Stellungnahme der Ad-hoc-Kommission In-vitro-Diagnostika der AWMF zur Umsetzung der Verordnung (EU) 2017/746 (IVDR) im Hinblick auf ...
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
OPEN ACCESS Stellungnahme
Stellungnahme der Ad-hoc-Kommission
In-vitro-Diagnostika der AWMF zur Umsetzung der
Verordnung (EU) 2017/746 (IVDR) im Hinblick auf
In-vitro-Diagnostika aus Eigenherstellung
Advisory opinion of the AWMF Ad hoc Commission In-vitro Diagnostic
Medical Devices regarding in-vitro diagnostic medical devices
manufactured and used only within health institutions established in
the Union according to Regulation (EU) 2017/746 (IVDR)
Abstract
In view of the approaching application date of Regulation (EU) 2017/746 Petra Hoffmüller1
(„IVDR“) and the resulting EU-wide, harmonized requirements for in
Monika Brüggemann2
vitro diagnostic medical devices (IVD) manufactured and used within
European health institutions, the Ad hoc Commission IVD“ of the German Thomas Eggermann1
Association of the Scientific Medical Societies (AWMF) takes a national Kamran Ghoreschi3
position on the details of the requirements and conditions related to
Detlef Haase2
the use of these IVD products.
The Ad hoc Commission IVD emphasizes the relevance of examination Jörg Hofmann4
procedures developed in medical laboratories, especially in the field of Klaus-Peter Hunfeld5,6
orphan diseases and new diagnostic markers. The IVDR sets an ad-
Katharina Koch7
equate regulatory framework for IVD manufactured and used within
health institutions as long as these requirements are fulfilled with reli- Christian Meisel8
ability and in accordance with the current state of the art in medical Patrick Michl9
laboratory sciences. At the same time, the IVDR requirements have to
Jens Müller10
be regarded under a pragmatic view and in accordance with the quality
management systems approved within the diffferent EU Member States. Carsten Müller11,12
On the one hand, the mandatory requirements of the RiLiBÄK play an Holger F. Rabenau4
essential role in Germany. On the other hand, elements of voluntarily Dirk Reinhardt13
applicable international standards may support the fulfilment of product
requirements for safety and performance according to Annex I of the Markus J.
IVDR. Both the complexity and possible solutions for the implementation Riemenschneider14
of the IVDR requirements are discussed on the basis of examples such Ulrich J. Sachs10
as the required documentation, performance evaluation and software
Ulrich Sack8
validation.
The Ad hoc Commission IVD recommends that, while aiming at a Albrecht Stenzinger15
preferably EU-wide harmonized interpretation of the IVDR requirements, Thomas Streichert12
the flexibility in medical laboratory diagnostics necessary for patient
Nils von Neuhoff13
care, including the use of IVDs from in-house production, should be
emphasized. Wilko Weichert15
Keywords: IVDR, laboratory-developed tests, in-house IVD, validation, Christof Weinstock16
performance evaluation, quality management Stefan Zimmermann5,17
Folker Spitzenberger18
Ad-hoc-Kommission
In-vitro-Diagnostika der
AWMF
GMS Zeitschrift zur Förderung der Qualitätssicherung in medizinischen
Laboratorien 2021, Vol. 12, ISSN 1869-4241 1/10Hoffmüller et al.: Stellungnahme der Ad-hoc-Kommission In-vitro-Diagnostika ...
Zusammenfassung 1 Deutsche Gesellschaft für
Humangenetik (GfH)
Vor dem Hintergrund des nahenden Geltungsbeginns der Verordnung 2 Deutsche Gesellschaft für
(EU) 2017/746 („IVDR“) und der damit EU-weit harmonisierten Anfor- Hämatologie und
derungen an In-vitro-Diagnostika (IVD) aus Eigenherstellung positioniert Medizinische Onkologie
sich die Ad-hoc-Kommission In-vitro-Diagnostika der Arbeitsgemeinschaft (DGHO)
der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF) im 3 Deutsche Gesellschaft für
Einzelnen zu den in der IVDR gestellten Anforderungen und Bedingungen Dermatologie (DDG)
zur Verwendung dieser Produkte.
Die Ad-hoc-Kommission IVD hebt die Bedeutung von in medizinischen 4 Gesellschaft für Virologie
(GfV)
Laboratorien eigenentwickelten Untersuchungsverfahren für die Patien-
tenversorgung vor allem im Bereich seltener Erkrankungen und neuer 5 Deutsche Gesellschaft für
diagnostischer Marker hervor. Die IVDR bildet für die Entwicklung und Hygiene und Mikrobiologie
Verwendung von IVD aus Eigenherstellung einen passenden regulatori- (DGHM)
schen Rahmen, sofern die Anforderungen zuverlässig entsprechend 6 Gesellschaft zur Förderung
dem Stand der medizinischen Wissenschaft und Technik, aber zugleich der Qualitätssicherung in
pragmatisch und in Übereinstimmung mit den in den Mitgliedstaaten medizinischen Laboratorien
bewährten Qualitätsmanagementsystemen umgesetzt werden. In (INSTAND e.V.)
Deutschland sind hier einerseits die verpflichtenden Anforderungen der 7 Gesellschaft für
RiLiBÄK zu nennen. Andererseits können Elemente von freiwillig anzu- Toxikologische und
wendenden internationalen Normen dazu dienen, die nach Anhang I Forensische Chemie
der IVDR umzusetzenden Anforderungen an Sicherheit und Leistung (GTFCh)
für IVD aus Eigenherstellung zu erfüllen. Sowohl die Komplexität als 8 Deutsche Gesellschaft für
auch Lösungskonzepte zur Umsetzung der Anforderungen werden u.a. Immunologie (DGfI)
am Beispiel der erforderlichen Dokumentation, der Leistungsbewertung 9 Deutsche Gesellschaft für
und der ggf. durchzuführenden Softwarevalidierung aufgezeigt. Gastroenterologie,
Die Ad-hoc-Kommission empfiehlt, bei einer möglichst weitreichend Verdauung und
harmonisierten Interpretation der Anforderungen gleichzeitig die für die Stoffwechsel (DGVS)
Patientenversorgung notwendige Flexibilität in der labordiagnostischen 10 Gesellschaft für Thrombose-
Versorgung einschließlich der Verwendung von IVD aus Eigenherstellung und Hämostaseforschung
zu gewährleisten. (GTH)
Schlüsselwörter: IVDR, IVD aus Eigenherstellung, In-Haus-Verfahren, 11 Deutsche Gesellschaft für
Validierung, Leistungsbewertung, Qualitätsmanagement Klinische Pharmakologie
und Therapie (DGKliPa)
12 Deutsche Gesellschaft für
Klinische Chemie und
Laboratoriumsmedizin
(DGKL)
13 Gesellschaft für
pädiatrische Onkologie und
Hämatologie (GPOH)
14 Deutsche Gesellschaft für
Neuropathologie und
Neuroanatomie (DGNN)
15 Deutsche Gesellschaft für
Pathologie (DGP)
16 Deutsche Gesellschaft für
Transfusionsmedizin und
Immunhämatologie (DGTI)
17 Paul-Ehrlich-Gesellschaft
(PEG)
18 Deutsche Gesellschaft für
Pharmazeutische Medizin
(DGPharMed)
GMS Zeitschrift zur Förderung der Qualitätssicherung in medizinischen
Laboratorien 2021, Vol. 12, ISSN 1869-4241 2/10Hoffmüller et al.: Stellungnahme der Ad-hoc-Kommission In-vitro-Diagnostika ...
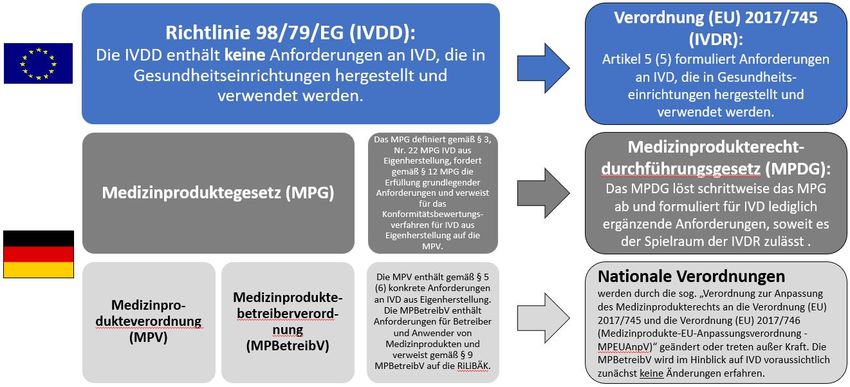
Einleitung geregelt, in Deutschland durch die Medizinprodukte-
betreiberverordnung (MPBetreibV) [5] und die MPV. Im
Die Arbeitsgemeinschaft der Wissenschaftlichen Medizi- Hinblick auf die grundlegenden Anforderungen an die
nischen Fachgesellschaften e.V. (AWMF), der deutsche Struktur- und Prozessqualität laboratoriumsmedizinischer
Dachverband von 179 medizinischen Fachgesellschaften, Untersuchungen ist in Deutschland die Richtlinie der
die wiederum rund 280.000 Mitglieder vertreten, bewer- Bundesärztekammer zur Qualitätssicherung laboratori-
tet die Harmonisierung des Rechtsrahmens für die Kon- umsmedizinischer Untersuchungen (RiliBÄK) anzuwenden
formitätsbewertung und das Inverkehrbringen von In-vitro- [6].
Diagnostika (IVD) in Europa grundsätzlich positiv. Mit der Die IVDR harmonisiert nun erstmals europaweit die Anfor-
Verordnung (EU) 2017/746 (IVDR) [1], deren Geltung am derungen an die Herstellung und die Verwendung von
26. Mai 2022 beginnt, werden die Ziele verfolgt, einen IVD aus Eigenherstellung (Abbildung 1). Die IVDR nimmt
reibungslos funktionierenden Binnenmarkt für IVD zu dagegen nicht in Anspruch, ärztlich-diagnostische Prozes-
gewährleisten und gleichzeitig ein hohes Maß an Qualität se zu regulieren. Dies steht im Einklang mit Artikel 168
von IVD im Sinne der Patientensicherheit durch sinnvolle des EU-Vertrages, gemäß dem bei der Tätigkeit der Euro-
standardisierte Maßnahmen zu sichern. päischen Union die Verantwortung der Mitgliedstaaten
IVD sind unerlässlich für die Früherkennung, Diagnose, für die Organisation des Gesundheitswesens und die
Prognose und Überwachung von Krankheiten, insbeson- medizinische Versorgung gewahrt wird.
dere von übertragbaren, seltenen und/oder genetisch Entsprechend der neuen EU-Verordnung dürfen in der
bedingten Krankheiten, für die Information über physio- Union ansässige Gesundheitseinrichtungen weiterhin ei-
logische/pathologische Zustände sowie zunehmend auch genentwickelte Produkte zur Diagnostik herstellen und
für die Allokation von Therapien im Sinne einer Präzisions- verwenden, sofern sie die Bedingungen des Art. 5 Abs. 5
medizin. Insbesondere für die Diagnostik seltener Erkran- der EU-Verordnung erfüllen. Der Umfang der zu erfüllen-
kungen („Orphan Diseases“) greifen medizinische Labo- den Anforderungen ist gegenüber der MPV jedoch in eini-
ratorien verschiedenster Fachrichtungen aus Mangel an gen Punkten gestiegen, so dass auf medizinische Labo-
kommerziell verfügbaren Diagnostika fast ausschließlich ratorien ein erheblicher Mehraufwand an Validierung und
auf eigenentwickelte Untersuchungsverfahren zurück. Dokumentation sowie ggf. die Umsetzung von normativen
Diese werden auch als „In-Haus-Verfahren“ bzw. im eng- Regelungen aus bislang fachfremden Bereichen zu-
lischen Sprachgebrauch als „Laboratory Developed Tests“ kommt. Des Weiteren hat die EU-Kommission bisher die
(LDT) bezeichnet. Chance versäumt, die Anforderungen an Gesundheitsein-
Aus medizinischer und ärztlicher Sicht ist die Sicherstel- richtungen, die IVD aus Eigenherstellung einsetzen, kon-
lung der Patientenversorgung von ebenso zentraler Be- kret zu präzisieren und unmissverständlich von den An-
deutung wie Regelungen zur Qualitätssicherung und forderungen an Wirtschaftsakteure, Händler, Hersteller
Produktsicherheit. Es ist davon auszugehen, dass die und Importeure abzugrenzen.
kommerzielle Vermarktung von Nischenprodukten wie
IVD für Orphan-Diseases-Diagnostik aufgrund der durch
die IVDR-Anforderungen an die Hersteller deutlich höhe- Empfehlungen der
ren Kosten für den Zulassungsaufwand und der zu erwar- Ad-hoc-Kommission
tenden niedrigen Einnahmen im Markt sich nicht rechnen
wird. Gerade deshalb sind Gesundheitseinrichtungen wie
In-vitro-Diagnostika (IVD)
medizinische Laboratorien gefordert und gezwungen, Die Ad-hoc-Kommission In-vitro-Diagnostika (IVD) der
weiterhin auf ihre IVD aus Eigenherstellung zurückzugrei- AWMF spricht sich dafür aus, sorgsam und zuverlässig,
fen, um einer Patientenunterversorgung vorzubeugen aber zugleich pragmatisch und mit Augenmaß, an die
und flexibel auf besondere Bedürfnisse oder unerwartete Umsetzung der zentralen Aspekte der IVDR-Forderungen
Situationen von Bedeutung für die individuelle oder öf- in medizinischen Laboratorien heranzugehen. Im Folgen-
fentliche Gesundheit zu reagieren. den werden die zu erfüllenden Anforderungen und Bedin-
gungen des Art. 5 Abs. 5 der IVDR erläutert, die Gesund-
heitseinrichtungen erfüllen müssen, wenn diese IVD-
Die neue IVDR Produkte selbst herstellen und verwenden. Gleichzeitig
werden die Komplexität und regulatorische Spielräume
In der bisher geltenden EG-Richtlinie 98/79/EG (IVD-Di-
dargestellt sowie pragmatische Lösungsansätze zur Um-
rektive, IVDD) werden für Produkte, die in Gesundheits-
setzung der Forderungen aufgezeigt.
einrichtungen zur Verwendung im selben Umfeld herge-
stellt werden, keine Anforderungen formuliert [2]. Auf-
grund des EU-Subsidiaritätsprinzips sind auf nationaler
Ebene in der Medizinprodukteverordnung (§ 5 Abs. 6
MPV) in Verbindung mit § 12 Medizinproduktegesetz
(MPG) seit 2007 Anforderungen an IVD aus Eigenherstel-
lung verankert [3], [4]. Der Betrieb und die Anwendung
aller Untersuchungsverfahren sind in nationalem Recht
GMS Zeitschrift zur Förderung der Qualitätssicherung in medizinischen
Laboratorien 2021, Vol. 12, ISSN 1869-4241 3/10Hoffmüller et al.: Stellungnahme der Ad-hoc-Kommission In-vitro-Diagnostika ...
Abbildung 1: Der europäische und nationale Rechtsrahmen für IVD aus Eigenherstellung. Die IVDD wird zum 26. Mai 2022
vollständig durch die IVDR abgelöst werden. Im Gegensatz zur IVDD ist die IVDR für alle Adressaten unmittelbar gültig und
enthält Anforderungen an IVD aus Eigenherstellung. Das MPG wird schrittweise durch das MPDG abgelöst, welches nur noch
Regelungen enthält, die im Handlungsspielraum des nationalen Gesetzgebers liegen. Die MPBetreibV wird für den Bereich IVD
nach bisherigem Kenntnisstand nicht geändert und wird weiter auf die RiLiBÄK verweisen. Die Aufzählung der nationalen
Verordnungen in der Abbildung ist nicht abschließend.
Forderung Art. 5 Abs. 5, letzter Satz: Dies erfordert allerdings nach entsprechender Rechtsauf-
fassung
Dieser Absatz gilt nicht für Produkte, die
1. eine breit angelegte Herstellung in großem Maßstab
im industriellen Maßstab hergestellt und
werden 2. nach einheitlichen Vorgaben, die
3. entsprechend industrielle Produktionsanlagen und
IVD aus Eigenherstellung sollen nur dann von der CE- -einrichtungen voraussetzt,
Kennzeichnungspflicht ausgenommen werden, wenn
diese nicht im sog. „industriellen Maßstab“ hergestellt was in Apotheken zumeist nicht gegeben ist [8], [9].
werden. Fazit: Die Ad-hoc-Kommission IVD der AWMF empfiehlt,
Diese Anforderung bzw. Einschränkung findet sich inhalts- dass auf europäischer (MDCG) oder nationaler Ebene
gleich in der aktuell noch gültigen Definition für IVD aus (AGMP bzw. die entsprechend zuständige FEG der ZLG)
Eigenherstellung im deutschen Medizinproduktegesetz der Begriff des „industriellen Maßstabs“ im Sinne der
MPG § 3, 22. Aufzählungspunkt. Sicherstellung der individuellen und flexiblen Patienten-
Sowohl die IVDR als auch das aktuell in Deutschland noch versorgung durch medizinische Laboratorien, die IVD aus
gültige MPG ermangeln einer Definition oder Interpretati- Eigenherstellung verwenden, einheitlich interpretiert wird.
on des Begriffs „industrieller Maßstab“. Als Intention einer Dabei kann die Analogie zu den Regelungen im Arzneimit-
solchen Regelung kann vermutet werden, dass für IVD telrecht als passende Grundlage dienen, um unter der
aus Eigenherstellung keine für kommerzielle Zwecke Herstellung im „industriellen Maßstab“
aufgebaute Produktionsausmaße mit entsprechend dafür 1. eine breit angelegte Herstellung in großem Maßstab,
nötigen technischen Produktionseinrichtungen und -anla- 2. nach einheitlichen Vorgaben zum Herstellungsprozess
gen vorliegen dürfen. Denn dies würde eine unmittelbare und -verfahren inkl. Abpackung, Kennzeichnung und
Vergleichbarkeit mit der Produktion von IVD bedeuten, Chargendokumentation,
die kommerziell durch entsprechende Konformitätsbe- 3. unter Verwendung industrieller Produktionsanlagen
wertungsverfahren mit anschließender CE-Kennzeichnung und -einrichtungen
in Verkehr gebracht werden.
Im Arzneimittelrecht (§ 4 (1) AMG) findet sich bisher eine zu verstehen, was in medizinischen Laboratorien nur
ähnliche Abgrenzung – nämlich über die Anwendung ei- selten der Fall sein dürfte.
nes „industriellen Verfahrens“ – zwischen den sog. Fer- Aus Sicht der Ad-hoc-Kommission IVD trifft für medizini-
tigarzneimitteln, die zulassungspflichtig sind, und den in sche Laboratorien eine Verwendung von IVD aus Eigen-
Apotheken hergestellten Rezepturarzneimitteln, die meist herstellung im industriellen Maßstab daher in aller Regel
keiner Zulassung bedürfen [7]. Arzneimittel, die nicht im nicht zu.
Voraus hergestellt werden, sind nämlich nur dann zulas-
sungspflichtige Fertigarzneimittel, wenn bei ihrer Zuberei-
tung ein „industrielles Verfahren“ zur Anwendung kommt.
GMS Zeitschrift zur Förderung der Qualitätssicherung in medizinischen
Laboratorien 2021, Vol. 12, ISSN 1869-4241 4/10Hoffmüller et al.: Stellungnahme der Ad-hoc-Kommission In-vitro-Diagnostika ...
Forderung Art. 5 Abs. 5 (a): Die Produkte medizinisches Laboratorium ein geeignetes Qualitäts-
managementsystem etablieren muss.
werden nicht an eine andere rechtlich In jedem Fall existieren mittlerweile europaweit gesetzli-
eigenständige Einrichtung abgegeben che Anforderungen bezüglich der Implementierung von
QM-Systemen in praktisch allen Formen von Gesundheits-
Nach aktuellem Verständnis und Auslegung dieser IVDR- einrichtungen, die in Deutschland u.a. durch die Regelun-
Anforderung dürfen Testverfahren, die im eigenen Labor gen nach § 135a Sozialgesetzbuch V schon jahrelang
entwickelt, validiert und hergestellt werden, nicht an eine umgesetzt werden. Fraglich ist allerdings, ob diese QM-
andere rechtlich eigenständige Einrichtung, scil. an eine Systeme stets für die Herstellung und Verwendung von
andere juristische Person, abgegeben werden. IVD aus Eigenherstellung geeignet sind.
Medizinische Laboratorien in Deutschland existieren in Da die Herstellung und Verwendung von IVD aus Eigen-
verschiedenen Organisations- und Rechtsformen. Sie herstellung aber praktisch vollständig medizinischen La-
sind als Einzel-Laboratorien, in einer BAG, Laborgemein- boratorien vorbehalten bleibt, für die in der Folge Anfor-
schaft oder in einem MVZ in der Rechtsform als GbR, derungen gemäß Artikel 5 Absatz 5c IVDR formuliert
PartG, GmbH oder als Laborverbund organisiert oder in werden, wird eine weitere Ausführung zu den Charakte-
größere Klinikverbünde mit den entsprechend diversen ristika des QM-Systems von Gesundheitseinrichtungen
Rechtsträgerschaften integriert. In Gemeinschaften, als nicht erforderlich erachtet.
Partnerschaften und Verbünden agieren medizinische Fazit: Die Ad-hoc-Kommission IVD der AWMF empfiehlt,
Laboratorien als selbstständige Einzelgesellschaften. dass die bereits bestehenden europäischen und nationa-
Die Sicherstellung einer guten medizinischen Versorgung len Regelungen der einzelnen Mitgliedstaaten bezüglich
setzt Versorgungsstrukturen voraus, die den Vorstellungen der Anforderungen an geeignete QM-Systeme für Gesund-
der Ärztinnen und Ärzte von ihrer Berufsausübung her heitseinrichtungen anerkannt und ggf. weiterentwickelt
Rechnung tragen. Aus diesem Grunde wurden neue werden. Eine Koexistenz bzw. eine Integration des
Rechts- und Praxisformen in Deutschland eingeführt, QM-Systems des medizinischen Laboratoriums in das
auch mit dem Ziel, Synergien zu bündeln und dabei eine QM-System der Gesundheitseinrichtung wird auf der
qualitativ hochwertige und qualitätsgesicherte sowie Grundlage der bisherigen Erfahrungen als grundsätzlich
kosteneffiziente Patientenversorgung zu realisieren. umsetzbar angesehen.
Dieser Intention entgegen wirkt eine mögliche Auslegung
der IVDR-Anforderung, dass medizinische Laboratorien, Forderung Art. 5 Abs. 5 (c): Das Labor
die vertraglich innerhalb einer Organisationsstruktur und
Rechtsform gebunden sind, jedoch eigenständig agieren,
der Gesundheitseinrichtung entspricht
weder ein in diesem Umfeld entwickeltes und hergestell- der Norm EN ISO 15189 oder ggf.
tes IVD-Produkt verwenden dürfen, noch von einer Mini- nationalen Vorschriften einschließlich
mierung des administrativen und dokumentarischen
Aufwands, den die Umsetzung der IVDR-Anforderungen
nationaler Akkreditierungsvorschriften
mit sich bringt, profitieren können. Die Anforderungen an die Qualität und die Kompetenz
Fazit: Die Ad-hoc-Kommission IVD der AWMF empfiehlt, der medizinischen Laboratorien, die Produkte innerhalb
dass medizinische Laboratorien innerhalb einer Organi- einer Gesundheitseinrichtung herstellen und verwenden,
sations- und Rechtsform als Einheit gesehen werden und werden aus Sicht der Ad-hoc-Kommission IVD der AWMF
deshalb ihre Produkte untereinander dort austauschen nach Artikel 5 Absatz 5c IVDR nicht eindeutig bzw. mit
dürfen, wo die Anforderung hinsichtlich des Verbleibs der großem Ermessensspielraum dargestellt. Zwar wird einer-
Produkte innerhalb einer Rechtsperson gewahrt bleibt seits eine Konformität der Laboratorien mit der Norm EN
und das IVD nicht kommerziell vermarktet oder anderwei- ISO 15189 [10] gefordert, aber durch den Begriff „oder“
tig in Verkehr gebracht wird. wird diese Norm nationalen Vorschriften einschließlich
Die Verpflichtung der Laboratorien zum Nachweis des nationaler Akkreditierungsvorschriften gleichgestellt.
erfolgreichen Transfers eines Untersuchungsverfahrens Die Situation in den Mitgliedstaaten der EU ist bezüglich
im Rahmen eines Technologie-Transfers bleibt davon der Anforderungen an QM-Systeme bzw. an die Akkredi-
unberührt. tierung für medizinische Laboratorien heterogen. Sie er-
streckt sich von einer gesetzlich vorgeschriebenen
Forderung Art. 5 Abs. 5 (b): Ein für „Vollakkreditierung“ nach EN ISO 15189 bis hin zu frei-
Herstellung und Verwendung der willigen Akkreditierungsprogrammen. In Deutschland ist
Produkte geeignetes seit dem Jahr 2008 mit der RiliBÄK und deren Veranke-
rung in der Medizinproduktebetreiberverordnung (MPBe-
Qualitätsmanagementsystem treibV) die Implementierung eines QM-Systems für alle
medizinische Laboratorien Pflicht. Gleichzeitig sind über
Die IVDR lässt im Rahmen dieser Bedingung offen, in
400 medizinische Laboratorien nach EN ISO 15189 (und
welcher Form die betreffende Gesundheitseinrichtung
vergleichbaren Normen wie der EN ISO 17020) meist
oder ggf. eine untergeordnete Organisationseinheit der
freiwillig akkreditiert; nur für wenige dieser Laboratorien
Gesundheitseinrichtung wie z.B. ein dort befindliches
GMS Zeitschrift zur Förderung der Qualitätssicherung in medizinischen
Laboratorien 2021, Vol. 12, ISSN 1869-4241 5/10Hoffmüller et al.: Stellungnahme der Ad-hoc-Kommission In-vitro-Diagnostika ...
besteht eine Akkreditierungspflicht (letzteres z.B. im Be- der Leistungsbewertung für kommerziell verfügbare In-
reich Neugeborenen-Screening). vitro-Diagnostika [12].
Die Anforderungen der EN ISO 15189 hinsichtlich der Die Bedingung nach Artikel 5 Absatz 5 d IVDR bezieht
Validierung, Verifizierung und der Qualitätssicherung von sich aber auf das „angezeigte Leistungsniveau“ oder die
Untersuchungsverfahren, zu denen auch „nicht genormte „spezifischen Erfordernisse der Patientenzielgruppe“.
Verfahren“ und „für das Laboratorium gestaltete oder Demnach kommt im Rahmen des Vergleichs eine Vielzahl
entwickelte Verfahren“ gehören, entsprechen dem Stand von analytischen und klinisch-diagnostischen Leistungs-
der medizinischen Wissenschaft und Technik und sorgen charakteristika der betreffenden IVD in Betracht, die zu-
für ein international vergleichbares Qualitätsniveau in dem durch das Methodenprinzip, das Design, verfügbare
diesem Bereich. Zusätzlich flankieren nach EN ISO 15189 Kontrollen, Turnaround-Zeiten, Erfahrungen aus der
anerkannte Elemente eines QM-Systems wie z.B. die An- Langzeitanwendung usw. bestimmt werden. Viele dieser
forderungen an die Dokumentation sowie die Durchfüh- Charakteristika werden für kommerziell verfügbare, CE-
rung von Korrektur- und Vorbeugemaßnahmen u.a. die gekennzeichnete IVD allerdings nicht veröffentlicht und
Anforderungen an die Untersuchungsverfahren. sind daher durch die verfügbare Literatur für medizinische
Ein großer Teil der Anforderungen der EN ISO 15189 wird Laboratorien, die IVD aus Eigenherstellung verwenden,
in der RiliBÄK reflektiert, jedoch bestehen auch Unter- größtenteils weder zugänglich noch auswertbar.
schiede. Teilweise, z.B. mit Bezug auf die Ergebnisqualität, Zudem müssen medizinische Laboratorien auf die Anfor-
werden in der RiliBÄK zudem Kriterien formuliert, die sich derungen der behandelnden Ärztinnen und Ärzte gezielt
in der EN ISO 15189 nicht finden. Ein Bezug zur EN ISO und in ihren Leistungsangeboten auf diese Anforderungen
15189 wird in der RiliBÄK nicht gezogen. spezifisch (und nicht „ähnlich“) eingehen, um die Anfor-
Fazit: Die Ad-hoc-Kommission IVD der AWMF empfiehlt, derungen einer komplexen Patientensituation und -ver-
dass Laboratorien für alle Tätigkeitsbereiche, die IVD aus sorgung zu erfüllen.
Eigenherstellung betreffen, die in den jeweiligen Mitglied- Fazit: Die Ad-hoc-Kommission IVD der AWMF empfiehlt,
staaten vorgesehenen und bewährten Konzepte für QM- dass auf europäischer (MDCG) oder nationaler Ebene
Systeme umsetzen. Die Umsetzung kann z.B. durch eine (AGMP bzw. die entsprechend zuständige FEG der ZLG)
freiwillige Akkreditierung des Laboratoriums nach inter- ein harmonisiertes Verständnis über die „Gleichartigkeit“
nationalen Normen oder durch eine Erfüllung der natio- von IVD und die dafür zu betrachtenden, möglichen
nalen Vorschriften wie der RiliBÄK nachgewiesen werden. Charakteristika erzielt und kommuniziert wird.
Unabhängig davon wird das QM-System im Rahmen der Zudem wird empfohlen, dass es zur Sicherstellung der
individuellen Patientenversorgung dann allein den medi-
Überwachung durch die zuständige Behörde (in
zinischen Laboratorien, die IVD aus Eigenherstellung
Deutschland: zuständige Behörden der Länder wie Gewer-
verwenden, vorbehalten sein sollte, die in den betreffen-
beaufsichtsämter, Landesamt für Soziale Dienste,
den Fällen jeweils zutreffenden Charakteristika für die
Eichämter usw.) inspiziert, sodass die Konformität mit
Beurteilung von Gleichartigkeit auszuwählen, diese
den entsprechenden Anforderungen auch im Rahmen
plausibel und nachvollziehbar zu dokumentieren und auf
dieser Überwachung dargelegt werden kann.
Anfrage der zuständigen Überwachungsbehörde vorzule-
gen.
Forderung Art. 5 Abs. 5 (d): Eine
Begründung, dass die spezifischen Forderung Art. 5 Abs. 5 (e):
Erfordernisse der Patientenzielgruppe Informationen für die zuständigen
nicht bzw. nicht auf dem angezeigten Überwachungsbehörden über das
Leistungsniveau durch ein gleichartiges Produkt inkl. Begründung für die
auf dem Markt befindliches Produkt Herstellung, Änderung und Verwendung
befriedigt werden können
Die IVDR verpflichtet die medizinischen Laboratorien zur
Weder in der IVDR noch in den bisher veröffentlichten Erstellung einer geeigneten Dokumentation, um ein Ver-
europäischen bzw. nationalen behördlichen Leitfäden ständnis der Herstellungsstätte und des -verfahrens, der
zur IVDR finden sich Erklärungen zum Begriff der Auslegung und der Leistungsdaten des Produkts ein-
„Gleichartigkeit“ (engl.: „equivalence“). Die IVDR verwen- schließlich der Zweckbestimmung der Laboratorien zu
det an mehreren Stellen die Begriffe „gleichartig“ (engl.: ermöglichen, welche der für sie zuständigen Überwa-
„equivalent“) und „ähnlich“ (engl.: „similar“), ohne hier chungsbehörde auf Ersuchen bereitgestellt werden muss.
erkennbar einen Unterschied zu machen. Zur Interpreta- Diese Informationen entsprechen größtenteils Inhalten
tion der Anforderung an die „Begründung“ liegt bisher einer Technischen Dokumentation zum Nachweis der
nur eine Stellungnahme der britischen Behörde MHRA Erfüllung der grundlegenden Sicherheits- und Leistungs-
vor [11]; ein Leitfaden der europäischen Vereinigung der anforderungen der IVDR.
Medizinproduktehersteller MedTech Europe interpretiert Die IVDR hält einen eigenen Anhang zur Technischen
das Gleichartigkeitsprinzip im Hinblick auf die Prinzipien Dokumentation (Anhang II) vor, der ihre umfangreichen
GMS Zeitschrift zur Förderung der Qualitätssicherung in medizinischen
Laboratorien 2021, Vol. 12, ISSN 1869-4241 6/10Hoffmüller et al.: Stellungnahme der Ad-hoc-Kommission In-vitro-Diagnostika ...
Bestandteile in klarer, organisierter, leicht durchsuchbarer Routine-Anwendung im Hause und ggf. daraus abzuleiten-
und eindeutiger Form vorgibt, um zu beschreiben, was de Korrekturmaßnahmen.
das Produkt leisten soll und wie sichergestellt ist, dass Diese Prozesse sind bereits integraler Bestandteil der für
diese Leistungen tatsächlich erbracht werden. Die Erstel- medizinische Laboratorien adäquaten EN ISO 15189 und
lung einer Technischen Dokumentation im vollen Umfang sind an vielen Stellen mit der Norm verknüpft, insbeson-
des Anhangs II wird nicht von Gesundheitseinrichtungen, dere bei der regelmäßigen Managementbewertung zur
die IVD-Produkte selbst herstellen und anwenden, einge- Eignung und klinischen Wirksamkeit von Untersuchungen
fordert. Die IVDR formuliert hier explizit keine Vorgaben. sowie der Sicherheit der Patienten, beim Ergreifen von
Fazit: Um – trotz föderaler Struktur – in Deutschland Korrektur- und Vorbeugemaßnahmen und bei externen
möglichst klare und einheitliche Vorgaben bezüglich des Beurteilungen sowohl bei einer Akkreditierung als auch
Formats und der Dimension der Dokumentation zu im Rahmen der Beteiligung an einer Leistungsbewertungs-
schaffen, strebt die Ad-hoc-Kommission IVD der AWMF prüfung.
an, eigens für Gesundheitseinrichtungen, die Produkte Die Ad-hoc-Kommission IVD der AWMF strebt an, für
aus Eigenherstellung verwenden, ein allgemeingültiges, medizinische Laboratorien einen Leitfaden zu den bereits
schlankes und zweckmäßiges „Dokumentenformat“ zur bestehenden Regularien der RiliBÄK und der EN ISO
Bereitstellung der geforderten Informationen zu erstellen. 15189 zur Verfügung zu stellen, der aufzeigt, welche
Dieses soll einerseits die Anforderungen der IVDR bedie- Methoden zur Auswertung der in der Routinediagnostik
nen, andererseits die Gesundheitseinrichtungen nicht erhaltenen Daten angewendet, welche Vorgaben als
vor die Herausforderung stellen, durch massiv erhöhten Entscheidungsgrundlage für ggf. einzuleitende CAPA-
Dokumentationsaufwand ihre eigentlichen Aufgaben, die Maßnahmen definiert und wie diese Prozesse und Ergeb-
Patienten- und Gesundheitsversorgung, nicht oder nur nisse für Überwachungsbehörden dokumentiert werden
eingeschränkt wahrnehmen zu können. können.
Forderung Art. 5 Abs. 5 (i): Begutachtung Forderung Art. 5 Abs. 5: Erfüllung der
der Erfahrungen, die aus der klinischen einschlägigen grundlegenden
Verwendung der Produkte gewonnen Sicherheits- und Leistungsanforderungen
wurden und Ergreifung aller gemäß Anhang I
erforderlichen Korrekturmaßnahmen
Anhang I Kap. 1: Risikomanagement
Kommerzielle IVD-Hersteller wurden bereits durch die
EG-Richtlinie 98/79/EG und Gesundheitseinrichtungen Die IVDR fordert gemäß Anhang I, Abschnitt 3 die Imple-
mit Eigenherstellung bereits durch die nationale MPV mentierung, Dokumentation und kontinuierliche Fort-
verpflichtet, ein systematisches Verfahren zu etablieren, schreibung eines Risikomanagementsystems. Dabei wird
das die Auswertung der Erfahrungen mit ihren IVD-Pro- das Risikomanagement als „kontinuierlicher, iterativer
dukten in der Anwendung und die Veranlassung ggf. er- Prozess während des gesamten Lebenszyklus eines
forderlicher Korrekturmaßnahmen ermöglicht. Über die Produkts“ verstanden, der eine „regelmäßige systemati-
Ausgestaltung eines solchen Post-Market Surveillance sche Aktualisierung“ erfordert.
Systems (PMS) gab weder die EU-Richtlinie noch die Kommerzielle Hersteller wenden im Kontext des Risiko-
deutsche Verordnung spezifizierte Vorgaben. Es lag in managements in der Regel die harmonisierte Norm
der Verantwortung des Herstellers/„Eigenherstellers“, EN ISO 14971:2013 an; diese Norm wurde kürzlich
ein dem IVD angepasstes Konzept zu implementieren. überarbeitet und – ohne Harmonisierung mit der IVDR –
In den Erwägungsgründen zur IVDR stellt die EU-Kommis- als EN ISO 14971:2020 publiziert. Entsprechend ihrem
sion nun dar, dass mit und über das QM-System ein In- Anwendungsbereich verwendet die Norm EN ISO 14971
strumentarium zur Überwachung der klinischen Wirksam- Begrifflichkeiten, die im Kontext eines medizinischen
keit und Sicherheit des am Markt und in der Anwendung Laboratoriums kaum Anwendung finden.
befindlichen IVD einzurichten ist, was für kommerzielle Speziell für die Anwendung des Risikomanagements auf
Hersteller detailliert in Kapitel VII Abschnitt I und medizinische Laboratorien wurde jedoch im September
Anhang III der IVDR reglementiert wird. Die PMS-Strategie 2020 die Norm EN ISO 22367 publiziert, die einen Risi-
soll Teil des QM-Systems sein, was die für Medizinproduk- komanagementprozess festlegt, anhand dessen medizi-
te-Hersteller maßgeblichen Normen EN ISO 13485 und nische Laboratorien mit medizinischen Untersuchungen
die EN ISO 14971 bereits verlangen [13], [14]. verbundene Risiken für Patienten, Labormitarbeiter und
Fazit: Für Gesundheitseinrichtungen, die eigenentwickelte Dienstleister erkennen und handhaben können [15]. Der
und -hergestellte Produkte zur Diagnostik einsetzen, ist Prozess umfasst die gemäß IVDR geforderte Erkennung,
ein derartiger PMS-Prozess vonseiten der IVDR ausdrück- Einschätzung, Bewertung, Kontrolle und Überwachung
lich nicht gefordert. Verlangt wird eine dokumentierte der Risiken und berücksichtigt darüber hinaus auch
Begutachtung der Erfahrungen mit den Produkten in der Aspekte der prä- und postanalytischen Phase.
GMS Zeitschrift zur Förderung der Qualitätssicherung in medizinischen
Laboratorien 2021, Vol. 12, ISSN 1869-4241 7/10Hoffmüller et al.: Stellungnahme der Ad-hoc-Kommission In-vitro-Diagnostika ...
Fazit: Die Ad-hoc-Kommission IVD der AWMF empfiehlt, diagnostischer Testbestecke alle die im Anhang I Kapitel 2
dass die Norm EN ISO 14971 keine Anwendung für me- der IVDR vorgegebenen Kriterien abdecken. Ausnahme
dizinische Laboratorien finden sollte. sind die zur Beurteilung der klinischen Leistungsmerkma-
Dagegen können passende Kriterien der Norm EN ISO le geforderten klinischen Leistungsstudien. Hier lässt die
22367:2020, die auf Untersuchungsverfahren, die medi- EU-Verordnung den Freiraum, auf eine Durchführung kli-
zinische Laboratorien für ihren eigenen Gebrauch entwi- nischer Leistungsstudien zu verzichten, wenn es ausrei-
ckeln, anwendbar sind, eine geeignete Grundlage für das chende Gründe dafür gibt, auf andere Quellen klinischer
nach der IVDR geforderte Risikomanagement darstellen. Leistungsdaten zurückgreifen zu können. Diese können
wissenschaftliche Literatur und auch Daten sein, die aus
Anhang I Kap. II (9): Leistungsmerkmale publizierten und im eigenen Labor bereits erhobenen
diagnostischen Routinetests gesammelt wurden.
Nach Definition der IVDR (Artikel 2 (39)) bezeichnet die Die Ad-hoc-Kommission IVD der AWMF empfiehlt, dass
Leistung eines Produkts seine Fähigkeit, die vom Herstel- Gesundheitseinrichtungen zunächst den Umfang des
ler angegebene Zweckbestimmung zu erfüllen. Diese klinischen Nachweises ihrer Produkte zur Begründung
besteht in der Analyseleistung und ggf. der klinischen der Erfüllung der grundlegenden Sicherheits- und Leis-
Leistung zur Erfüllung dieser Zweckbestimmung. Beide tungsanforderungen in einer in ihrem QM-System inte-
Leistungsmerkmale zur Auslegung und Herstellung eines grierten Dokumentation spezifizieren und begründen
Produkts sind in Anhang I aufgezeigt. Weitergehende sollten. Für die Leistungsbewertung des IVD-Produkts
Anforderungen an Umfang und Nachweis von Leistungs- werden dann die Ergebnisse zur wissenschaftlichen,
merkmalen finden sich dort erstmal nicht. analytischen und klinischen Leistung in einem Bericht
Die IVDR weitet jedoch die Anforderungen zum Leistungs- zusammengefasst und das positive Nutzen-Risiko-Verhält-
nachweis aus, denn es heißt in Artikel 5 IVDR: Zum nis belegt.
Nachweis der Erfüllung der Anforderungen der grundle-
genden Sicherheits- und Leistungsanforderungen gemäß Anhang I Kap. II (16): Software
Anhang I muss u.a. eine Leistungsbewertung durchgeführt
werden. Die Leistungsbewertung (clinical evidence) wie- Unter IVD-Software ist Software als eigenständiges IVD
derum ist definiert als die Beurteilung und Analyse von (stand-alone software), als Teil eines IVD (embedded
Daten zur Feststellung oder Überprüfung der wissenschaft- software) oder als Zubehör eines IVD zu verstehen. Da-
lichen Validität (scientific validity), der Analyseleistung runter fallen Software-Lösungen zur Auswertung der
(analytical performance) und gegebenenfalls der klini- Messergebnisse von Testverfahren, Anwendungen zur
schen Leistung (clinical performance) eines Produkts Berechnung und Befundinterpretation und ggf. Labor-
(Artikel 2 (44), IVDR). Es wird hier eine weitere Begrifflich- informationssysteme, die meist an die Laborprozesse
keit eingeführt, die wissenschaftliche Validität, welche und weitere IT-Infrastruktur (Schnittstellen) angepasst
die Assoziation von Analyt und Krankheitsbild bzw. phy- werden müssen, um im medizinischen Labor genutzt
siologischem Zustand veranschaulicht. Mit der Leistungs- werden zu können. Hier sind durchaus Kombinationen
bewertung wird dann der klinische Nachweis (clinical von Software üblich, um den Gesamtprozess abzubilden.
benefit) erbracht, dass ein Produkt sicher ist und den So kann ein Analyse-Gerät als IVD mit einer Middleware
beabsichtigten klinischen Nutzen erzielt. oder einem Laborinformationssystem kommunizieren,
Neben der Validierung des analytischen Verfahrens, das welches die Daten (ggfs. modifiziert) wieder an ein Klinik-
experimentelles Design, Plan und Bericht mit Bewertung Informationssystem (ggfs. ein Medizinprodukt) senden
umfasst, werden klinische Leistung und wissenschaftliche kann.
Validität in den Fokus gestellt. Die klinische Leistung de- Gerade im Bereich hochmoderner High-Throughput-Me-
monstriert die diagnostische Genauigkeit des IVD und thoden, in welchen die Geschwindigkeit der technischen
hängt von der Zweckbestimmung und somit von einer Entwicklung und der wissenschaftlichen wie klinischen
definierten Patientenpopulation ab. Zur Beurteilung der Erkenntnisse immens hoch ist (z.B. NGS-Technologie),
klinischen Leistungsmerkmale als Teil des klinischen sind medizinische Laboratorien auf Parametrierung,
Nachweises fordert die IVDR umfangreiche Leistungsstu- Konfiguration, Customizing und auf vollständig selbstent-
dien. Und im Rahmen der wissenschaftlichen Validität, wickelte Software-Tools angewiesen; dabei sind die
in welcher die Assoziation des Analyten zu einem bestimm- Grenzen zu kommerzieller Software (IVD oder Medizinpro-
ten physiologischen Zustand bzw. Krankheitsbild belegt dukt) fließend. Zudem gibt es gerade bei bioinformati-
wird, ist eine systematische Literaturrecherche unerläss- schen Pipelines (Calling, Annotation, Variantenbewertung)
lich, die Literaturdaten, Expertengutachten, Stellungnah- Hybridformen, in denen Software-Komponenten kommer-
men und Leitlinien von Fachgesellschaften umfassen zieller Hersteller übernommen, Nutzung externer Daten-
kann. Die Dokumentation soll nachvollziehbar Suchstra- quellen sowie Daten von Dienstleistern mit eigenentwi-
tegie und Algorithmen, verwendete Quellen und Kriterien ckelten Systemen und frei verfügbar (Open-Source) Soft-
der Datenauswahl enthalten. ware verwendet werden.
Fazit: Für medizinische Laboratorien gelten zum Leistungs- Die IVDR und der MDCG-Leitfaden 2019-11 [16] adres-
nachweis die Anforderungen der RiliBÄK und der Norm sieren die Thematik von in einer Gesundheitseinrichtung
EN ISO 15189, die für eine vollumfängliche Validierung hergestellten und in Betrieb genommenen Software nicht
GMS Zeitschrift zur Förderung der Qualitätssicherung in medizinischen
Laboratorien 2021, Vol. 12, ISSN 1869-4241 8/10Hoffmüller et al.: Stellungnahme der Ad-hoc-Kommission In-vitro-Diagnostika ...
und lassen viele Fragestellungen hinsichtlich der Abgren- • LDT: Laboratory Developed Test
zungen zwischen Eigenherstellung und Inverkehrbringen • MDCG: Medical Device Coordination Group
offen. Ferner wird Aspekten wie der immer mehr an Be- • MHRA: Medicines and Healthcare products Regulatory
deutung zunehmenden Bioinformatik keine Rechnung Agency
getragen. • MPBetreibV: Medizinproduktebetreiber-Verordnung
Gleichermaßen gelten für Software die bereits genannten • MPG: Medizinprodukte-Gesetz
Voraussetzungen, u.a. dass kein vergleichbares Software- • MPV: Medizinprodukte-Verordnung
produkt auf dem Markt existiert, die grundlegenden Si- • MVZ: Medizinisches Versorgungszentrum
cherheits- und Leistungsanforderungen des Anhang I zu • NGS: Next Generation Sequencing
erfüllen und eine technische Dokumentation anzufertigen • PartG: Partnerschaftsgesellschaft
sind. Nach Anhang I Kapitel 2 (16) muss die Software • PMS: Post-Market Surveillance
unter Berücksichtigung des Software-Lebenszyklus-Pro- • RiliBÄK: Richtlinie der Bundesärztekammer zur Quali-
zesses, des Risikomanagements einschließlich der Infor- tätssicherung laboratoriumsmedizinischer Untersu-
mationssicherheit, der Verifizierung und der Validierung chungen
entwickelt werden. Weiterhin wird die Festlegung von • ZLG: Zentralstelle der Länder für Gesundheitsschutz
Mindestanforderungen an Hardware, Eigenschaften von bei Arzneimitteln und Medizinprodukten
IT-Netzen und IT-Sicherheitsmaßnahmen inklusive des
Schutzes vor unbefugtem Zugriff verlangt. In der geforder-
ten Dokumentation sollen neben der Risikoklassifizierung Anmerkungen
die Entwicklungsstufen der Software dargelegt, die Da-
tenverarbeitung und Datenauswertung insbesondere der Interessenkonflikte
verwendeten Algorithmen beschrieben und unter Berück-
sichtigung der Verwendungsumgebung, Hardware-Konfi- Die Autoren erklären, dass sie keine Interessenkonflikte
gurationen und möglichen Betriebssysteme die Verifizie- in Zusammenhang mit diesem Artikel haben.
rung und Validierung zusammengefasst werden.
Fazit: Die Ad-hoc-Kommission IVD der AWMF empfiehlt
eine Überarbeitung des bestehenden MDCG-Leitfadens Literatur
zur Qualifizierung und Klassifizierung von Software unter
Integration der bislang nicht berücksichtigten zur Diagno- 1. Verordnung (EU) 2017/746 des Europäischen Parlaments und
des Rates vom 5. April 2017 über In-vitro-Diagnostika und zur
stik genutzten Software, wie Bioinformatik-Pipelines oder Aufhebung der Richtlinie 98/79/EG und des Beschlusses
Kombinationen von Software. Weiterhin wird speziell für 2010/227/EU der Kommission.
Software-Eigenherstellungen von Gesundheitseinrichtun- 2. Richtlinie 98/79/EG des Europäischen Parlaments und des
gen eine Klärung gefordert, die Rates vom 27. Oktober 1998 über In-vitro-Diagnostika.
• zwischen den hohen Anforderungen zur Leistungsbe- 3. Medizinprodukte-Verordnung vom 20. Dezember 2001 (BGBl. I
wertung von Software oder Pipelines an kommerzielle S. 3854), die zuletzt durch Artikel 3 der Verordnung vom 27.
September 2016 (BGBl. I S. 2203) geändert worden ist.
Hersteller und an Software ohne Inverkehrbringung
von Gesundheitseinrichtungen differenziert, 4. Medizinproduktegesetz in der Fassung der Bekanntmachung
vom 7. August 2002 (BGBl. I S. 3146), das zuletzt durch Artikel
• die Grenzen von eigenentwickelter, parametrierter und
223 der Verordnung vom 19. Juni 2020 (BGBl. I S. 1328)
konfigurierter Software unmissverständlich definiert, geändert worden ist.
• den besonderen Bedürfnissen medizinischer Labore
5. Medizinprodukte-Betreiberverordnung in der Fassung der
bei der Integration von eigenentwickelter Software Bekanntmachung vom 21. August 2002 (BGBl. I S. 3396), die
oder Softwaremodulen in kommerziell bezogenen zuletzt durch Artikel 9 der Verordnung vom 29. November 2018
Systemen Beachtung schenkt. (BGBl. I S. 2034) geändert worden ist.
6. Bundesärztekammer. Richtlinie der Bundesärztekammer zur
Qualitätssicherung laboratoriumsmedizinischer Untersuchungen;
Abkürzungen Gemäß des Beschlusses des Vorstands der Bundesärztekammer
in seiner Sitzung am 18.10.2019. Deutsches Ärzteblatt; 2019.
DOI: 10.3238/arztebl.2019.rili_baek_QS_Labor20192312
• AGMP: Arbeitsgruppe Medizinprodukte der ZLG
• AMG: Arzneimittel-Gesetz 7. Arzneimittelgesetz in der Fassung der Bekanntmachung vom
12. Dezember 2005 (BGBl. I S. 3394), das zuletzt durch Artikel
• AWMF: Arbeitsgemeinschaft der Wissenschaftlichen
5 des Gesetzes vom 9. Dezember 2020 (BGBl. I S. 2870)
Medizinischen Fachgesellschaften geändert worden ist. Verfügbar unter: https://www.gesetze-im-
• BAG: Berufsausübungsgemeinschaft internet.de/amg_1976/AMG.pdf
• FEG: Fachexpertengruppen der ZLG 8. Prinz W. Die Herstellung von Rezepturarzneimitteln für
• GbR: Gesellschaft bürgerlichen Rechts Apotheken. PharmR. 2008 Aug;8:364 ff.
• GmbH: Gesellschaft mit beschränkter Haftung 9. Prütting D, Saalfrank V, Stollmann F, Wesser S, editors.
• IVD: In-vitro-Diagnostika Kloesel/Cyran Arzneimittelrecht Kommentar. Deutscher
• IVDD: EU-Richtlinie über In-vitro-Diagnostika (98/79/EG) Apotheker Verlag. § 4 Erl. 8 unter Hinweis auf den
• IVDR: Europäische Verordnung über In-vitro-Diagnosti- Ausschussbericht zur 14. AMG-Novelle.
ka ((EU) 2017/746)
GMS Zeitschrift zur Förderung der Qualitätssicherung in medizinischen
Laboratorien 2021, Vol. 12, ISSN 1869-4241 9/10Hoffmüller et al.: Stellungnahme der Ad-hoc-Kommission In-vitro-Diagnostika ...
10. DIN EN ISO 15189 Medizinische Laboratorien – Anforderungen Korrespondenzadresse:
an die Qualität und Kompetenz (ISO 15189:2012, korrigierte
Fassung 2014-08-15); Deutsche Fassung EN ISO 15189:2012.
Prof. Dr. Folker Spitzenberger
Technische Hochschule Lübeck, Mönkhofer Weg 239,
11. Medicines & Healthcare products Regulatory Agency. MHRA
23562 Lübeck, Deutschland
guidance on the health institution exemption (HIE) – IVDR and
MDR (Northern Ireland). 2021. Verfügbar unter: https:// folker.spitzenberger@th-luebeck.de
www.gov.uk/government/publications/mhra-guidance-on-the-
health-institution-exemption-hie-ivdr-and-mdr-northern-ireland Bitte zitieren als
12. Medtech Europe. Clinical Evidence Requirements for CE Hoffmüller P, Brüggemann M, Eggermann T, Ghoreschi K, Haase D,
certification under the in-vitro Diagnostic Regulation in the Hofmann J, Hunfeld KP, Koch K, Meisel C, Michl P, Müller J, Müller C,
European Union. 1st ed. 2020 May. Rabenau HF, Reinhardt D, Riemenschneider MJ, Sachs UJ, Sack U,
Stenzinger A, Streichert T, von Neuhoff N, Weichert W, Weinstock C,
13. DIN EN ISO 13485 Medizinprodukte – Zimmermann S, Spitzenberger F, Ad-hoc-Kommission
Qualitätsmanagementsysteme – Anforderungen für In-vitro-Diagnostika der AWMF. Stellungnahme der Ad-hoc-Kommission
regulatorische Zwecke (ISO 13485:2016); Deutsche Fassung In-vitro-Diagnostika der AWMF zur Umsetzung der Verordnung (EU)
EN ISO 13485:2016. 2017/746 (IVDR) im Hinblick auf In-vitro-Diagnostika aus
Eigenherstellung. GMS Z Forder Qualitatssich Med Lab.
14. DIN EN ISO 14971 Medizinprodukte – Anwendung des
2021;12:Doc03.
Risikomanagements auf Medizinprodukte (ISO 14971:2019);
DOI: 10.3205/lab000043, URN: urn:nbn:de:0183-lab0000432
Deutsche Fassung EN ISO 14971:2019.
15. DIN EN ISO 22367 Medizinische Laboratorien – Anwendung des Artikel online frei zugänglich unter
Risikomanagements auf medizinische Laboratorien (ISO https://www.egms.de/en/journals/lab/2021-12/lab000043.shtml
22367:2020); Deutsche Fassung EN ISO 22367:2020.
16. Medical Device Coordination Group. MDCG-2019-11. Guidance Veröffentlicht: 25.05.2021
on Qualification and Classification of Software in Regulation (EU) Veröffentlicht mit Erratum: 28.05.2021
2017/745 – MDR and Regulation (EU) 2017/746 – IVDR. 2019.
Copyright
©2021 Hoffmüller et al. Dieser Artikel ist ein Open-Access-Artikel und
Erratum steht unter den Lizenzbedingungen der Creative Commons Attribution
4.0 License (Namensnennung). Lizenz-Angaben siehe
http://creativecommons.org/licenses/by/4.0/.
Ein Autor wurde entfernt.
GMS Zeitschrift zur Förderung der Qualitätssicherung in medizinischen
Laboratorien 2021, Vol. 12, ISSN 1869-4241 10/10Sie können auch lesen

















































