PRÄKLINISCHE DOKUMENTATION UND INVESTIGATOR S BROCHURE - BFARM
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

25.11.2020
Präklinische Dokumentation und
Investigator´s Brochure
Dr. Gunther Boos
Absender | Titel | 25.11.2020 | Seite 1
Inhaltsverzeichnis
1. Einleitung
Ziele der präklinischen Testung, Übersicht Arzneimittelentwicklung
2. Präklinische Dokumentation
Regulatorischer Hintergrund
3. Investigator´s Brochure (IB) = Prüferinformation
Definition, generelle Anforderungen, Inhalt
4. ICH M3 (R2)
Zentrale Guideline ‐ regelt Umfang und Timing der präklinischen Testung
5. Beispiele präklinischer Studientypen
Genotoxizität, toxikologische Studien mit wiederholter Verabreichung
Absender | Titel | 25.11.2020 | Seite 2
1
25.11.2020
1. Einleitung
Ziele der präklinischen Testung
Übersicht Arzneimittelentwicklung
Absender | Titel | 25.11.2020 | Seite 3
Damit die Sicherheit der Teilnehmer von
klinischen Prüfungen gewahrt wird,
prüfen die Wissenschaftler des BfArM
jede klinische Prüfung. Dabei bewerten
sie die Unterlagen zur pharmazeutischen
Herstellung der untersuchten
Arzneimittel, die Angemessenheit und
die Ergebnisse der pharmakologischen
und toxikologischen Vorprüfungen sowie
den Prüfplan, der genau beschreibt, wie
die Studie durchgeführt werden soll.
Bereits in der Planungsphase klinischer
Studien unterstützt das BfArM mit einem
breiten Spektrum wissenschaftlicher
Beratung.
Absender | Titel | 25.11.2020 | Seite 4
2
25.11.2020
Einleitung
Primäre Ziele der präklinischen Testung:
Charakterisierung der pharmakologisch‐toxikologischen Eigenschaften einer
Prüfsubstanz zu dem Zweck eine initiale therapeutische Dosis und sichere
Startdosis bei FiH‐Studien zu definieren.
Des weiteren werden potentielle toxische Dosen und Effekte (inklusive deren
Reversibilität) und Targetorgane für Toxizität iden fiziert → um in Konsequenz
ein adäquates klinisches Monitoring etablieren zu können.
Die relative Bedeutung der präklinischen Daten nimmt im Laufe der
Arzneimittelentwicklung mit Zunahme der klinischen Daten immer weiter ab.
Absender | Titel | 25.11.2020 | Seite 5
Einleitung ‐ Arzneimittelentwicklung
Präklinische Studien (sicherheitsrelevant, GLP)
Z
Sicherheitspharmakologie, Genotoxizität, Toxizität bei wiederholter Gabe (2 Wo)
u
Toxizität bei wiederholter Gabe (4 Wo, 3 Mo) l
Toxizität bei wiederholter Gabe (6 - 9 Mo), Reproduktionstoxizität, Kanzerogenität
a
s
Klinische Entwicklungsphase s
Phase I Kinetik, Verträglichkeit, 20‐50 Gesunde, sehr kurze Exposition
u
n
Phase II Therapeutisch explorativ, 50‐100 Patienten, 1‐3 Monate
g
Phase III Therapeutisch konfirmativ > 1000 Patienten,
Monate bis Jahre
6 Absender | Titel | 25.11.2020 | Seite 6
Bundesinstitut für Arzneimittel und Medizinprodukte | Das BfArM ist ein Bundesinstitut im Geschäftsbereich des Bundesministeriums für Gesundheit
3
25.11.2020
2. Präklinische Dokumentation
Regulatorischer Hintergrund
Absender | Titel | 25.11.2020 | Seite 7
Präklinische Dokumentation (1)
Gesetzliche / regulatorische Verankerung (1)
Richtlinie 2001/20/EG DES EUROPÄISCHEN PARLAMENTS UND DES RATES vom 4. April 2001 zur
Angleichung der Rechts‐ und Verwaltungsvorschriften der Mitgliedstaaten über die Anwendung der guten
klinischen Praxis bei der Durchführung von klinischen Prüfungen mit Humanarzneimitteln, AMG ‐ § 40 sowie die
zugehörige GCP Verordnung dienen der nationalen Umsetzung diese Richtlinie 2001/20/EG
ICH E8 GENERAL CONSIDERATIONS FOR CLINICAL TRIALS: Before any clinical trial is carried out, results of
non‐clinical investigations or previous human studies should be sufficient to indicate that the drug is acceptably
safe for the proposed investigation in humans. The purpose and timing of animal pharmacology and toxicology
studies intended to support studies of a given duration are discussed in ICH M3. The role of such studies for
biotechnology products is cited in ICH S6.
ICH E8(R1) Draft version Endorsed on 8 May 2019: In preparing a development plan, the non‐clinical information that is required for the drug
should be addressed. Non‐clinical information may include toxicology, carcinogenicity, pharmacology, and pharmacokinetics to support clinical trials (e.g., ICH
Safety (S) Guidelines and M3 Nonclinical Safety Studies). Important considerations for determining the necessary non‐clinical studies, and their timing with respect
to clinical studies, depend on the physiological and toxicological characteristics of the drug. These characteristics can include the drug’s chemical or molecular
properties (e.g., small‐molecule, biologic/cellular/gene therapy, complex drug, and vaccine); pharmacological basis of principal effects (mechanism of action);
route(s) of administration; absorption, distribution, metabolism, and excretion (ADME); physiological effects on organ systems; dose/concentration‐response
relationships; half‐life; duration of action; and indication. Use of the drug in special populations (e.g., pregnant or breast‐feeding women, children, elderly) may
require additional toxicological assessments. Before proceeding to studies in humans, there should be sufficient information to support selection of the initial
human dose and safe duration of exposure, and to provide a preliminary assessment of physiological and toxicological effects of the drug.
regeln / regulieren
Voraussetzung Genehmigungsfähigkeit: unter anderem
1. Vorherige Durchführung einer pharmakologisch‐toxikologischen Prüfung (nicht‐klinischen oder
präklinischen) Testung entsprechend aktueller wissenschaftlicher Standards
Absender | Titel | 25.11.2020 | Seite 8
4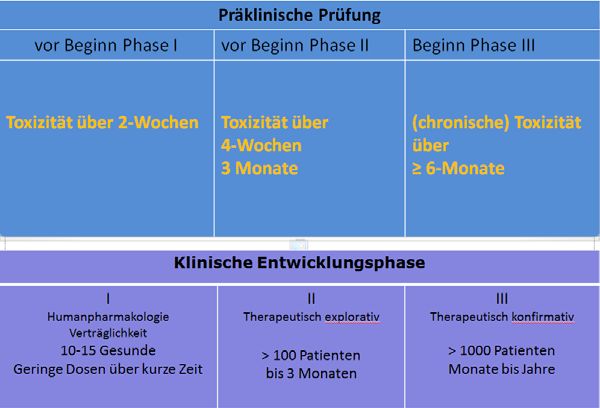
25.11.2020
Präklinische Dokumentation (2)
EU‐Verordnung 536/2014 zu Klinischen Prüfungen mit Arzneimitteln
Ablösung der Richtlinie 2001/20/EG
Richtlinie 2001/20/EG erlaubt nationale Besonderheiten
Ziel: EU weites harmonisiertes Verfahren ‐ Einführung eines gemeinsamen,
koordinierten Assessments bei multinationalen klinischen Prüfungen
wenig Einfluss auf die präklinische Dokumentation und deren
inhaltliche Bewertung
Absender | Titel | 25.11.2020 | Seite 9
Präklinische Dokumentation (3)
Gesetzliche Verankerung (2)
1. Guidance CT‐1, March 2010 (EU Commission)
2. ICH E6 (R2) ‐ Guideline for Good Clinical Practice
3. EU‐Richtlinie 2005/28/EC „Good Clinical Practice“
4. ICH M3 (R2) ‐ Non‐clinical safety studies for the conduct of human clinical trials for
pharmaceuticals
regeln / regulieren
Was? Wie? Wann? Wo?
Hier sind detailliertere Informationen zu Qualität, Umfang, Struktur und Bewertung der
vorzulegenden präklinischen Daten zu finden.
IMPD (gesamte Informationen zum Prüfpräparat) – fast alle Sponsoren verweisen auf
Investigator's Brochure (IB)
Absender | Titel | 25.11.2020 | Seite 10
5
25.11.2020
3. Investigator´s Brochure
Aufbau/Inhalt
Abschnitt Nonclinical Studies
Darstellung und Interpretation der präklinischen Daten
Absender | Titel | 25.11.2020 | Seite 11
Investigator´s Brochure (1)
= IB = Prüferinformation = Prüfarztbroschüre
Definition (Guideline E6(R2)): Zusammenstellung der für die Untersuchungen mit
Prüfpräparaten am Menschen relevanten klinischen und nicht‐klinischen Daten über
das betreffende Präparat.
Umfang ca. 60–120 Seiten, Extrakt/Konzentrat aus sehr viel umfangreicheren
Datenmengen.
Generelle Anforderungen
→ Informa on des Prüfarztes zum besseren Verständnis des Studienprotokolls:
(Rationale, Dosierungen, Dosischema, Ein‐ Ausschlusskriterien, Monitoring)
→ Info in objektiver, klarer und verständlicher Form
→ IB ist zu aktualisieren wenn neue relevante Daten vorliegen, mind. 1 x / Jahr
→ Falls Prüfsubstanz zugelassen: IB kann durch SmPC ersetzt werden (Phase 4)
→ Abschnitt nicht‐klinische Daten ist durch einen „Präkliniker“ zu genehmigen
Absender | Titel | 25.11.2020 | Seite 12
6
25.11.2020
Investigator´s Brochure (2)
Inhalt geregelt in Guideline for good clinical practice E6(R2)
Absender | Titel | 25.11.2020 | Seite 13
Investigator´s Brochure (3)
NONCLINICAL STUDIES Pharmacology & Toxicology ‐ Struktur, Gliederung
• Primäre (gewünschte pharmakologische Aktivität) Pharmakodynamik
• Sekundäre Pharmakodynamik
• Pharmakokinetik (ADME)
• Sicherheitspharmakologie (Eimalgabe, Herz, Atmung, ZNS)
• Toxikokinetik (Bestimmung AUC und Cmax in toxikologischen Studien)
• Toxikologie (Applikationsart, Anwendungsdauer müssen der am Menschen geplanten entsprechen):
Toxizität nach Einmalgabe („single dose“ einer hohen Dosis, vormals LD50)
Toxizität nach Mehrfachgabe („repeat dose“, 2 Spezies, Targetorgan)
Genotoxizität (Detektion von genotoxischen/mutagenen Effekten)
Reproduktionstoxikologie (Ratte, Kaninchen, Fertilität, Embryotox, Prä‐Postnatal)
Kanzerogenität (zur Zulassung, Ratte Maus 2 Jahre)
Lokale Toleranz (fallweise, in „repeat dose“ Studien integrieren)
Photosafety (fallweise, Absorption, Verteilung, in vitro 3T3 Neutralrot uptake Assay)
Immunotoxizität (fallweise)
Juvenile Tierstudien (fallweise, Adultprogramm und Reprotox komplett, PIP)
Absender | Titel | 25.11.2020 | Seite 14
725.11.2020
Investigator´s Brochure (4)
NONCLINICAL STUDIES Pharmacology & Toxicology ‐
Allgemeine Darstellung der Daten
‐ Übersicht über alle relevanten Studien (GLP‐Status, Dosierung, Applikationsart,
Expositionsdauer, Konzentration, Speziesbegründung, Zelllinie, Ergebnisse)
‐ Bei Erstanwendung/Anwendung in früher Phase neuer Arzneimittel am
Menschen wird eine kritische Diskussion anhand folgender Kriterien erwartet:
Neuer bzw. nicht genau bekannter Wirkmechanismus
Neue oder nicht genau bekannte Struktur und biologische Funktion des
Zielmoleküls
Relevanz der Tierversuche
‐ Zusammenfassung der einzelnen Studien
‐ Diskussion und Schlussfolgerung (klinische Relevanz, Sicherheitsabstände)
‐ Tabellarische Formate/Auflistungen sollten nach Möglichkeit verwendet
werden, um die Übersichtlichkeit der Präsentation zu verbessern.
Absender | Titel | 25.11.2020 | Seite 15
Investigator´s Brochure (5)
NOAEL = No Observed Adverse Effect Level.
Höchste Dosis in Studien mit wiederholter
Gabe, bei der keine signifikant erhöhten
NONCLINICAL STUDIES Pharmacology
schädigenden & Toxicology
behandlungsbedingten Befunde
beobachtet wurden.
Spezifische Daten zu einzelnen Studien
1. Spezies Exposition am NOAEL / geschätzte bzw.
2. Dosis gemessene Exposition beim Menschen
=
3. „noteworthy findings“ (z.B. histologische Befunde)
Sicherheitsabstand, MOE (Margin of Exposure)
4. Reversibilität (ja/nein)
5. NOAEL, Toxikokinetik: (AUC, Cmax)
6. Sicherheitsabstände zur Anwendung beim Menschen
7. Tabellen Startdosis FIH [mg/Proband]: i.d.R.
NOAEL [mg/kg] / Speziesfaktor x 60 kg x SF1
1FDA Guidance for Industry. Estimating the MRSD in Initial Clinical Therapeutics in Adult Healthy Volunteers.
Absender | Titel | 25.11.2020 | Seite 16
825.11.2020
Investigator´s Brochure (6)
Tabellarische Darstellung präklinischer Studien, Beispiel Repeated dose Toxicology
Species/St Duration/Route/N Dose Exposure Major findings NOAEL
udy ID/GLP umber/ Sex/ (mg/kg Cmax (mg/kg/day)
Group KG) (ng/ml)
AUC Exposure margin to
(ng*h/ml) planned clinical
dose, based on
exposure (MOE)
Rat Wistar/ 4‐weeks/oral 0, 1, 3, 10 Day 28: 1 mg/kg (NOEL): ‐ NOAEL: 3 mg/kg
GBL‐R13/ (gavage)/10 /sex
GLP: Yes /group 1 mg/kg: 3 mg/kg (NOAEL): AUC Klinik: 30
Cmax: 25 death: one male (start dose 50
Recovery 2 weeks AUC: 75 animal; liver mg/day)
enzymes slightly ↑, MOE = 7 (210/30 )
3 mg/kg: reversibel
Cmax: 75 AUC Klinik: 120
AUC: 210 10 mg/kg: liver (max. human 250
enzymes mg/day)
10 mg/kg: moderately ↑, liver MOE = 1,8 (210/120)
Cmax: 250 histopathological
AUC: 1200 findings
17 Absender | Titel | 25.11.2020 | Seite 17
Bundesinstitut für Arzneimittel und Medizinprodukte | Das BfArM ist ein Bundesinstitut im Geschäftsbereich des Bundesministeriums für Gesundheit
Investigator´s Brochure (7)
Diskussion/Bewertung der Befunde im Hinblick auf:
‐ Schweregrad / Reversibilität / Dosisabhängigkeit
‐ Mechanistische Erklärungen
‐ Spezies‐Spezifität
‐ Human‐Relevanz
‐ Abzuleitende Überwachungsparameter
Risikoabschätzung
Absender | Titel | 25.11.2020 | Seite 18
925.11.2020
Investigator´s Brochure (8)
Diskussion (unter Einbeziehung aller präklinischen Infos):
1 mg/kg: keine auffälligen Befunde → NOEL
3 mg/kg: Tod: ein ♂ der mittleren Dosisgruppe (Applikationsfehler). Leberenzyme
leicht ↑, Leber ohne histopathologische Befunde, vermutlich adaptiv da
Prüfsubstanz durch Leber metabolisiert wird. Rückkehr zu Normalwerten nach
Absetzen der Substanz → NOAEL
MOE zur Startdosis 7‐fach (AUCNOAEL Ratte/AUC Human: 210/30)
MOE zur klinischen Maximaldosis 1,8‐fach (AUC RatteNOAEL/AUC Human: 210/120).
10 mg/kg: Lebertoxizität (Leberenzmye deutlich ↑, histopath. Befunde). Leichte
Tendenz zur Reversibilität nach Absetzen der Substanz. Exposition nicht mehr
dosisproportional. Veränderte Metabolisierung durch Sättigungsprozesse. Keine
entsprechenden Befunde aus zweiter Tierspezies (Hund) und bei bisherigen
(frühen) Studien am Menschen. Eventuell nagerspezifischer Befund.
Risiko akzeptabel. Vorsichtige Dosiserhöhung. Engmaschige Überwachung der
Leberenzyme. Definition von Abbruchkriterien. Weitergehende/begleitende in
vivo PK‐Untersuchungen am Menschen.
Absender | Titel | 25.11.2020 | Seite 19
4. Guideline ‐ ICH M3 (R2)
regelt Umfang der präklinischen Testung
Timing zur klinischen Entwicklungsphase
Absender | Titel | 25.11.2020 | Seite 20
1025.11.2020
ICH M3(R2) Guideline (1)
Absender | Titel | 25.11.2020 | Seite 21
ICH M3(R2) Guideline (2)
• International International Council for Harmonisation of Technical
Requirements for Pharmaceuticals for Human Use, d.h. gilt verbindlich in
Grüderregionen EU, Japan und USA nun beisitzend China, Brasilien, (Süd‐)
Korea, Kanada, Taiwan, Schweiz, Singapore. (www.ich.org)
• Empfehlenden Charakter „soft law“
• Regelt Umfang der präklinische Entwicklung (abhängig von Indikation,
Behandlungsdauer, Art der Substanz, experimentelle Ansätze)
• Verweist auf weitere spezifischere Guidelines (u.a. Advanced cancer (ICH
S9), Lokale Toleranz (EMA/CHMP/SWP/2145/2000 R1), Phototox (ICH S10),
juvenile Tox (EMEA/CHMP/SWP/169215/2005), Reprotox (ICH S5 R2)
• Tierversuche gesetzlich vorgeschrieben. Allerdings klare Empfehlung zur
Reduktion des Einsatzes von Versuchstieren entsprechend dem 3R‐Prinzip
(Reduce, Refine, Replace)
• Präklinische Exposition > klinische Exposition (Dauer und Dosis)
• Timing zur klinischen Entwicklung
Alle Guidelines sind auf den Homepage der European Medicines Agency (EMA) (http://www.ema.europa.eu/ema/) und/oder der „International Conference
on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use“ (http://www.ich.org/) zu finden
Absender | Titel | 25.11.2020 | Seite 22
1125.11.2020
ICH M3(R2) Guideline (3)
zeitliches Timing der Präklinik zur klinischen Phase
• Primäre Pharmakodynamik (vor FIH/Phase 1)
• Pharmakokinetik (in vitro, ADME)
• Sicherheitspharmakologie („core battery“ vor FIH/Phase 1)
• Toxizität nach Einmalgabe (vor FIH/Phase 1)
• Toxizität nach Mehrfachgabe (14 Tage vor FIH/Phase 1)
• Toxizität nach Mehrfachgabe (4 Wochen ‐ 3 Monate, vor Phase 2)
• Toxizität nach Mehrfachgabe (≥ 6 Monate, i.d.R. vor Phase 3)
• Genotoxizität (in vitro vor FIH/Phase 1)
• Genotoxizität (in vivo vor Phase 2)
• Reproduktionstoxikologie (WOCBP, Fertilität zu Phase 3)
• Kanzerogenität (zur Zulassung)
• Lokale Toleranz (in repeat dose Studien, fallweise)
• Photosafety (fallweise)
• Immunotoxizität (fallweise)
• Juvenile Tierstudien (fallweise)
Absender | Titel | 25.11.2020 | Seite 23
5. Beispiele präklinischer Studientypen
Prüfung auf Genotoxizität
Toxikologische Studien mit wiederholter Verabreichung
Absender | Titel | 25.11.2020 | Seite 24
1225.11.2020
Genotoxizität (1)
Warum Testung auf Genotoxizität?
DNA‐Schaden → Mutation →→→→ Krebs
Effekt häufig irreversibel, in vielen Fällen keine Wirkschwelle
definierbar, Kanzerogener Langzeitteffekt, d.h. praktisch (und
auch aus ethischer Sicht) nicht in klinischen Studien nachweisbar,
Schädigung der Keimzellen in den Reproduktionsorganen
(Vererbung auf Nachkommen)
Wann Testung auf Genotoxizität?
Sehr früh in der Entwicklung, d.h. vor erstmaliger klinischer Gabe
Absender | Titel | 25.11.2020 | Seite 25
Genotoxizität (2)
Bild Beispiel „groß“
Präklinische Prüfung
vor Beginn Phase I vor Beginn Phase II Beginn Phase III
Genotoxizität in vitro Genotoxizität in vivo
Klinische Entwicklungsphase
I II III
Humanpharmakologie Therapeutisch explorativ Therapeutisch konfirmativ
Verträglichkeit
10‐15 Gesunde > 100 Patienten > 1000 Patienten
Geringe Dosen über kurze Zeit bis 3 Monaten Monate bis Jahre
26 Absender | Titel | 25.11.2020 | Seite 26
Bundesinstitut für Arzneimittel und Medizinprodukte | Das BfArM ist ein Bundesinstitut im Geschäftsbereich des Bundesministeriums für Gesundheit
1325.11.2020
Genotoxizität (3)
Definitionen
Genotoxizität: Allgemeine Schädigung oder Veränderung des
genetischen Materials (DNA) durch biologische, chemische oder
physikalische Noxen.
Mutagenität: Bezeichnet die Induktion dauerhafter, vererbarer
Veränderungen des genetischen Materials.
Indikatortest
(Messung primärer DNA‐Schäden)
Mutationstest
(Messung irreversibler DNA‐Schäden)
Absender | Titel | 25.11.2020 | Seite 27
Genotoxizität (4)
• Es gibt eine ganze Reihe von genotoxischen Endpunkten wie Genmutationen,
Chromosomen‐Mutationen und Genomutationen (Chromosomenzahl). Kein
gängiges Testsystem das zuverlässig alle diese Endpunkte erfasst. Zudem wird der
wichtigste Mutagenitätstest in Bakterien durchgeführt ‐ Unterschiede zwischen
prokaryontischen und eukaryontischen Zellen.
Beispiel einer Testbatterie:
• A test for gene mutation in bacteria ‐ Ames test
• An in vitro test with cytogenetic evaluation of chromosomal damage with
mammalian cells or an in vitro mouse lymphoma tk assay.
• An in vivo test for chromosomal damage using rodent hematopoietic cells ‐
Mikronukleustest
• Compounds giving positive results in the standard test battery may, depending on
their therapeutic use, need to be tested more extensively ‐ Comet‐Assay
Guideline: ICH S2 (R1): Guidance on Genotoxicity Testing and Data Interpretation for Pharmaceuticals intended for Human use
Absender | Titel | 25.11.2020 | Seite 28
1425.11.2020
Genotoxizität (5)
Ames Test ‐ Prinzip
Hohe Zahl an
Testsubstanz (Mutagen) Rückwärtsmutanten
his‐ zu his+
Plattieren Medium ohne Histidin Inkubation
Salmonella Stamm
(Histidinabhängig =
his‐) + S9‐Mix
Spontanmutanten
Kontrolle (negativ)
OECD guideline 471
29 Absender | Titel | 25.11.2020 | Seite 29
Bundesinstitut für Arzneimittel und Medizinprodukte | Das BfArM ist ein Bundesinstitut im Geschäftsbereich des Bundesministeriums für Gesundheit
Genotoxizität (6)
Mikrokerntest (in vitro oder in vivo)
Prinzip: Mikronuklei als kleine runde von
Kernmembran umschlossene, DNA
enthaltende Partikel sichtbar. Als Marker
für die mutagene Wirkung eines Agens
dient die Häufigkeit von Zellen mit
Mikrokernen.
MK
In vivo MNT (Erytrocyten):
Nager, hohe Dosen, IP oder IV,
MK Expositionsmessungen. Kernausstoß
während Erythrozytenreifung, nicht aber
Interphasenkerne mit Mikrokernen (MK) von Mikrokernen
OECD guidelines 474, 487
30 Absender | Titel | 25.11.2020 | Seite 30
Bundesinstitut für Arzneimittel und Medizinprodukte | Das BfArM ist ein Bundesinstitut im Geschäftsbereich des Bundesministeriums für Gesundheit
1525.11.2020
Genotoxizität (7)
Komet‐Assay (Einzellgelelektrophorese)
• Detektion von DNA‐Strangbrüchen auf Einzelzellniveau
• Agarosegelelektroporetische Auftrennung nach Molekülgröße
• DNA‐Strangbrüche führen dazu, dass die DNA in Fragmenten in Richtung Anode wandert
• DNA‐Färbung mit Ethidiumbromid
• Länge und Intensität des Kometen direkt proportional zum Ausmaß der DNA‐Schädigung
Kathode (-) Anode (+) Kathode (-) Anode (+)
Negativkontrolle Positivkontrolle
OECD guideline 489
31 Absender | Titel | 25.11.2020 | Seite 31
Bundesinstitut für Arzneimittel und Medizinprodukte | Das BfArM ist ein Bundesinstitut im Geschäftsbereich des Bundesministeriums für Gesundheit
Toxizitätsstudien mit wiederholter Gabe (1)
Identifikation potentieller Zielorgane der toxischen Wirkungen und Definierung des
Übergangs von nichttoxischen zu toxischer Dosen (NOAEL). Die Toxizitätsstudien mit
wiederholter Gabe beim Tier sind so strukturiert, dass sie über eine vergleichbare oder
längere Expositionsdauer als die beabsichtigte Dauer der KP beim Menschen durchgeführt
werden. So würden Toxizitätsstudien mit zwei Spezies (Nager und Nicht‐Nager) für eine
Mindestdauer von zwei Wochen im Allgemeinen jede KP von einer Dauer von bis zu zwei
Wochen unterstützen. KPs von längerer Dauer sollen von Toxizitätsstudien von mindestens
gleicher Dauer unterstützt werden. Chronische Studien über 6‐Monate (Nager) und 9‐
Monate (Nicht‐Nager) unterstützen im Allgemeinen eine zeitlich unlimitierte Behandlung
in klinischen Prüfungen (ICH M3 Guideline).
Absender | Titel | 25.11.2020 | Seite 32
1625.11.2020
Toxizitätsstudien mit wiederholter Gabe (2)
Absender | Titel | 25.11.2020 | Seite 33
Toxizitätsstudien mit wiederholter Gabe (3)
• Identifikation potentieller Zielorgane der toxischen Wirkungen und Def.
des Übergangs von nichttoxischen zu toxischer Dosen (NOAEL)
• Behandlungsgruppen: Kontrollgruppe (negativ), sowie drei Dosisgruppen
(Definition eines NOAELs, toxischer Dosen)
• Monitoring verschiedener Parameter während der Studie: Futteraufnahme,
Verhalten, KG, Hämatologie, klinische Chemie
• Es gibt eine vorgeschriebene Anzahl von > 40 Gewebearten, die histologisch
untersucht werden sollen, in Abhängigkeit von den gefundenen Zielorganen
kann dies umfangreicher sein
• begleitende toxikokinetische Untersuchungen (Exposition am
NOAEL/Klinischen Exposition = Sicherheitsfaktor)
Absender | Titel | 25.11.2020 | Seite 34
1725.11.2020
Toxizitätsstudien mit wiederholter Gabe (4)
• Untersuchung der Reversibilität (Recoverygruppen)
• jeweils 2 Säuger (Nager und Nichtnager, Spezieswahl anhand ähnlicher PK
und PD zum Menschen, Affe nur wenn zwingend erforderlich)
• Untersuchung an beiden Geschlechtern
• Zusätzliche Tiere für toxikokinetische Untersuchungen (Mikrosampling,25.11.2020
Vielen Dank für Ihre
Aufmerksamkeit!
Kontakt
Bundesinstitut für Arzneimittel und Medizinprodukte
Klinische Prüfung
Präklinik und pharmazeutische Qualität
Kurt‐Georg‐Kiesinger‐Allee 3
53175 Bonn
Ansprechpartner
Dr. Gunther Boos
Gunther.Boos@bfarm.de
www.bfarm.de
Folien auf der BfArM Homepage (https://www.bfarm.de/DE/Service/Veranstaltungen/Ringvorlesungen/2020‐Winter/_node.html )
Guidelines sind auf den Homepages der EMA (http://www.ema.europa.eu/ema/ ) und ICH (http://www.ich.org/ ) zu finden
Absender | Titel | 25.11.2020 | Seite 37
19Sie können auch lesen

















































