Problempunkte der Medizinprodukte-Verordnung: Klassifizierung, Konformitätsbewertung, klinische Bewertung
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Problempunkte der Medizinprodukte-Verordnung:
Klassifizierung, Konformitätsbewertung, klinische
Bewertung
12. Augsburger Forum für Medizinprodukterecht
Augsburg, 6. Oktober 2016
Dr. iur. Dr. med. Adem Koyuncu
Partner, Head of Life Sciences Germany
Problempunkte der Medizinprodukte-Verordnung
Gliederung
Kurzer Rückblick und Ausblick: Der Weg bis zum
aktuellen Verordnungsentwurf und das weitere
Gesetzgebungsverfahren
Problempunkte bei der Klassifizierung von
Medizinprodukten
Problempunkte bei der Konformitätsbewertung von
Medizinprodukten
Problempunkte bei der klinischen Bewertung von
Medizinprodukten
2Kurzer Rückblick und Ausblick
3Rückblick: Der Weg bis zum aktuellen Verordnungsentwurf
Bisherige Rechtslage auf EU-Ebene:
Richtlinie 93/42/EWG über Medizinprodukte
Richtlinie 90/385/EWG über aktive implantierbare medizinische
Geräte
Richtlinie 98/79/EG über In-vitro-Diagnostika
2010: Der PIP-Skandal wird publik
26. September 2012: Veröffentlichung von zwei Verordnungs-
Entwürfen durch die Europäische Kommission
Verordnung über Medizinprodukte (sonstige sowie aktive
implantierbare Medizinprodukte)
Verordnung über In-vitro-Diagnostika
→ Unmittelbar innerhalb der EU geltende Verordnungen
→ Umgestaltung des Medizinprodukte-Rechtsrahmens
4Rückblick: Der Weg bis zum aktuellen Verordnungsentwurf
Quelle: Präsentation von Wilfried Reischl, BVMed-Symposium, 16.06.2016)
5Rückblick: Der Weg bis zum aktuellen Verordnungsentwurf
Wesentliche Stationen des bisherigen Verfahrens
2013/2014: 1. Lesung des Europäischen Parlaments mit
Beschluss von über 600 Änderungsanträgen
19. Juni 2015: Beschluss des Rats für Beschäftigung, Soziales,
Gesundheit und Verbraucherschutz (EPSCO) zur
Verhandlungsposition des Rates zu den Verordnungsentwürfen
5. Oktober 2015: EPSCO unter luxemburgischer Präsidentschaft
einigt sich auf einen Gemeinsamen Standpunkt
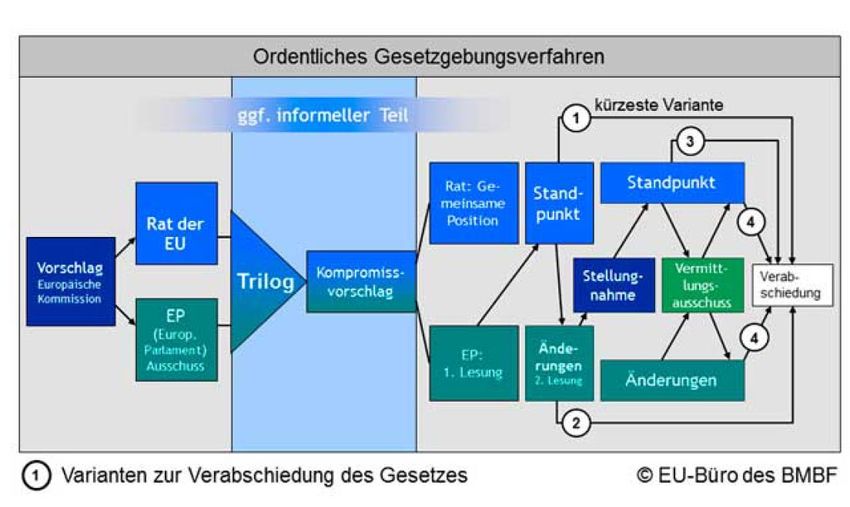
13. Oktober 2015: Beginn der Trilog-Verhandlungen zwischen
Rat, Europäischem Parlament und Europäischer Kommission auf
Grundlage des Gemeinsamen Standpunkts des Rates und der
Änderungsanträge des Europäischen Parlaments
Juni 2016: Einigung auf Trilog-Kompromiss (aktueller
Verordnungsentwurf per 27. Juni 2016)
6Struktur der Medizinprodukte-Verordnung
1. Kapitel: Geltungsbereich und Definitionen
2. Kapitel: Bereitstellung und Inbetriebnahme von Produkten, Pflichten der
Wirtschaftsakteure, Aufbereitung, CE-Kennzeichnung, freier Verkehr
3. Kapitel: Identifizierung und Rückverfolgbarkeit von Produkten,
Registrierung von Produkten und Wirtschaftsakteuren, Kurzbericht über
Sicherheit und klinische Leistung, Europäische Datenbank für
Medizinprodukte
4. Kapitel: Benannte Stellen
5. Kapitel: Klassifizierung und Konformitätsbewertung
6. Kapitel: Klinische Bewertung und klinische Prüfungen
7. Kapitel: Überwachung von Inverkehrbringen, Vigilanz und
Marktüberwachung
8. Kapitel: Kooperation der Mitgliedsstaaten, Koordinierungsgruppe
Medizinprodukte („Medical Device Coodination Group“) und
Produktverzeichnisse
9. Kapitel: Vertraulichkeit, Datenschutz, Finanzierung, Sanktionen
10.Kapitel: Schlussbestimmungen (Übergangsfristen etc.)
Anhänge: I - XVI
Umfang: 97 (+ 32) Artikel zzgl. 16 Anhänge, 352 Seiten
7Ausblick: Das weitere Gesetzgebungsverfahren
Anvisierter Zeitplan für die nächsten Schritte auf EU-Ebene
(Angaben ohne Gewähr)
Überprüfung des Verordnungsentwurfs durch die Sprachjuristen
der Europäischen Kommission im Herbst 2016 (läuft derzeit)
Ende 2016: Verabschiedung durch den Rat in erster Lesung
Zweite Lesung des Europäischen Parlaments Mitte Januar 2017
Anfang 2017 („early 2017“): Veröffentlichung der Verordnung
(gemeinsam mit der Verordnung über In-vitro-Diagnostika) im
Amtsblatt der Europäischen Union
8Problempunkte bei der
Klassifizierung von Medizinprodukten
9Problempunkte bei der Klassifizierung von
Medizinprodukten
Rechtsgrundlage:
„Klassifizierung von Medizinprodukten“ geregelt in Art. 41
MDR
Systematisch ist diese Vorschrift Teil des Kapitel V
„Klassifizierung und Konformitätsbewertung“ – darin:
Abschnitt 1 - Klassifizierung
Klassifizierungskriterien in Anhang VII speziell niedergelegt
10Problempunkte bei der Klassifizierung von
Medizinprodukten
Einstufung in die Klassen I, IIa, IIb und III erfolgt an Hand von
Zweckbestimmung des Herstellers und damit verbundenen Risiken
Klassifizierung erfolgt konkret gemäß den in Anhang VII der
Verordnung niedergelegten Kriterien und den nun 23 Regeln
Recht der EU Kommission mittels Durchführungsrechtsakten
(„implementing acts“) abweichende Entscheidungen für bestimmte
Produkte, Produktkategorien oder Produktgruppen zu treffen (vgl.
Art. 41 Abs. 3, 3a MDR)
Bestimmung der Einstufung durch Anwendung der
Klassifizierungskriterien
Einstufung in andere Klasse aus Gründen der öffentlichen Sicherheit
(wegen neuester wissenschaftlicher Erkenntnisse, Vigilanz-
Erkenntnissen)
Zur Sicherstellung einheitlicher Anwendung der Klassifizierungskriterien
11Problempunkte bei der Klassifizierung von
Medizinprodukten
Klassifizierungsregeln haben Auswirkungen auf „Neu-
Klassifizierung“ einiger Produkte
Höherklassifizierung bestimmter Implantate von Klasse IIb zu
Klasse III bringt zusätzlichen Aufwand für
Medizinproduktehersteller
Ergänzend dazu: Durch Eingruppierung in Klasse III müssen
diese implantierbaren Produkte nun mitunter auch noch das
„Scrutiny-Verfahren“ durchlaufen (dazu gleich mehr)
12Problempunkte bei der Klassifizierung von
Medizinprodukten
Spezielle Produktgruppe 1: Software/Apps als Medizinprodukt
Vorfrage: Einstufung von Software als Medizinprodukt? Abgrenzung zu
Lifestyle-Produkten → vgl. Recital 18a, Art. 2 Abs. 1
Recital 18a:
Es muss eindeutig festgelegt werden, dass Software als solche,
wenn sie vom Hersteller speziell für einen oder mehrere der in der
Definition von Medizinprodukten genannten medizinischen Zwecke
bestimmt ist, als Medizinprodukt gilt, während Software für
allgemeine Zwecke, auch wenn sie in Einrichtungen der
Gesundheitsversorgung eingesetzt wird, sowie Software, die für
Anwendungen in den Bereichen Lebensstil und Wohl-befinden
eingesetzt wird, kein Medizinprodukt ist. Die Einstufung der
Software, entweder als Produkt oder als Zubehör, ist unabhängig
von ihrem Ort und von der Art ihrer Verbindung mit einem Produkt.
Software ist „aktives Medizinprodukt“
Und die Klassifizierung?
13Problempunkte bei der Klassifizierung von
Medizinprodukten
Spezielle Produktgruppe 1: Software/Apps als Medizinprodukt
Klassifizierung von (stand-alone) Software gem. Anhang VII, Regel 10a
Software, die dazu bestimmt ist, Informationen zu liefern, die zu Entscheidungen
für diagnostische oder therapeutische Zwecke herangezogen werden, gehört zur
Klasse IIa, es sei denn, diese Entscheidungen haben Auswirkungen, die direkt
oder indirekt Folgendes verursachen können:
den Tod oder eine irreversible Verschlechterung des Gesundheitszustands; in
diesem Fall wird sie der Klasse III zugeordnet;
eine schwerwiegende Verschlechterung des Gesundheitszustands oder einen
chirurgischen Eingriff; in diesem Fall wird sie der Klasse IIb zugeordnet.
Software, die für die Kontrolle von Körperfunktionen bestimmt ist, gehört zur
Klasse IIa, es sei denn, sie ist für die Kontrolle von vitalen physiologischen
Parametern bestimmt, bei denen die Art der Änderung zu einer unmittelbaren
Gefahr für den Patienten führen könnte; in diesem Fall wird sie der Klasse IIb
zugeordnet.
Sämtliche andere Software wird der Klasse I zugeordnet.
Tendenziell Höherklassifizierung mit regulatorischen Folgen
Software, die ein Produkt steuert oder dessen Anwendung beeinflusst, wird
automatisch derselben Klasse zugerechnet wie das Produkt.
14Problempunkte bei der Klassifizierung von
Medizinprodukten
Spezielle Produktgruppe 2: Stoffliche Medizinprodukte
Bestimmte stoffliche Medizinprodukte (solche, die dazu bestimmt
sind, in den menschlichen Körper eingeführt zu werden, und die
vom Körper aufgenommen oder lokal im Körper verteilt werden)
müssen weiterhin arzneimittelrechtliche Anforderungen erfüllen
Entgegen ursprünglichem Kommissionsentwurf werden stoffliche
Medizinprodukte nicht allgemein und automatisch in Klasse III
eingestuft.
Exkurs:
Behandlung stofflicher Medizinprodukte in der nationalen
verwaltungsgerichtlichen Rechtsprechung („stets auch
Präsentationsarzneimittel“) streitbehaftet.
15Problempunkte bei der Klassifizierung von
Medizinprodukten
Spezielle Produktgruppe 2: Stoffliche Medizinprodukte
Regel 21 (Anhang VII):
Produkte, die aus Stoffen oder Kombinationen von Stoffen bestehen, die dazu
bestimmt sind, durch eine Körperöffnung in den menschlichen Körper
eingeführt oder auf die Haut aufgetragen zu werden, und die vom Körper
aufgenommen oder lokal im Körper verteilt werden, werden wie folgt
zugeordnet:
der Klasse III, wenn sie oder ihre Metaboliten systemisch vom
menschlichen Körper aufgenommen werden, um ihre Zweckbestimmung
zu erfüllen;
der Klasse III, wenn sie ihre Zweckbestimmung im Magen oder im unteren
Magen-Darm-Trakt erfüllen und wenn sie oder ihre Metaboliten systemisch
vom menschlichen Körper aufgenommen werden;
der Klasse IIb in allen anderen Fällen, es sei denn, sie werden auf die
Haut aufgetragen; in diesem Fall werden sie der Klasse IIa zugeordnet,
oder
der Klasse IIa, wenn sie in der Nasenhöhle oder der Mundhöhle bis zum
Rachen angewandt werden und ihre Zweckbestimmung in diesen Höhlen
erfüllen.
16Problempunkte bei der
Konformitätsbewertung von Medizinprodukten
17Problempunkte bei der Konformitätsbewertung von
Medizinprodukten
Rechtsgrundlage:
„Konformitätsbewertung von Medizinprodukten“ geregelt in
Art. 42-48 MDR
Systematische Stellung in Kapitel V „Klassifizierung und
Konformitätsbewertung“ – darin: Abschnitt 2
Anhänge VIII-XI regeln die Einzelheiten der verschiedenen
Konformitätsbewertungsverfahren
18Problempunkte bei der Konformitätsbewertung von
Medizinprodukten
Definition gemäß Art. 1 Abs. 1 Nr. 28:
„Konformitätsbewertung“ bezeichnet das Verfahren, nach dem
festgestellt wird, ob die Anforderungen dieser Verordnung an ein
Produkt erfüllt worden sind → Voraussetzung für Inverkehrgabe des
Produkts (Art. 42)
Konformitätsbewertungsverfahren für Produkte der Klasse I bleibt
generell in der alleinigen Verantwortung des Herstellers, jedoch
eingeschränkte Beteiligung der Benannten Stelle bei
Produkten, deren Inverkehrgabe in sterilem Zustand erfolgt
Wiederverwendbare chirurgische Instrumente
Produkte mit Messfunktion
Konformitätsbewertungsverfahren für Produkte der Klasse IIa, IIb und III
erfordert die Mitwirkung der Benannten Stelle
Bestandteile des Konformitätsbewertungsverfahrens sind stark
ausdifferenziert und von Art/Klassifizierung des Medizinprodukts
abhängig → Detailregelung in Anhängen VIII bis XI
19Problempunkte bei der Konformitätsbewertung von
Medizinprodukten
Klasse Anforderungen an Konformitätsbewertungsverfahren
I • Technische Dokumentation, EU-Konformitätserklärung
• Nur bei vorgenannten Ausnahmen Beteiligung der Benannten Stelle
IIa • Beteiligung der Benannten Stelle
• Prüfung des Qualitätsmanagementsystems und Bewertung der technischen
Dokumentation zumindest eines repräsentativen Produkts jeder
Produktkategorie
• Alternativ: Erstellung der technischen Dokumentation (Anhang II) in
Kombination mit Produktkonformitätsprüfung
IIb • Beteiligung der Benannten Stelle
• Prüfung des Qualitätsmanagementsystems und Bewertung der technischen
Dokumentation zumindest eines repräsentativen Produkts jeder generischen
Produktgruppe
• Gesonderte Bewertung der technischen Dokumentation für implantierbare
Produkte (aber: Rückausnahmen für bestimmter Produkte vorgesehen)
• Alternativ: Baumusterprüfung in Kombination mit Produktkonformitätsprüfung
20Problempunkte bei der Konformitätsbewertung von
Medizinprodukten
Klass Anforderungen an Konformitätsbewertungsverfahren
e
III • Beteiligung der Benannten Stelle
• Prüfung des Qualitätsmanagementsystems und Bewertung der technischen
Dokumentation
• Alternativ: Baumusterprüfung in Kombination mit Produktkonformitätsprüfung
• Ergänzend dazu Sonderregeln für
• Arzneimittel-Medizinprodukt-Kombinationen, bei denen der Stoff
integraler Bestandteil ist
• aus nicht lebensfähigen Transplantaten und menschlichem/tierischem
Gewebe hergestellte Medizinprodukte
• Produkte, die aus Stoffen oder Kombinationen von Stoffen bestehen, die
dazu bestimmt sind, durch eine Körperöffnung oder durch Anwendung
auf der Haut in den menschlichen Körper eingeführt zu werden, und die
vom menschlichen Körper aufgenommen oder lokal im Körper verteilt
werden
Sonder Bei Sonderanfertigungen wenden die Hersteller das Verfahren nach Anhang XI an und
anfertig stellen vor Inverkehrbringen des Produkts die Erklärung gem. Abschnitt 1 Anhang XI
ungen aus (abweichend für implantierb. Sondernanfertigungen der Klasse III).
21Problempunkte bei der Konformitätsbewertung von
Medizinprodukten
Scrutiny-Verfahren (Art. 43a MDR)
Einführung des „Scrutiny-Verfahrens“ als Teil des
Konformitätsbewertungsverfahrens der Benannten Stelle
Zusätzliches Prüfverfahren durch Experten-Komitee - unter
Überwachung der Europäischen Kommission - für bestimmte
Hochrisiko-Medizinprodukte:
Implantierbare Produkte der Klasse III;
Aktive Produkte der Klasse IIb, die dazu bestimmt sind, ein
Arzneimittel gemäß Anhang VII Abschnitt 5.3 (Regel 11) an den
Körper abzugeben und/oder aus dem Körper zu entfernen (z.B.
Insulinpumpen)
Entbehrlichkeit des Verfahrens nur in bestimmten Ausnahmefällen:
Re-Zertifizierungen
Unter bestimmten Bedingungen bei bloßen Modifikationen
Unter bestimmten Bedingungen bei Vorliegen gemeinsamer
Technischer Spezifikationen
22Problempunkte bei der Konformitätsbewertung von
Medizinprodukten
Details des Scrutiny-Verfahrens sind in Anhang VIII, Kapitel I, Ziffer
6.0 niedergelegt
Transparenz und Mitteilungspflichten beim Scrutiny-Verfahren
Berichtspflichten der Benannten Stelle über Anwendung des
Scrutiny-Verfahrens gegenüber zuständigen Behörden und der
Europäischen Kommission
Europäische Kommission erstellt Jahresbericht über Produkte,
die Scrutiny-Verfahren durchlaufen haben → Vorlage des
Berichts u.a. an Europäisches Parlament und Rat
Benannte Stellen melden zuständigen Behörden alle
ausgestellten Prüfbescheinigungen für Produkte, bei denen
Scrutiny-Verfahren Teil des Konformitätsbewertungsverfahrens
war → zuständige Behörden und ggf. Europäische Kommission
können bei begründeten Bedenken Überprüfungsverfahren bzw.
Maßnahmen einleiten (Art. 44 Abs. 1aa)
23Problempunkte bei der Konformitätsbewertung von
Medizinprodukten
Überwachungsbewertung
Bei Produkten der Klassen IIa, IIb und III erfolgt eine (gem. Anhang
VIII, Kapitel 1., Ziff. 4) Überwachungsbewertung, wofür detaillierte
Anforderungen an Audits durch Benannte Stellen festgelegt werden
Regelmäßige Audits mindestens alle 12 Monate, einschließlich
Audits in den Betriebsstätten des Herstellers und gegebenenfalls
den Betriebsstätten seiner Zulieferer und/oder Subunternehmer
Unangekündigte Vor-Ort-Audits nach dem Zufallsprinzip, aber
mindestens einmal alle 5 Jahre beim Hersteller und seinen
Zulieferern und/oder Subunternehmern → einschließlich
Stichprobenentnahme aus Produktion oder Herstellungsprozess
Stichprobenentnahme von auf dem Markt befindlichen Produkten
24Problempunkte bei der Konformitätsbewertung von
Medizinprodukten
Art. 13 MDR: Person responsible for Regulatory Compliance (Für
die Einhaltung der Regulierungsvorschriften zuständige Person)
Diese Person ist mindestens dafür verantwortlich, dass
a) die Konformität der Produkte in angemessener Weise gemäß dem
Qualitätsmanagementsystem geprüft wird, in dessen Rahmen die
betreffenden Produkte hergestellt werden, bevor ein Produkt freigegeben
wird;
b) die technische Dokumentation und die Konformitätserklärung erstellt
und auf dem neuesten Stand gehalten werden;
ba) die Verpflichtungen zur Überwachung nach dem Inverkehrbringen
gemäß Artikel 8 Absatz 6 erfüllt werden;
c) die Berichtspflichten gemäß den Artikeln 61 bis 66 erfüllt werden;
d) im Fall von Prüfprodukten die Erklärung gemäß Anhang XIV Kapitel II
Abschnitt 4.1 abgegeben wird.
Sind mehrere Personen benannt, müssen ihre jeweiligen
Aufgabenbereiche schriftlich festgehalten (abgegrenzt) werden.
25Problempunkte bei der
klinischen Bewertung von Medizinprodukten
26Problempunkte bei der klinischen Bewertung von
Medizinprodukten
Rechtsgrundlage:
„Klinische Bewertung von Medizinprodukten“ geregelt in Art.
49 MDR
Systematische Stellung in Kapitel VI „Klinische Bewertung
und klinische Prüfungen“
Anhang XIII: „Klinische Bewertung und klinische
Weiterverfolgung nach dem Inverkehrbringen“
27Problempunkte bei der klinischen Bewertung von
Medizinprodukten
Art. 8 Abs. 1b: „Die Hersteller führen eine klinische Bewertung nach
Maßgabe der in Artikel 49 und in Anhang XIII festgelegten
Anforderungen durch, die auch eine klinische Weiterverfolgung nach
dem Inverkehrbringen umfasst.“
→ Einbeziehung von Post-Market-Daten
→ Erhöhte Bedeutung des Post-Market-Monitorings
Definition gemäß Art. 1 Abs. 1 Nr. 32:
„Klinische Bewertung“ bezeichnet einen systematischen und
geplanten Prozess zur kontinuierlichen Generierung, Sammlung,
Analyse und Bewertung der klinischen Daten zu einem Produkt, mit
dem Sicherheit und Leistung , einschließlich des klinischen
Nutzens, des Produkts bei bestimmungsgemäßer Verwendung
nach Angabe des Herstellers überprüft werden.
28Problempunkte bei der klinischen Bewertung von
Medizinprodukten
Art. 49 Abs. 1 MDR:
„Der Nachweis, dass die in Anhang I genannten allgemeinen
Sicherheits- und Leistungsanforderungen und gegebenenfalls
relevante Anforderungen nach Anhang IIa bei normalem
bestimmungsgemäßem Einsatz des Produkts erfüllt werden, sowie
die Beurteilung unerwünschter Nebenwirkungen und der
Annehmbarkeit des Nutzen-/Risiko-Verhältnisses gemäß Anhang I
Abschnitte 1 und 5 erfolgen auf der Grundlage klinischer Daten, die
einen ausreichenden klinischen Nachweis bieten.“
29Problempunkte bei der klinischen Bewertung von
Medizinprodukten
Gemäß Art. 49 Abs. 2 erfolgt eine klinische Bewertung nach einem genau
definierten und methodisch soliden Verfahren, das sich auf folgende
Grundlagen stützt:
a) eine kritische Bewertung der einschlägigen derzeit verfügbaren wissenschaftlichen
Literatur über Sicherheit, Leistung, Konzeptionsmerkmale und Zweckbestimmung des
Produkts; dabei müssen folgende Bedingungen erfüllt sein:
– das Produkt, das Gegenstand der klinischen Bewertung für die Zweckbestimmung ist,
ist dem Produkt, auf das sich die Daten beziehen, gemäß Anhang XIII Teil A Abschnitt 4a
nachgewiesenermaßen gleichwertig
und
– die Daten zeigen in geeigneter Weise die Übereinstimmung mit den einschlägigen
Sicherheits- und Leistungsanforderungen;
b) eine kritische Bewertung der Ergebnisse aller verfügbaren klinischen Prüfungen, wobei
genau darauf geachtet wird, ob die Prüfungen gemäß den Artikeln 50 bis 60 und Anhang
XIV durchgeführten wurden;
d) eine Prüfung der gegebenenfalls derzeit verfügbaren alternativen Behandlungsoptionen
für diesen Zweck.
30Problempunkte bei der klinischen Bewertung von
Medizinprodukten
Deutliche Verschärfung: Durchführung von klinischen Prüfungen für
implantierbare Produkte und Produkte der Klasse III ganz weitgehend
erforderlich
→ nur wenige Ausnahmen für diese Produktgruppen vorgesehen
(Modifikation eines bereits vermarkteten Produkts, Äquivalenz mit
vermarktetem Produkt eines anderen Herstellers, bestimmte
„Altfälle“ unter bestehendem Rechtsrahmen, bestimmte
Einzelprodukte wie Nahtmaterial, Zahnkronen etc.)
Für bestimmte Produkte ohne medizinischen Verwendungszweck
(z.B. Kontaktlinsen) kann auf die Durchführung klinischer Prüfungen
verzichtet werden → Bezugnahme auf klinische Daten äquivalenter
Medizinprodukte (Kriterien für Äquivalenz finden sich in Anhang XIII)
31Problempunkte der Medizinprodukte-Verordnung
Zusammenfassung und Ausblick
Klassifizierung von Medizinprodukten
Konformitätsbewertung von Medizinprodukten
Klinische Bewertung von Medizinprodukten
32Problempunkte der Medizinprodukte-Verordnung
Vielen Dank!
--------------------------------------------------------------------------------
Referent:
Dr. iur. Dr. med. Adem Koyuncu
Rechtsanwalt und Arzt
Zugelassen als RA in Brüssel und Düsseldorf
Lehrbeauftragter u.a. der Bucerius Law School und Uni Düsseldorf
Mitglied der Ethikkommission der TU Dresden
Partner der Kanzlei COVINGTON & BURLING LLP
Kunstlaan 44, 1040 Brüssel
Tel.: +32.2.549.5240
Mobil: +49.173.543.4308
E: akoyuncu@cov.com
www.cov.com
33Sie können auch lesen

















































