PRAKTIKUM TRENNMETHODEN SOSE 2021 - PROTOKOLL
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Praktikum Trennmethoden
SoSe 2021
Protokoll
DNA-Analytik
Betreuer: Prof. Dr. Norbert Sträter
Franz Thiemann (3750567)
Georg Müller ()
Versuchsdurchführung: 26.07.2021
Protokollabgabe: 24.09.2021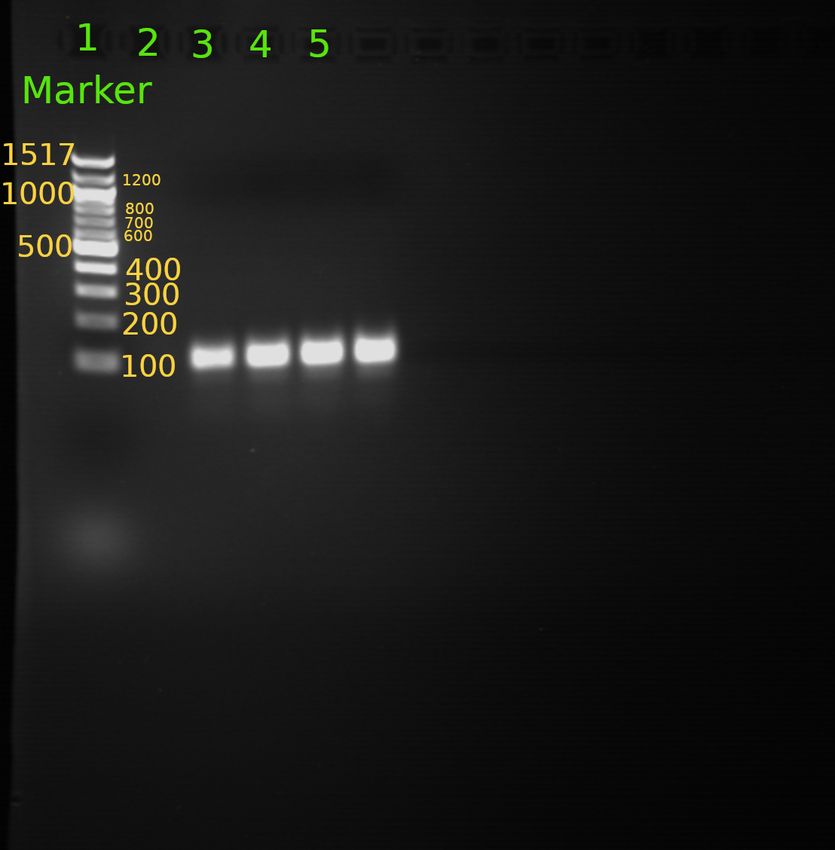
Inhaltsverzeichnis
1 Aufgabenstellung 3
2 Chemikalien 3
3 Geräte und Materialien 3
4 Durchführung und Beobachtung 4
4.1 Probennahme . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
4.2 DNA-Extraktion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
4.3 Polymerase-Kettenreaktion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
4.4 Restriktionsverdau . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
4.5 Herstellen des Agarose-Gels . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
4.6 Gelelektrophorese . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
5 Auswertung 6
5.1 Ergebnisse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
5.2 PCR-Produkte . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
5.3 Verdaute und unverdaute genomische DNA . . . . . . . . . . . . . . . . . . . . . . . 8
5.4 Diskussion des Laufverhaltens und der Verdauung . . . . . . . . . . . . . . . . . . . 8
5.5 Geschnittene DNA-Vektoren . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
5.6 Unterschiede der mit BamHI verdauten Proben . . . . . . . . . . . . . . . . . . . . . 9
6 Fehlerbetrachtung 10
7 Literatur 10
2DNA-Analytik 4.1
1 Aufgabenstellung
Genomische DNA aus Haarfollikel und einer Tomate soll mittels Phenol-Chloroform-Extraktion
isoliert werden. Die DNA aus den Haarfollikeln soll einer PCR-Reaktion unterzogen werden. An-
schließend wird das Laufverhalten der Proben zusammen mit Proben plasmidischer DNA in der
Agarose-Gelelektrophorese untersucht.
2 Chemikalien
• eine Tomate
• FastDigest, BamHI, NdeI
• Tris
• Essigsäure
• EDTA
• Glycerol
• TAE-Puffer, FastDigest-Puffer, CutSmart-Puffer
• Agarose
• Ethidiumbromid-Lösung
• Marker, Proben
• Dithiothreitol
3 Geräte und Materialien
• Trichter, Filter, Pinzette
• Gelkammer, Gelschlitten, Spacer, Gelkamm, Stromversorgungsgerät
• Mikrowelle
• Flakons, Eppendorfgefäße
• Eppendorf-Pipetten verschiedener Größen mit Spitzen
• Thermoschüttler, Tischzentrifuge, Vortexer
3DNA-Analytik 4.4
4 Durchführung und Beobachtung
4.1 Probennahme
Zur Entnahme der menschlichen DNA wurden 5 Barthaare von Herrn Müller mit einer Pinzette
entfernt und 1 cm hinter der Wurzel gekürzt. Die Haare wurden in einem Eppendorfgefäß mit
947 µl Puffer X1, 40 µl ein-molare Dithiothreitol-Lösung und 12,5 µl Proteinase K versetzt. Der
Inhalt wurde kurz im Vortexer durchmischt und mit der Tischzentriufuge am Boden gesammelt.
Anschließend wurde das Eppendorfgefäß bei 56 °C für zwei Stunden in den Thermoschüttler gestellt.
Zur Entnahme der DNA-Probe einer Tomate wurde eine halbe Tomate in Stückchen geschnitten
und im Mörser mit 15 ml Puffer X2 verrieben. Die Mischung wurde filtriert und 0,5 ml davon in
ein Eppendorfgefäß gefüllt.
4.2 DNA-Extraktion
Die Extraktion der DNA erfolgte mittels Phenol-Chloroform-Extraktion und wurde für die Haar-
probe, die Tomatenprobe und eine E.coli -Probe durchgeführt. Dabei wird genutzt, dass Lipide
sich in der organischen Phase lösen und DNA sowie RNA in der wässrigen Phase. Die denatu-
rierten Proteine sammeln sich in der Interphase, was vor allem bei der Tomatenprobe deutlich als
weißer Niederschlag erkennbar war. So können diese abgetrennt werden. Dabei wurde nach dem
Praktikumsskript1 vorgegangen. Bei der Haarprobe konnte keine vollständige Phasenseperation be-
obachtet werden. Es wurde daher nur einmal extrahiert und anschließend mit der überstehenden,
wässrigen Phase weitergearbeitet.
Die Messung der DNA-Konzentration erfolgte mit einem NanoDrop UV-VIS-Spektrometer. Dabei
OD260
kann die Reinheit der DNA über das OD280 -Verhältnis bestimmt werden, welches 1.8 betragen soll-
te. Aus der Reinheit und der Absorbtion bei 260 nm lässt sich die DNA-Konzentration abschätzen.
Die DNA-Konzentration in der Tomaten-Probe (Probe 10) wurde mit 249,1 ng µl−1 und die der
E.coli -Probe mit 468,9 ng µl−1 ausgegeben. Die Proben zeigten dabei eine relativ hohe Reinheit,
OD260
was daran zu erkennen war, dass das OD280 -Verhältnis nur leicht unter 1.8 lag.
4.3 Polymerase-Kettenreaktion
Die Polymerase-Kettenreaktion dient zur Amplifikation der DNA der PV92 Alu Site, welche auf
das Vorhandensein eines SINEs untersucht werden soll. Dazu wurden dem wässrigen Überstand der
Haarprobe (4.2) 2 µl Reaktionspuffer B sowie dNTP-Lösung, 1 µl pv92 Vorwärts- und Rückwärtsprimer,
1,8 µl MgCl2-Lösung und 1 µl DNA-Polymerase zugegeben. Anschließend wurde das Eppendorf-
Gefäß in den Thermocycler eingesetzt und das Temperaturprogamm gestartet.
4.4 Restriktionsverdau
Zur Herstellung der E.coli -Proben (Probe 7 und 8) wurden je 12,7 µl doppelt destilliertes Was-
ser, 2 µl des zehnfach-Cutsmart-Puffers, sowie 4,27 µl (2 µg) der E.coli -Plasmid-DNA zugegeben. In
Probe 7 wurde 1 µl EcoRI-HF als Restriktionsenzym zugegeben, wähernd in Probe 8 1 µl HindIII-
HF zugeben wurde. Für den Verdau der Tomaten-Probe wurden 9 µl ddH2O 2 µl des zehnfach
Cutsmart-Puffers, sowie 8,0 µl (2 µg)der extrahierten Tomaten-DNA gemischt. Als Restriktionsen-
4DNA-Analytik 5.1
zym wurde 1 µl HindIII-HF verwendet. Die Proben wurden durch leichtes auf und ab Pipettieren
mit einer schräg angeschnittenen Pipettenspitze gemischt und anschließend bei 37 °C für eine Stun-
de incubiert.
Proben 4 und 5 wurden nach der im Praktikumsskript1 angegebenen Tabelle gemischt und 15 min
bei 37 °C inkubiert.
4.5 Herstellen des Agarose-Gels
Es sollte ein 40 ml 0,8 % Gel herstellt werden. Dazu wurde die Gelkammer montiert, 0,3 g Aga-
rose eingewogen und in 40 ml TAE-Puffer gelöst. Anschließend wurde das Becherglas für 90 s in
der Mikrowelle bei 900 W erhitzt. Nach ca. 30 s war ein starkes Überkochen festzustellen. Nach
Rücksprache mit dem Assistenten wurde das Erhitzen mit 600 W fortgesetzt. Nach einem kurzen
Abkühlen von ca. 5 Minuten wurden 0,5 µl Ethidiumbromid-Lösung zugegeben. Es war zu beachten,
dass ab diesem Zeitpunkt alle Arbeiten am Gel nur mit blauen Nitril-Handschuhen durchgeführt
werden dürfen. Die Lösung wird durch Schwenken durchmischt, in den Gelschlitten gegossen und
der Gelkamm eingesetzt. Die vollständige Polymerisation ist nach ca. 25 min abgeschlossen.
4.6 Gelelektrophorese
Der Gelkamm wurde entfernt und die Elektrophoresekammer bis zur Markierung mit TAE-Puffer
gefüllt. Zu den Proben wird vor der Zugabe 4 µl 6xDNA-Ladepuffer zugegeben. Dieser erhöht die
Dichte der Probe und sorgt dafür, dass diese in die Probentasche sinkt. Die Proben wurden geord-
net nach Probennummer (Tabelle 1) mit der Mikropipette in die Taschen gefüllt. Der Deckel wird
aufgesetzt und die Elektrophorese gestartet. Dabei wird zunächst eine Spannung von 100 V verwen-
det, die nach zehn Minuten auf 120 V erhöht wird. Nach 40 min wurde die Elektrophorese beendet
und das Gel mit dem Schlitten aus der Elektrophoresekammer entnommen. Die Dokumentation
erfolgt in Form eines UV-Bildes.
Tabelle 1: Auf dem Gel aufgetragene Proben.
Probennummer Volumen DNA Zusatz / Verarbeitung
1 10 µl Marker 1kbp -
2 12 µl Plasmid unverdaut
3 12 µl Plasmid mit FastDigest BamHI verdaut (16h)
4 20 µl Plasmid mit FastDigest BamHI verdaut (15min)
5 20 µl Plasmid mit FastDigest BamHI und NdeI verdaut (15min)
6 20 µl gDNA E.coli -
7 20 µl gDNA E.coli mit EcoRI-HF verdaut (1h)
8 20 µl gDNA E.coli mit HindIII-HF verdaut (1h)
9 12 µl gDNA Tomate -
10 20 µl gDNA Tomate mit HindIII verdaut (1h)
Die PCR Versuche wurden auf einem seperaten Gel durchgeführt.
5DNA-Analytik 5.2
5 Auswertung
5.1 Ergebnisse
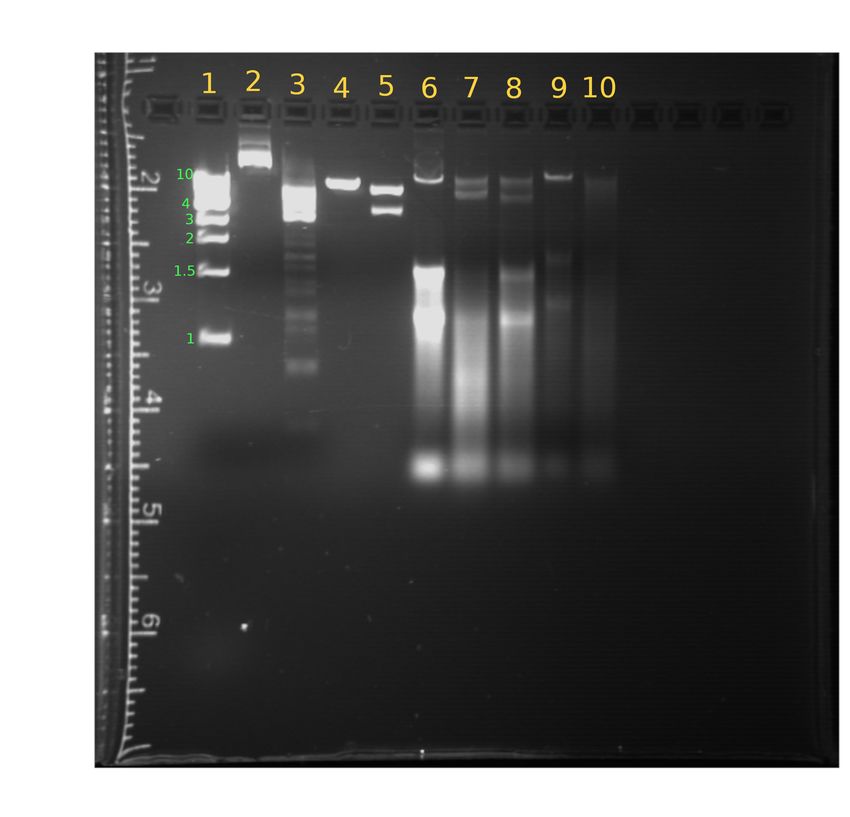
Abbildung 1: Beschriftete UV-Aufnahme des Agarosegels.
5.2 PCR-Produkte
Die Polymerase-Produkte wurden für alle Versuchsgruppen auf einem seperaten Gel getrennt. Dieses
1,5 %-Agarosegel ist dichter und kann so die kürzeren PCR-Produkte gut auftrennen. Auf dem Gel
wurde außerdem ein 100bp Marker verwendet.
6DNA-Analytik 5.2
Abbildung 2: Agarose-Gel des Versuchstags.
Wie auf Abbildung 2 zu erkennen ist, befinden sich auf dem Gel keine Banden, was darauf schließen
lässt, das die PCR-Reaktion für alle Gruppen nicht funktioniert hat. Dies ist vermutlich auf eine
Verunreinigung im Reaktionspuffer zurückzuführen und wird näher in Kapitel 6 diskutiert.
Daher wurde zur Auswertung ein Muster-Gel zur Verfügung gestellt:
Abbildung 3: Beschriftetes PCR-Mustergel zur Auswertung.
Auf dem Elektrophorese-Gel wurde in Spur 1 eine 100bp DNA-Leiter eingesetzt, um die Größe der
PCR-Produkte bestimmen zu können. Die Proben der Spuren 3, 4, 5 und 6 zeigen jeweils das gleiche
Laufverhalten und können mit Hilfe der DNA-Leiter einer Länge von ungefähr 130bp zugeordnet
werden. Die Proben sollten dabei auf das Vorhandensein einer pv92-SINE-Insertion untersucht
werden. Dazu wurde der entsprechende Bereich der DNA in einer PCR-Reaktion amplifiziert. Falls
die Insertion vorhanden ist, ist eine Länge von 450 bp zu erwarten. Falls keine Insertion vorhanden
7DNA-Analytik 5.4
sein sollte, sollte das DNA-Stück eine Länge von nur 130 bp besitzen. Da alle Proben eine Länge von
130 bp zeigen, kann geschlussfolgert werden, dass in allen Proben die pv92-Insertion nicht vorhanden
ist.
5.3 Verdaute und unverdaute genomische DNA
Die unverdaute genomische DNA der E.coli Bakterien in Spur 6 zeigt eine Bande mit einer Laufstre-
cke von 5,9 mm. Des Weiteren befindet sich ab einer Laufstrecke von 14,0 mm eine sehr breit gelaufe-
ne Bande, die sich bis zum Ende durchzieht und vermutlich auf eine Verunreinigung zurückzuführen
ist. Nach Verdau mit EcoRI-HF (Spur 7) für eine Stunde ist zu erkennen, dass sich die Bande in
zwei schwächere Banden mit den Laufstecken 6,0 mm und 7,36 mm aufspaltet. Dies zeigt, dass
die genomische DNA der Tomate mindestens eine (5’-GAATTC-3’) Sequenz in ihrer DNA enthält
und so spezifisch von EcoRI geschnitten werden kann. Die Verunreinigung, welche sich ab einer
Laufstrecke von 14,0 mm zeigt, ist auch in dieser Probe erkennbar. Auch die mit HindIII verdaute
genomische E.coli -DNA zeigt zwei Fragmente mit ähnlichen Längen, allerdings scheint das kürzere
Fragment mit einer Laufstrecke von 7,5 mm eine minimal kürzere Länge. Diese Abweichung liegt
jedoch innerhalb der Messgenauigkeit (6).
Die unverdaute, genomische DNA der Tomate (Spur 9) zeigt eine intensive Bande bei 5,5 mm.
Des Weiteren sind schwächere, breitere Banden bei 13,0 mm und 17,0 mm erkennbar. Nach dem
Verdau mit HindIII sind keine Banden mehr auf dem Gel zu erkennen, was u.a. auf eine zu hohe
Verdünnung zurückzuführen sein kann.
5.4 Diskussion des Laufverhaltens und der Verdauung
Das unverdaute Plasmid in Spur 2 zeigt zwei Banden: Eine intensive Bande mit einer Laufstrecke
von 4,5 mm und eine deutlich schwächere Bande mit 2,9 mm Laufstrecke. Ungeschnittene Plasmid-
DNA liegt typischerweise in der Supercoiled-Form vor, in welcher der DNA-Strang in sich verdreht
ist. Sie kann aber auch in der Relaxed-Circular Form vorliegen wenn sie durch Enzyme wie Topoi-
somerase umgewandelt wird. In der Supercoiled-Form ist der DNA-Strang kleiner und legt damit
eine größere Laufstrecke im Gel zurück als in der Relaxed-Circular-Form. Daher kann die erste
intensivere Bande auf die Supercoiled-Form und die zweite, schwächere Bande auf die in kleinen
Anteilen vorliegende Relaxed-Cirular-Form zurückgeführt werden. In Spur 3 wurde das Plasmid mit
BamHI über einen Zeitraum von 16 Stunden verdaut. Die beiden in Frage kommenden Plasmide
besitzen nur eine Position, an welcher BamHI selektiv schneiden kann. Es wird also nur ein Produkt
erwartet, welches der linearisierten Form des Plasmids entspricht. Neben der Hauptbande mit einer
Laufstrecke von 8,3 mm, was einer Länge von ca. 4,4 kbp entspricht, existieren viele schwächere Ban-
den, welche kürzeren DNA Fragmenten zuzuordnen sind. Dies weist auf eine hohe Star-Aktivität
hin. Die hohe Star-Aktivität kann zum einen durch die mit 16 Stunden sehr lange Inkubationszeit
als auch durch das Verwenden der nicht HF-Variante (high fidility-Variante) der BamHI Restrikti-
onsendonuclease erklärt werden. In Spur 4 wurde das Plasmid für einen deutlich kürzeren Zeitraum
von nur 15 min inkubiert. Dies resultiert in einer deutlich geringeren Star-Aktivität, was am Fehlen
der schwächeren Banden bei hohen Laufstrecken zu erkennen ist. Die Hauptbande ist des Weiteren
deutlich schärfer, liegt mit einer Laufstrecke von 6,6 mm aber fast an der gleichen Position, da die
gleiche Restriktionsendonuclease verwendet wurde. In Spur 5 wurde das Plasmid sowohl mit BamHI
8DNA-Analytik 5.6
als auch mit NdeI für 15 min inkubiert. Da beide in Frage kommenden Plasmide je eine Nucleotid-
sequenz für BamHI und eine für NdeI besitzen, ist zu erwarten, dass zwei Fragmente entstehen. Es
wurden zwei Banden in der Spur beobachtet: Die erste, stärkere Bande bei einer Laufstrecke von
6,9 mm und eine zweite, leicht schwächere bei 8,7 mm.
5.5 Geschnittene DNA-Vektoren
Anhand der Laufstrecken der Banden sind die Längen der DNA-Fragmente abschätzbar. Aus die-
sen Längen kann auf das vorliegende Plasmid geschlossen werden. Im Fall von Plasmid pVitro2
EGFP ergibt sich ein Fragment der Größe 2367 bp und eines der Größe 4662 bp, während sich bei
Plasmid pEGFP-N1 ein Fragment der Größe 426 bp und eines der Größe 4307 bp bildet. Über die
Größe dieser Fragmente kann das vorliegende Peptid bestimmt werden. Es wurden zwei Banden in
der Spur beobachtet: Die erste, stärkere Bande bei einer Laufstrecke von 6,9 mm und eine zweite,
leicht schwächere bei 8,7 mm. Die Banden lassen sich eher den Längen 4662 bp (erste Bande) und
2367 bp (zweite Bande) zuordnen. Damit liegt Plasmid pVitro2 EGFP vor.
Abbildung 4: Intercalation von Ethidiumbromid in DNA2
Etidiumbromid bindet durch eine Intercalation zwischen den Basenpaaren über π-Stapel-Wechselwirkungen
mit der DNA. Die Anzahl der Bindungsstellen ist somit proportional zur Anzahl der Basenpaare
und somit zur Masse der DNA. Beim Verdau mit BamHI und NdeI entstehen gleiche Stoffmengen
der beiden unterschiedlich langen Fragmente. Das kürzere Fragment, welches den längeren Laufweg
zeigt, besitzt eine kleinere molare Masse und so auch eine kleinere Gesamtmasse als das längere
Fragment. Dementsprechend ist auch die Fluoreszenzintensität der Bande (mit der längeren Lauf-
strecke) geringer als die der ersten Bande, obwohl die gleiche Stoffmenge vorliegt.
5.6 Unterschiede der mit BamHI verdauten Proben
Wie bereits in Kapitel 5.4 erwähnt, zeigen die Proben eine annähernd gleiche Hauptbande mit einer
Laufstrecke von 8,3 mm. Neben der Hauptbande enthält die 16 Stunden lang verdaute Probe viele
schwächere Banden mit längeren Laufstrecken, die kürzeren DNA Fragmenten entsprechen. Diese
Banden sind in der nur 15 min verdauten Probe nicht zu erkennen. Die schwächeren Banden in Probe
3 lassen sich daher auf die Star-Aktivität der Restriktionsendonuclease zurückführen. Star-Aktivität
ist ein Maß für die Selektivität des DNA-Verdaus durch Restriktionsendonucleasen. Dabei bindet
die Restriktionsendonuclease an eine Sequenz der DNA, die nicht ihrer Standardsequenz entspricht
9DNA-Analytik 7.0
und spaltet diese. Eine hohe Star-Aktivität entspricht einem hohen Anteil solcher unspezifischer
Verdauungen. Da Probe 4 diese Banden nicht enthält, ist davon auszugehen, dass die Star-Aktivität
wesentlich langsamer abläuft als die spezifische Verdauung.
6 Fehlerbetrachtung
Wie in 5.2 erwähnt, waren auf dem dichteren Agarose-Gel zur Trennung der PCR-Produkte (Ab-
bildung 2) keine Banden zu erkennen. Es ist also davon auszugehen, dass die PCR-Reaktion nicht
abgelaufen ist oder nicht funktioniert hat. Nach Rücksprache mit den Assistenten ist der nahelie-
genste Grund hierfür ein verunreinigter Reaction-Puffer B. Schon eine kleine Verunreinigung dieses
Puffers reicht aus, um die Funktion der FirePol-Polymerase zu stören. Aber auch eine Verunreini-
gung der FirePol-Polymerase selbst ist nicht auszuschließen. In jedem Fall muss die Verunreinigung
in den zur Verfügung gestellten Substanzen bereits vorgelegen haben, da die PCR-Reaktion bei
keiner Versuchsgruppe erfolgreich war. Eine weitere Möglichkeit ist, dass die eingesetzte DNA aus
Haarwurzeln nicht extrahiert oder bei der Extraktion zerstört wurde. Aber auch die Elektrophorese
selbst lief nicht optimal: Die DNA-Leiter ist nur schwach erkennbar, zeigt aber, dass die Laufzeit
unzureichend war, da die längste Laufstrecke der 100 bp-Markierung nur 23,3 mm zurücklegte.
Auch das Gel der anderen Proben hätte eine längere Elektrophorese-Zeit benötigt. Die längste
Laufstrecke beträgt 31,7 mm und erstreckt sich damit nur über die Hälfte des verfügbaren Gels.
Dies führt dazu, dass die Banden der DNA-Leiter ab einer Länge von 4 kbp überlappen und nicht
zugeordnet werden können. Die kurze Laufstrecke führt außerdem dazu, dass die Länge eines DNA-
Fragments nur mit einer sehr hohen Unsicherheit bestimmt werden kann, da die Breite einer Bande
sich bereits über einen Bereich von mehreren Kilobasenpaaren erstreckt.
7 Literatur
Literatur
1. Sträter, N. Trennmethoden Praktikum Teil II (Zugegrifffen am 20.07.2021), https://moodle2.
uni-leipzig.de/pluginfile.php/2253121/mod_resource/content/1/Praktikumsskript_
2021.pdf.
2. Bipweb Biochemische Praktika für Studierende der Medizin, W. Ethidiumbromid (Zugegriff-
fen am 21.09.2021), http : / / biochem . ch / Bipweb _ all / bipweb / lexikon / metaboliten /
ethidiumbromid/ethidiumbromid.html.
10Sie können auch lesen

















































