige Arbeitsgruppen mit verschiedenen Shank-Knockout-
oder -Knockin-Mausmodellen beschäftigt und deren morphologische Auffälligkeiten
studiert haben, um den Einfluss der ProSAP/Shank-Proteine auf die neuronale
Plastizität aufzuzeigen. Im Folgenden sollen ausgewählte Mausmodelle vorgestellt
und ihre morphologischen Charakteristika erläutert werden.
15EINLEITUNG
1.4.1 Shank1
Hung et al. publizierten 2008 Untersuchungen an einer Shank1-/--Maus, welcher
durch die Deletion von Exon 14 und 15 sämtliche Isoformen des Proteins fehlten.
Es wurden die Spines und Synapsen in der CA1-Region des Hippocampus, Stratum
radiatum, in adulten Tieren untersucht. Im Vergleich zum Wildtyp zeigten sich die
Spines in ihrer Zahl reduziert und insgesamt verkleinert, was mit einer geringeren
glutamatergen Transmission korrelierte. Weiterhin fanden sich niedrigere Level an
Homer und GKAP. Hinsichtlich ihres Verhaltens zeigten die Shank1-/--Mäuse ein
gesteigertes Angstempfinden und eine beeinträchtigte Langzeitgedächtnisleistung
(61). 2014 führten Wöhr et al. am selben Mausmodell eine Verhaltensstudie im
Hinblick auf die soziale Interaktion von Shank1-/--Mutanten durch: Auch hier zeigten
die KO-Mäuse typisch autistische Verhaltensweisen und eine gestörte
Kommunikation mit den gesunden Kontrolltieren (136).
1.4.2 Shank2
Schmeisser et al. untersuchten 2012 die morphologischen Eigenschaften von
adulten Shank2ex7-/--Mäusen in der CA1-Region des Hippocampus, Stratum
radiatum. Mit der Deletion von Exon 7 war keine der Protein-Isoform mehr
nachweisbar (Abbildung 3). Morphologisch zeigten die Mutanten weniger Spines als
ihre gesunden Geschwister. Dies ging einher mit einer geringeren Übertragungsrate
elektrischer Signale zwischen den Synapsen. Auf molekularer Ebene fanden sich
erhöhte Level an AMPA- und NMDA-Rezeptoren in Cortex, Hippocampus und
Striatum. Weiterhin konnten Hyperaktivität, repetitive Verhaltensweisen und
Störungen in Kommunikation und sozialer Interaktion beobachtet werden.
Interessanterweise fand sich auf molekularer Ebene der Shank2ex7-/--Mäuse eine
Hochregulation der ProSAP2/Shank3-Level, dem – so wurde vermutet – ein
kompensatorischer Mechanismus zu Grunde lag (111).
Ein weiteres Mausmodell wurde durch die Arbeitsgruppe von Won et al. vorgestellt.
Es handelte sich hierbei um Mutanten, denen Exon 6 und 7 und damit ebenfalls
sämtliche Shank2-Isoformen genommen wurden. Die Tiere zeigten auch hier
autistische Verhaltensweisen wie gestörte soziale Interaktion und Kommunikation
sowie ein gehäuftes Auftreten repetitiver Verhaltensmuster. Zwar konnten
morphologisch keine signifikanten Unterschiede zu den Wildtypen bezüglich
16EINLEITUNG
Neuronendichte und PSD-Parametern in der CA1-Region, Stratum radiatum,
herausgearbeitet werden, doch ergab sich eine reduzierte Funktion der NMDA-
Rezeptoren, welche durch direkte und indirekte Stimulation der Rezeptoren wieder
aufgehoben werden konnte. Dies führte zu einer Verbesserung der sozialen
Interaktion der Shank2ex6+7-/--Mäuse (137).
17EINLEITUNG
Shank 2e
Shank 2a
Shank 2b
Shank 3a
Shank 3b
Shank 3c
Shank 3d
Shank 3e
Shank 3f
Abbildung 3: Schematische Darstellung der ProSAP/Shank-Isoformen
a, ProSAP1/Shank2-Isoformen Shank 2e, a-b. b, ProSAP2/Shank3-Isoformen Shank 3a-f. a-b, N
(N-terminal), ANK (Ankyrin repeats), SH3 (Scr homology 3), PDZ (PSD-95/DLG-/ZO1), Pro (Prolin-
reiche Domäne), Hbs (Homer binding site), SAM (sterile alpha motif), C (C-terminal); eigene
Darstellung in Anlehnung an Jiang und Ehlers (2013).
18EINLEITUNG
1.4.3 Shank3
Bozdagi et al. publizierten 2010 eine Studie, in welcher der Effekt einer Shank3-
Haploinsuffizienz auf die synaptische Funktion und Plastizität wie auch auf soziale
Verhaltensmuster untersucht wurde. Damit übertrugen sie den Genotyp von am
Phelan-McDermid-Syndrom erkrankten Menschen auf ein Mausmodell. Durch
Deletion der Exone vier bis neun konnte die Ankyrin-Domäne nicht translatiert und
somit die Isoformen Shank3a und Shank3b nicht gebildet werden (Abbildung 3). Es
entstand eine heterozygote Shank3ex4-9+/--Maus. Diese zeigte in der CA1-Region
des Hippocampus, Stratum radiatum, eine reduzierte elektrische Übertragung an
glutamatergen Synapsen und auch die Kommunikation der Neurone zwischen CA3-
und CA1-Region – welche die sogenannte Schaffer-Kollaterale darstellen – fiel
geringer aus als die Kontrollen in gesunden Tieren. Ebenfalls war die
Langzeitpotenzierung (LTP) in den Mutanten beeinträchtigt: Während die Spines
bei den gesunden Probanden im Rahmen der LTP langfristig expandierten, war
dieser Effekt bei den Shank3ex4-9+/--Mäusen nur vorübergehend. Weiterhin zeigte
sich die Kontaktaufnahme zu ihren gesunden Wurfgeschwistern in einer offenen
fünfminütigen Testperiode reduziert (24).
Die Arbeitsgruppe um Wang et al. entwickelte eine homozygote Shank3ex4-9-/--Maus,
um den Einfluss dieses Proteins auf Form und Funktion der Synapsen in vivo zu
untersuchen. Weiterhin führten sie Verhaltenstests durch, in welchen die Tiere
erneut Autismus-typische Muster zeigten. Da männliche Mutanten bezüglich der
Bewegungskoordination stärker beeinträchtigt zu sein schienen als ihre weiblichen
Geschwister, konnte hier sogar eine geschlechtsspezifische Aussage getroffen
werden. Morphologisch fand sich in der CA1-Region des Hippocampus, Stratum
radiatum, bei jüngeren KO-Tieren (vier Wochen) eine reduzierte Spinedichte,
welche sich nach zehn Wochen wieder angeglichen hatte. Die Länge der Spines
war sowohl bei den jungen als auch bei den älteren Mutanten im Vergleich zum
Wildtyp erhöht. Weiterhin konnten Wang et al. eine veränderte
Proteinzusammensetzung der PSD feststellen. So waren nicht nur die Shank3-
Level erniedrigt, sondern auch Homer1b/c, GKAP und GluA1 schwächer exprimiert
(132).
In dem Mausmodell von Peca et al. wurden durch den Verlust der PDZ-Domäne
lediglich die Isoformen Shank3e und Shank3f noch exprimiert, während Shank3a-d
fehlten (Abbildung 3). Es wurden somit mehr Isoformen verloren als in den zwei
19EINLEITUNG
zuvor beschriebenen Modellen. Übereinstimmend mit den vorangegangenen
Arbeiten stießen auch Peca et al. in ihren Tests auf Autismus-typisches Verhalten.
Shank3B-/--Mäuse zeigten repetitive Verhaltensweisen in Form von exzessiver
Fellpflege und scheuten die soziale Interaktion mit ihren Wurfgeschwistern.
Morphologische Untersuchungen wurden hier erstmals im dorsolateralen Striatum
durchgeführt. Zunächst fanden sich in den Mutanten längere Dendriten, welche
komplexere Dendritenbäume mit mehr Verzweigungen ausbildeten. Darüber hinaus
ergab sich eine Abnahme der Spinedichte im Vergleich zum Wildtyp; Länge und
Breite der Spines blieben hingegen unverändert. Weiterhin wurden die PSDs
vermessen – auch hier zeigten sich kürzere und schmalere PSDs in den
Shank3B-/--Mäusen. Abschließend konnte in elektrophysiologischen Tests eine
Verringerung elektrischer Potenziale an der Postsynapse in den Mutanten im
Vergleich zu den Kontrolltieren herausgearbeitet werden (91).
Schmeisser et al. nutzten in ihrer Studie neben den Shank2ex7-/--Mäusen ein
Shank3-Mausmodell, welches die Isoformen Shank3a bis einschließlich Shank3c
nicht exprimierte, da die SH3-Domäne des Shank-Proteins zerstört wurde
(Abbildung 3). Ziel war ein Vergleich der morphologischen, elektrophysiologischen
und behavioralen Analysen beider Mutanten. Die morphologische Analyse wurde in
der CA1-Region des Hippocampus durchgeführt. Unterschiede zwischen den
Shank3ab-/--Mäusen und ihren gesunden Wurfgeschwistern fanden sich hierbei
nicht, sowohl die Spinedichte als auch Länge und Breite der PSDs zeigten keine
signifikanten Abweichungen. Auf molekularer Ebene verhielten sich die Shank3-
Mutanten umgekehrt zu den Shank2-Mutanten: AMPA- und NMDA-Rezeptoren
waren in Cortex, Hippocampus und Striatum schwächer exprimiert als im Wildtyp,
und in den Shank3ab-/--Mäusen fiel eine kompensatorische Zunahme an
ProSAP1/Shank2 auf (111).
Einen gänzlich anderen Ansatz verfolgten Han et al. 2012 in einer Shank3-
Überexpressionsstudie. Dabei kam durch das Einsetzen eines Transgens eine
Erhöhung der Isoformen Shank3a bis Shank3c um das 1,2- bis 2-fache zustande.
Insgesamt ergab sich eine Zunahme der Shank-Proteine um 50 %. Die veränderten
Mäuse zeigten ein manisches, hyperaktives Verhalten und erlitten epileptische
Anfälle. Als Korrelat auf molekularer Ebene fand sich eine Zunahme des
Strukturproteins F-Aktin in Shank3+/+-Mäusen (57). Die erhöhte Shank3-
Proteindosis führt zu einer verstärkten Interaktion mit dem Arp2/3-Proteinkomplex.
20EINLEITUNG
Dieser bindet an das F-Aktin und initiiert die dendritische Nukleation sowie das
sogenannte „barbed end branching“, ein Verzweigen der Aktinfilamente (39).
Morphologisch zeigte sich eine erhöhte Spinedichte im Hippocampus (CA1, Stratum
radiatum). Interessanterweise konnte das hyperaktive Verhalten der transgenen
Mäuse durch das Antikonvulsivum Valproat therapiert werden (57).
Auch Mei et al. nutzten im Jahr 2016 ein konditionales Knockin-(KI)-Mausmodell
und erweiterten damit die aus der gleichen Arbeitsgruppe stammende Studie von
Peca et al. im dorsalen Striatum (Lokalisation nicht näher bezeichnet): Mit Hilfe des
Rekombinationssystems Cre/loxP flankierten sie die PDZ-Domäne mit loxP in
Inversionsstellung. In Abwesenheit des Rekombinase-Enzyms Cre verhielt sich die
Shank3fx/fx-Maus genotypisch wie ein Knockout-Modell und zeigte – analog zu den
Daten von Peca et al. – zum einen die bereits bekannten Autismus-typischen
Verhaltensweisen, zum anderen eine geringere synaptische Transmission sowie
eine reduzierte Spinedichte im dorsalen Striatum. Durch Aktivierung von Cre mittels
Tamoxifen in ausgewachsenen Mäusen konnten die Shank3-Level wieder auf ein
gesundes Niveau angehoben werden konnte, wie es in den Kontrolltieren zu
messen war. Hiernach wurden die Experimente wiederholt und bestätigten
schließlich die Rescue-Hypothese: Die Knockin-Mäuse Shank3fx/fx:Cre legten die
zuvor gezeigte übermäßige, repetitive Fellpflege ab und interagierten in normalem
Maß mit ihren Geschwistern. Auch die gestörte synaptische Übertragung striataler
Neurone normalisierte sich und hinsichtlich der Spinedichte überstiegen die
Mutanten die Kontrollgruppe sogar (84).
Im Gegensatz dazu arbeiteten Kouser et al. mit einem homozygoten Mausmodell,
dem durch Deletion von Exon 21 inklusive der Homer-Bindungs-Domäne sämtliche
Shank3-Isoformen fehlten. Hier zeigten die Shank3DC/DC-Mäuse wieder das bereits
bekannte Verhalten mit minimaler sozialer Interaktion, einem ausgeprägten
Vermeidungsverhalten in Bezug auf neue Situation und Beeinträchtigung in
Bewegung und Koordination. Darüber hinaus führte der Verlust am Shank3-Protein
verglichen mit dem Wildtyp zu einer herabgesetzten NMDA- und AMPA-Rezeptor-
vermittelten synaptischen Transmission im Hippocampus (CA1, Stratum radiatum).
Auch fanden sich erhöhte mGluR5-Level an der PSD. Die Spinedichte blieb
hingegen unverändert (72).
Eine Studie von Uppal et al. fasste überwiegend die neuronale Ultrastruktur von
Shank3-Mutanten ins Auge. Ausgehend von einem heterozygoten Mausmodell,
21Sie können auch lesen

















































