Themenheft Familiärer Darmkrebs - LADR
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

Themenheft Familiärer Darmkrebs
Ihr Labor vor Ort
Kiel/ Oldenburg
Flintbek
Eutin Güstrow
Lübeck Grevesmühlen
Hamburg
Ratzeburg Parchim
Bremerhaven
Wittstock
Leer Geesthacht
Pritzwalk Neuruppin
Bremen Rotenburg
Kyritz Bernau
Oldenburg Dannenberg
Seehausen Hennigsdorf
Verden Berlin
Schüttorf
Braunschweig Rüdersdorf
Hannover
Steinfurt
Magdeburg
Münster Salzgitter
Borken Paderborn
Recklinghausen
Göttingen Leipzig
Dortmund
Dresden
Köln Homberg Erfurt
LADR Facharztlabore
Frankfurt Basislabore
Krankenhauslabore
Intermed – unser
Partner für Logistik
Idar-Oberstein und medizinischen
Handel
Saarbrücken Nürnberg
Stuttgart
Baden-Baden
Stand 05/2019
Ulm
München
Freiburg
Der LADR Laborverbund Dr. Kramer & Kollegen ist ärztlich und inhabergeführt. Um die 17 regionalen
Facharztlabore herum sind bundesweit mehr als 3 000 Mitarbeiter tätig, davon über 170 Laborärzte,
Humangenetiker, Mikrobiologen, Pathologen und Naturwissenschaftler sowie Spezialisten aus
klinischen Fachgebieten. Seit 75 Jahren steht der LADR Laborverbund mit ärztlicher Tradition
für labormedizinische Qualität und Beratung. Die LADR Fachlabore versorgen bundesweit gemeinsam
mit den kooperierenden Laborgemeinschaften mehr als 20 000 Ärztinnen und Ärzte im Interesse
der Patienten. Darüber hinaus vertrauen über 370 Kliniken ihre Analytik den Laboratorien des
LADR Laborverbundes an.
Themenheft
Familiärer Darmkrebs
Stand 01/2020
Inhalt
Genetische Beratung und Diagnostik als Schlüssel zur gezielten 4
Krebsfrüherkennung und Prävention!
• Welche Formen von erblichem Darmkrebs werden 6
unterschieden?
• Wann besteht der Verdacht auf erblichen Darmkrebs? 6
• Wie wird familiärer (erblicher) Darmkrebs vererbt? 7
Lynch-Syndrom / Hereditary Nonpolyposis Colorectal Cancer (HNPCC) 8
• Definition des HNPCC-/Lynch-Syndroms 8
• Genetik 9
• Pathologie 9
• Ablauf der HNPCC-Diagnostik 10
• Wem sollte eine molekularpathologische bzw. 12
genetische Mutationsanalyse angeboten werden?
• Erkrankungsrisiken für Genträger/-innen 12
• Vorsorgeprogramm für Patienten/-innen mit HNPCC 13
• Vorsorgeprogramm 14
Familiäre adenomatöse Polyposis und Polyposis-Syndrome 15
• Familiäre adenomatöse Polyposis (FAP), attenuierte 15
FAP (AFAP), MUTYH-assoziierte Polyposis (MAP)
• Vorsorgeprogramm und Therapie 16
• Nicht-adenomatöse Polyposis-Syndrome 18
• Vorsorgeprogramme 19
Umfassende interdisziplinäre Beratung betroffener 20
Familien ist von entscheidender Bedeutung
Wie wird eine genetische Untersuchung veranlasst? 22
Fachliteratur 23
3
LADR Themenheft Familiärer Darmkrebs
Genetische Beratung und Diagnostik als
Schlüssel zur gezielten Krebsfrüherkennung
und Prävention!
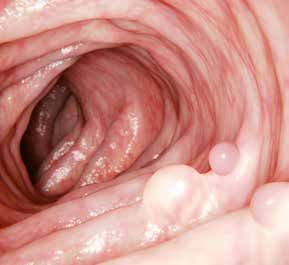
Das kolorektale Karzinom ist bei Frauen nach In ca. 25 % der Familien von Patienten
dem Brustkrebs der zweit- und bei Männern mit Darmkrebs finden sich weitere betrof-
nach Prostata- und Lungenkrebs der dritt- fene Familienmitglieder (Abb. 1). Die Basis der
häufigste maligne Tumor in den deutschspra- familiären Häufung bei 20 % ist im Einzelfall
chigen Ländern. Jährlich werden in Deutschland bis heute nur unvollständig verstanden. Hierbei
ungefähr 60 000 Neuerkrankungen diagnosti- können z. B. Veränderungen in einer größeren
ziert, insgesamt sind über 450 000 Menschen Anzahl disponierender Erbanlagen (Polygenie)
betroffen. Das mittlere Erkrankungsalter liegt im Zusammenwirken mit exogenen Einfluss-
zwischen 70 – 75 Jahren. Da Personen mit faktoren verantwortlich sein. Wir gehen heute
familiärem Darmkrebs häufig bereits im frühen davon aus, dass in ca. 5 % aller Fälle die Erkran-
Erwachsenenalter erkranken, ist die Identifizie- kung erblich ist und wesentlich als Folge einer
rung von Risikopersonen, also Familienmitglie- zugrunde liegenden Mutation in einem hierfür
dern mit hohem Erkrankungsrisiko, von beson- verantwortlichen Gen entstanden ist. Aufgrund
derer Bedeutung. Für Darmkrebs gilt, dass eine der großen Häufigkeit von Darmkrebs kann es
möglichst frühe Diagnosestellung die Prognose jedoch in einer einzelnen Familie auch zu einer
entscheidend beeinflusst. Die gezielte Erken- zufälligen Häufung sporadischer Fälle kommen.
nung von Krebsvorstufen bei Risikopersonen (Abb. 1).
ermöglicht die Vermeidung einer malignen
Entartung und erlaubt speziell bei Darmkrebs
eine effiziente Krebsprävention.
4LADR Themenheft Familiärer Darmkrebs
MAP** andere
0,5 % Formen
sporadisch FAP* ca. 0,5 %
75 % 1%
familiäre
Häufung
Abb. 1:
20 %
Genetische Basis
von Darmkrebs
erblicher
Darmkrebs
ca. 5 %
Lynch-Syndrom
3%
Ca. 75 % aller Darmkrebsfälle werden als Daten modifiziert u. a. nach National Institute of
sporadisch angesehen und zeigen in der Regel Health, National Cancer Institute.
keine familiäre Häufung. In ca. 20 % sind weitere
nahe Verwandte betroffen. In ca. 5 % aller Fälle * FAP: Familiäre adenomatöse Polyposis coli
liegt erblicher Darmkrebs als Folge einer verän- ** MAP: MUTYH-assoziierte Polyposis coli
derten Erbanlage mit sehr hohem Erkrankungs-
risiko vor. Die Häufigkeitsangaben divergieren
in der Literatur zum Teil erheblich, sie stellen
Anhaltswerte dar.
5LADR Themenheft Familiärer Darmkrebs
Welche Formen von erblichem Wann besteht der Verdacht auf
Darmkrebs werden unterschieden? erblichen Darmkrebs?
Wichtigster Vertreter unter den erblichen Typische Hinweise für erbliche Krebserkran-
Formen von Darmkrebs ist mit ca. 3 % von allen kungen, die analog prinzipiell auch für andere
kolorektalen Karzinomen (KRK) das HNPCC erbliche Krebserkrankungen gelten, sind:
(Hereditary Nonpolyposis Colorectal Cancer)-/
Lynch-Syndrom. Es folgen die Polyposis-
Syndrome, von denen die klassische familiäre • Familiäre Häufung von Darmkrebs
adenomatöse Polyposis coli (FAP) mit ca. 1 % bzw. assoziierter Tumore
der Hauptvertreter ist. Klinisch wird davon • Typisches Tumorspektrum
eine attenuierte (abgeschwächte) Form (AFAP) • Frühes Erkrankungsalter
mit Mutationen im gleichen Gen (APC-Gen) • Mehrere Tumore bei einer
unterschieden. Patienten mit der seltenen betroffenen Person
MUTYH-assoziierten Polyposis (MAP) weisen
Mutationen im MUTYH-Gen auf. MAP folgt als
einzige unter den erblichen Darmkrebsformen
einem autosomal rezessiven Erbgang. Während
für die genannten Krankheitsbilder adeno-
matöse Polypen typisch sind, existiert eine
weitere Gruppe von Polyposis-Syndromen mit
vorwiegend hamartomatösen Polypen wie das
Peutz-Jeghers-Syndrom, die seltene familiäre
juvenile Polyposis, und das Cowden-Syndrom.
Abb. 1 sowie Tab. 3 (Seite 15) und Tab. 5 (Seite 18)
fassen die wesentlichen Formen zusammen.
6LADR Themenheft Familiärer Darmkrebs
Wie wird familiärer (erblicher) z. B. beim Lynch-Syndrom, unterschiedliches
Darmkrebs vererbt? Erkrankungsalter). Eine wichtige Ausnahme von
Erblicher Darmkrebs folgt in der Regel einem der autosomal dominanten Vererbung erblicher
autosomal dominanten Erbgang. Hierbei führt Darmkrebserkrankungen stellt die MUTYH-
das Vorliegen einer pathogenen Mutation eines assoziierte Polyposis dar, die einem autosomal
Risikogens auf einem der paarigen Chromo- rezessiven Erbgang folgt. Hierbei erkranken
somen zur Erkrankung. Hieraus lässt sich in der Regel nur Personen einer Generation
ableiten, dass Kinder betroffener Personen (Geschwister), die die pathogenen Mutationen
bzw. bisher noch nicht erkrankte Anlageträ- in beiden Kopien des MUTYH-Gens aufweisen.
ger/-innen ein statistisches Risiko von 50 % Ein Erkrankungsrisiko für Kinder von MAP-
aufweisen, ebenfalls wieder Anlageträger zu Patienten besteht hierbei in der Regel nicht
sein und damit selbst auch ein erhöhtes Erkran- bzw. ist äußerst klein. Aus diesem Grunde ist
kungsrisiko zu tragen. Die mit einer Mutation die molekulargenetische Aufklärung als Voraus-
verbundenen individuellen Erkrankungsrisiken setzung für die genetische Beratung vor allem
können von den statistischen durchschnittlichen bei weniger stark ausgeprägten Polyposis-
Risiken für Anlageträger/-innen abweichen. Ein Syndromen essentiell.
Teil der Anlageträger/-innen entwickelt lebens-
lang keine Krebserkrankung (unvollständige
Penetranz). Innerhalb einer Familie kann die
Erkrankung darüber hinaus sehr unterschied-
lich ausgeprägt sein (variable Manifestationen
Abb. 2:
Kolon-Ca. 44 J. Stammbaum einer
Endometrium-Ca. 47 J. Familie mit Lynch-
Syndrom
R Kolon-Ca. Kolon-Ca.
31 J. 45 J.
R R
Kolon-Ca.
27 J.
R: Risikoperson
7LADR Themenheft Familiärer Darmkrebs
Lynch-Syndrom / Hereditary Nonpolyposis
Colorectal Cancer (HNPCC)
Definition des HNPCC-/Lynch-Syndroms pathogenen Mutation in einem Mismatch-Repair
Das HNPCC-Syndrom ist die wichtigste Form (MMR)-Gen im Tumor. Es wird heute (siehe
des erblichen Darmkrebs und häufigste geneti- auch AWMF-Leitlinie Kolorektale Karzinome)
sche Krebsdisposition überhaupt. Da Patienten favorisiert, die Erkrankung bei Anlageträgern
mit Lynch-/HNPCC-Syndrom in der Regel nur mit einer nachgewiesenen pathogenen Muta-
einzelne kolorektale Adenome oder Karzinome tion in einem MMR-Gen als Lynch-Syndrom
aufweisen, die sich klinisch nicht von spora- zu bezeichnen, während für Personen (bisher)
dischen Formen unterscheiden, wurden die ohne nachgewiesene Mutation, die die
anamnestischen Kriterien zur Identifizierung Amsterdam-/Bethesda-Kriterien erfüllen, die
des HNPCC-Syndroms (Amsterdam-Kriterien Bezeichnung HNPCC verwendet werden sollte.
und die weiter gefassten Bethesda-Kriterien) Die Begriffe werden jedoch häufig synonym
definiert. Die Diagnose HNPCC kann mithin gebraucht. Die Bezeichnung Lynch-Syndrom
allein auf Basis dieser anamnestischen Krite- macht darüber hinaus eher deutlich, dass die
rien gestellt werden (Tab. 1). Hierzu zählen Erkrankung nicht nur auf kolorektale Karzinome
daher auch Familien ohne Nachweis einer beschränkt ist.
Tab. 1: Amsterdam-Kriterien
Amsterdam-Kriterien Alle Kriterien müssen erfüllt sein
und revidierte • Mindestens drei Familienangehörige mit histologisch gesichertem kolorektalem Karzinom
Bethesda-Richtlinie des Endometriums, Dünndarms, Ureters oder Nierenbeckens, davon einer mit den beiden
anderen erstgradig verwandt; FAP muss ausgeschlossen sein
• Wenigstens zwei aufeinanderfolgende Generationen betroffen
• Bei mindestens einem Patienten Diagnosestellung vor dem Alter von 50 Jahren
Revidierte Bethesda-Richtlinien
Mindestens eines der genannten Kriterien muss erfüllt sein
• Patienten mit kolorektalem Karzinom vor dem 50. Lebensjahr
• Patienten mit synchronem oder metachronem kolorektalem Karzinom oder anderen
HNPCC-assoziierten Tumoren*, unabhängig vom Alter
• Patienten mit kolorektalem Karzinom mit MSI-H-Histologie** vor dem 60. Lebensjahr
• Patient mit kolorektalem Karzinom (unabhängig vom Alter), der einen Verwandten
1. Grades mit einem kolorektalen Karzinom oder einem HNPCC-assoziierten Tumor
vor dem 50. Lebensjahr hat
• Patient mit kolorektalem Karzinom (unabhängig vom Alter), der mindestens
zwei Verwandte 1. oder 2. Grades hat, bei denen ein kolorektales Karzinom oder
ein HNPCC-assoziierter Tumor (unabhängig vom Alter) diagnostiziert wurde.
* zu den HNPCC-assoziierten Tumoren gehören Tumoren in: Kolorektum, Endometrium, Magen,
Ovarien, Pancreas, Urothel, Gallengang, Dünndarm und Gehirn (meist Glioblastome wie bei
Turcot-Syndrom) sowie Talgdrüsenadenome und Keratoakanthome (bei Muir-Torre-Syndrom)
** Vorliegen von Tumor infiltrierenden Lymphozyten, Crohn-ähnlicher lymphozytärer Reaktion,
muzinöser/Siegelring-Differenzierung oder medullärem Wachstumsmuster
8LADR Themenheft Familiärer Darmkrebs
Genetik Pathologie
Das Lynch-Syndrom wird durch Mutationen in HNPPC-assoziierte Kolonkarzinome sind meist
einem der DNA-Mismatch-Reparatur (MMR)- muzinöse Tumoren, die bevorzugt im rechten
Gene verursacht. Etwa jede 500. Person ist Hemikolon auftreten.
Träger einer pathogenen Mutation in einem
MMR-Gen. Erst wenn in einer Zelle beide Kopien Mikrosatelliteninstabilität (MSI): Zeichen der
eines der verantwortlichen Gene eine Mutation gestörten DNA-Reparatur sind Verlängerungen
aufweisen, kann der Tumor entstehen. Im Falle kurzer DNA-Wiederholungssequenzen, der
von erblichen Krebserkrankungen liegt bereits Mikrosatelliten (sog. MSI). Sie findet sich bei
in jeder Körperzelle eine Mutation in einer 10 – 15 % aller Kolonkarzinome und bei 15 – 20 %
Genkopie vor, so dass jede auftretende Mutation aller Endometrium-Karzinome im Tumor
in der zweiten intakten Kopie des jeweiligen material. In Verbindung mit Erkrankungs-
Gens in einer Zelle die Tumorentstehung auslöst alter und Familienbefund sind sie jedoch ein
(Knudsonsche Zwei-Treffer-Hypothese). Da das konkreter Hinweis für das Vorliegen eines
Vorliegen einer Keimbahnmutation die statisti- HNPCC-Syndroms. Bei Patienten mit Lynch-
sche Wahrscheinlichkeit für das Auftreten des Syndrom findet sich in Abhängigkeit von dem
Funktionsverlustes beider Kopien eines Gens beteiligten Gen bei mehr als 80 % eine MSI.
(Loss of Heterozygosity, LOH) deutlich erhöht,
lassen sich die klinischen, für erbliche Tumor- Immunhistochemie: In Abhängigkeit vom betei-
erkrankungen typischen Hinweise (siehe Seite ligten Gen lässt sich bei ca. 95 % der Patienten
6: Wann besteht der Verdacht auf erblichen mit Lynch-Syndrom immunhistochemisch ein
Darmkrebs?) hierdurch erklären. Pathogene Ausfall von Reparaturproteinen nachweisen.
Mutationen werden vor allem in den Genen Da die Genprodukte von MLH1 und PMS2 sowie
MLH1 (50 %), MSH2 (40 %), MSH6 (7 – 10 %), PSM2 MSH2 und MSH6 jeweils einen Proteinkomplex
(< 5 %) und EPCAM (1 – 3 %) nachgewiesen. bilden, führen z. B. Mutationen im MLH1-Gen
Die Entwicklung des Karzinoms setzt jedoch in der immunhistochemischen Analyse neben
weitere Mutationsereignisse in einer Zelle (z. B. dem Ausfall von MLH1 auch zum Ausfall seines
von K-ras, TP53) voraus. Partnerproteins PMS2.
Ein Ausfall von MLH1 und PMS2 kann jedoch
auch – meist infolge einer Methylierung des
MLH1-Promotors – im Tumorgewebe auftreten
und das MLH1-Gen funktionell inaktivieren. Eine
MLH1-Promotormethylierung kann jedoch auch
Folge einer bestimmten somatischen Mutation
(V600E) im BRAF-Gen sein. Da diese Mutation
praktisch nie bei einem erblichen Tumor beob-
achtet wird, ist das Vorliegen dieser Mutation
ein starker Hinweis für einen nicht erblichen
Tumor (Abb. 3, Seite 10).
9LADR Themenheft Familiärer Darmkrebs
Ablauf der HNPCC-Diagnostik chemische/molekularpathologische Analyse bei
Die Diagnose HNPCC-/Lynch-Syndrom erfolgt positiven Amsterdam-/Bethesda-Kriterien folgt
meist unter Berücksichtigung sowohl klinischer, einem molekularpathologischen Algorithmus
familienanamnestischer, pathologischer und (Abb. 3), dem sich meist eine genetische Analyse
genetischer Informationen. Eine immunhisto- anschließt.
Abb. 3:
Ablauf der der Positive Amsterdam-/revidierte Bethesda-Kriterien
molekularpatholo-
gischen Abklärung Immunhistochemie Tumorgewebe (Biopsie/Resektat)
eines Mismatch-
Reparaturdefektes bei
klinischem Verdacht
auf HNPCC-/Lynch- MLH1/PMS2 MSH2 oder
MLH1
Syndrom (modifiziert MSH2/MSH6 MSH6 oder
fehlt
nach AWMF-Leitlinie vorhanden PMS2 fehlt
Kolorektales Karzi-
nom, 2019). Amster-
dam-/Bethesda-
Kriterien siehe Tab. 1
(Seite 8) MSI-Testung BRAF-Mutationsanalyse
Keine Nachweis
Stabil Instabil Mutation der V600E
(Wildtyp) Mutation
Sporadisches
Mikrosatelliten- V. a. HNPCC /
Mikrosatelliten-
stabiles KRK Lynch-Syndrom
instabiles KRK
10LADR Themenheft Familiärer Darmkrebs
Abb. 4 zeigt den Algorithmus zum Ablauf Amsterdam-/Bethesda-Kriterien abhängt. Die
der genetischen Diagnostik bei Patienten prädiktive Testung von Risikopersonen in der
mit Verdacht auf HNPCC-/Lynch-Syndrom. Familie setzt jedoch eine genetische Analyse
Wichtig ist, dass die Inanspruchnahme der voraus.
HNPCC-Vorsorge allein von dem Zutreffen der
Abb. 4:
Positive Amsterdam-/revidierte Bethesda-Kriterien bei Schema des Ablaufs
nachgewiesener Mikrosatelliteninstabilität* der genetischen Dia-
Keimbahnmutationsanalyse** gnostik und Vorsorge
bei Patienten mit
Verdacht auf HNPCC
(modifiziert nach
Nachweis einer pathogenen Kein Nachweis einer pathogenen AWMF-Leitlinie Kolo-
MMR-Genmutation MMR-Genmutation rektale Karzinome)
Diagnose: Lynch-Syndrom Diagnose: HNPCC-Syndrom
Prädiktive Testung***
Prädiktive Testung***
weiterer Familienmitglieder
weiterer Familienmitglieder
nicht möglich
Ausschluss der Nachweis der
pathogenen pathogenen HNPCC-Vorsorge
Mutation Mutation
Vorsorge entspr.
asympt. Bevölkerung
* Bei hochgradigem Verdacht auf (V. a.) HNPCC/Lynch-Syndrom und Nichtvorhandensein von Tumor-
gewebe kann auch direkt eine Mutationsanalyse erfolgen.
** Eine diagnostische Keimbahnuntersuchung erfordert eine Aufklärung und Einwilligung nach dem
Gendiagnostikgesetz (GenDG). Es sollte das Angebot einer genetischen Beratung erfolgen.
*** Eine prädiktive genetische Diagnostik muss im Rahmen einer genetischen Beratung erfolgen, die
eine Qualifikation voraussetzt.
11LADR Themenheft Familiärer Darmkrebs
Wem sollte eine molekularpathologische Erkrankungsrisiken für Genträger/-innen
bzw. genetische Mutationsanalyse angeboten Wie der Name Lynch-Syndrom auch zum
werden? Ausdruck bringen soll, handelt es sich dabei
Patienten mit HNPCC/Lynch-Syndrom (die die um eine Erkrankung, die mit deutlich erhöhten
Amsterdam-/Bethesda-Kriterien erfüllen) kann Risiken für Karzinome auch anderer Organ-
sowohl eine molekularpathologische (Abb. 3, systeme verbunden ist. Das Risiko von
Seite 10) als auch Mutationsanalyse (Abb. 4, HNPCC-Anlageträgern, mindestens ein HNPCC-
Seite 11) angeboten werden. Die Qualitätssiche- assoziiertes Karzinom zu entwickeln, beträgt
rungsvereinbarung Molekulargenetik definiert 80 – 90 %. KRK treten im Rahmen von HNPCC
als Voraussetzung für die Berechnung der im Mittel im 44. Lebensjahr auf, vor dem 25.
Gebührenordnungsposition 11431 (Hereditäres Lebensjahr sind KRK bei HNPCC-/Lynch-
non-polypöses kolorektales Karzinom, HNPCC) Syndrom sehr selten.
für eine Mikrosatellitenanalyse die Erfüllung Abb. 5 fasst die relevanten Erkrankungs-
der Bethesda-Kriterien. Die Voraussetzung für risiken zusammen. Die Einzelwerte geben
die Berechnung der Gebührenordnungsposition Mittelwerte an, die sowohl individuell wie auch
11432 (Hereditäres non-polypöses kolorektales abhängig von den beteiligten Genen abweichen
Karzinom, HNPCC) für die direkte Analyse der können.
HNPCC-Gene (MLH1, MSH2, MSH6, PMS2) ist
80
gegeben, wenn alle Amsterdam-Kriterien erfüllt
sind (Tab. 1, Seite 8).
65 %
Abb. 5:
Erkrankungsrisiken
für Träger/-innen
50 %
einer HNPCC-/Lynch-
Syndrom-Mutation. Mutations-
Die Risiken variieren träger
70
für einzelne MMR-Ge- Normal-
60
ne. Das KRK-Risiko ist bevölkerung
für Männer
50 ca. 10 %
höher als40
für Frauen.
30
20 7,5 %
5,5 % 5% 6%
10 2,5 % 3%
1% 1% 0% 0%
0
Kolorektales Endometr. Ovarial Magen Dünndarm Urothel
Ca. Ca. Ca. Ca. Ca. Ca.
12LADR Themenheft Familiärer Darmkrebs
Vorsorgeprogramm für
Patienten/-innen mit HNPCC
Nicht in allen Familien gelingt der Nachweis
einer pathogenen Mutation. Die AWMF-Leit-
linie Kolorektales Karzinom führt aus, dass
eine molekulargenetische Abklärung auch dann
erfolgen kann, wenn keine Mikrosatelliteninsta-
bilität untersucht bzw. nachgewiesen werden
konnte. Abb. 5 fasst den Ablauf der genetischen
Abklärung und Vorsorge zusammen.
Patienten und Risikopersonen, die die
klinischen Amsterdam-/revidierten
Bethesda-Kriterien (bei nachgewie-
sener MSI) erfüllen, sollen sich entspre-
chend der AWMF-Leitlinie Kolorektales
Karzinom auch ohne erfolgten Mutations-
nachweis dem HNPCC-Vorsorgeprogramm
unterziehen. Eine genetische
Beratung zwischen
dem 18. und 25.
Risikopersonen (nicht erkrankte nahe Lebensjahr ist
Verwandte betroffener Personen) für HNPCC-/ Risikopersonen zu
Lynch-Syndrom ist mit Erreichen der Einwil- empfehlen.
ligungsfähigkeit (in der Regel ab dem
18. Lebensjahr), jedoch vor dem 25. Lebensjahr
eine genetische Beratung zu empfehlen. Sobald
die krankheitsverursachende Mutation in der
betreffenden Familie bekannt ist, sollten Risiko-
personen auf die Möglichkeit einer prädiktiven
Testung hingewiesen werden.
13LADR Themenheft Familiärer Darmkrebs
Vorsorgeprogramm
Entsprechend der AWMF-Leitlinie Kolorektales
Karzinom wird das nachfolgende Krebsfrüh-
erkennungsprogramm für HNPCC-Patienten
empfohlen (Tab. 2):
Tab. 2:
Empfohlenes HNPCC- Altersangabe Untersuchung
Krebsfrüherken-
Ab dem 25. Lebensjahr Körperliche Untersuchung Jährlich
nungsprogramm
(nach der AWMF- Koloskopie Jährlich
Leitlinie Kolorektales Gynäkologische Untersuchung einschließlich
Jährlich
Karzinom, 2019) transvaginaler Sonographie
Ab dem 35. Lebensjahr ÖDG Regelmäßig
Endometriumbiopsie Jährlich
ÖDG: Ösophago- Duodeno-Gastroskopie
Die früher empfohlene Ultraschallunter-
suchung des Abdomens und die Urinzytologie
als Hinweis auf ein Urothel-Ca. sind nicht mehr
Bestandteil der aktuellen AWMF-Leitlinie Kolo-
rektales Karzinom.
Bei Patientinnen mit HNPCC-Syndrom
sollte mit 40 Jahren, bzw. fünf Jahre vor dem
frühesten Erkrankungsalter in der Familie, eine
prophylaktische Hysterektomie und ggf. Ovarek-
tomie besprochen werden.
14LADR Themenheft Familiärer Darmkrebs
Familiäre adenomatöse Polyposis
und Polyposis-Syndrome
Familiäre adenomatöse Polyposis (FAP), Die attenuierte FAP (AFAP) ist typischer-
attenuierte FAP (AFAP), MUTYH-assoziierte weise durch weniger als 100 kolorektale
Polyposis (MAP) Adenome und/oder das im Vergleich zur klassi-
Die familiäre adenomatöse Polyposis coli schen FAP etwa 10 – 15 Jahre spätere Auftreten
(FAP) ist die bekannteste und häufigste kolo- von Adenomen und kolorektalen Karzinomen
rektale Polyposis (Tab. 3). Die Diagnose kann gekennzeichnet. In etwa 15 – 30% der Familien
mit Nachweis von oft mehr als 100 Polypen, die lassen sich Mutationen im APC-Gen nachweisen.
meist in der 2. Lebensdekade entstehen, in der Im Einzelfall sind die Übergänge zur klassi-
Regel leicht gestellt werden. In vielen Fällen ist schen FAP fließend.
die Familienanamnese zusätzlich wegweisend. Die MUTYH-assoziierte Polyposis (MAP)
Klinisch entwickeln praktisch alle Patienten ein ist eine wichtige Differenzialdiagnose der FAP.
KRK und ca. 75 % extrakolonische intestinale Sie entspricht klinisch derjenigen der AFAP.
Manifestationen, von denen Duodenal- bzw. Aufgrund des autosomal rezessiven Erbgangs
Papillenadenome als Präkanzerosen einzu- ist sie in der Regel nicht mit wesentlich
ordnen sind. Magenadenome sind bei deutlich erhöhten Risiken für Kinder verbunden. Eine
weniger als 10 % der Patienten weniger bedeu- MAP wird bei 15 – 20% der APC-Mutations-nega-
tend. Sie haben darüber hinaus keine potentiell tiven Patienten diagnostiziert. Die Polypen und
präneoplastische Potenz. Weitere extraintes- Karzinome bei den Anlageträgern treten meist
tinale Manifestationen sind Desmoidtumoren, später und häufig im proximalen Kolon auf. Bei
Schilddrüsen-Karzinome, Hepatoblastome ca. 0,5 % aller Darmkrebspatienten lassen sich
sowie harmlose Osteome, Epidermoidzysten Mutationen in beiden Kopien des MUTYH-Gens
oder Pigmentanomalien der Retina (CHPRE). nachweisen (Abb. 1, Seite 5). Die Heterozygoten-
Bei bis zu 95 % der Patienten mit dem klas- frequenz für eine pathogene MUTYH-Mutation
sischen Bild einer FAP können Mutationen im in der Normalbevölkerung beträgt ca. 1 – 2%.
APC-Gen nachgewiesen werden. Bei 20 – 25 % Heterozygote Anlageträger/-innen tragen ein
der Patienten liegt eine Neumutation vor. leicht erhöhtes KRK-Risiko.
Darüber hinaus sind Mosaikbefunde bekannt.
Tab. 3:
Polypen Lebens- Hereditäre gastro
Polypen-
Krankheit Gen Lokalisa- zeitrisiko weitere Symptome intestinale Krank-
zahl
tion für KRK
heitsbilder mit
Familiäre APC 1 000 bis Dickdarm, 100 % Desmoide, Osteome, CHPRE, vorwiegend adeno
adenomatöse > 5 000 Duodenum, Epidermoidzysten, Hepatoblastom, matösen Polypen
Polyposis (FAP) (Magen) Medulloblasom (modifiziert nach
Aretz, 2010)
Attenuierte FAP APC 10 bis 100 Dickdarm, 80 – 100 % Selten
(AFAP) Duodenum,
(Magen)
MUTYH-assoziierte MUTYH 20 bis Dickdarm, 80 – 100 % Erhöhte Inzidenz extraintestinaler
Polyposis (MAP) Hunderte Duodenum, Malignome, selten Talgdrüsen
(Magen) tumore
15LADR Themenheft Familiärer Darmkrebs
Vorsorgeprogramm und Therapie
Verwandte eines FAP-Patienten, die aufgrund Risikopersonen bezeichnet. In der AWMF-Leit-
des autosomal-dominantem Erbgangs als Muta- linie Kolorektales Karzinom wird das nachfol-
tionsträger in Betracht kommen, werden als gende Vorsorgeprogramm empfohlen (Tab. 4):
Tab. 4:
Therapie-/Vorsorge- • Bei Risikopersonen sollte ab dem Patienten mit erhaltenem Rektumstumpf
progamm bei fami- 10. Lebensjahr im Anschluss an eine soll regelmäßig eine Rektoskopie durch-
liärer adenomatöser humangenetische Beratung der Familie geführt werden, wobei das Untersuchungs-
Polyposis coli (nach eine prädiktive genetische Diagnostik intervall 12 Monate nicht überschreiten soll.
der AWMF-Leitlinie empfohlen werden, soweit die zugrunde • Eine Ösophago-Gastro-Duodenoskopie
Kolorektales Karzi- liegende APC-Keimbahnmutation in der (ÖGD) und Duodenoskopie (Seitblickoptik)
nom, 2019) Familie identifiziert werden konnte. mit besonderer Inspektion der Papillen-
• Risikopersonen, bei denen die Mutation region sollte spätestens ab dem 25. – 30.
bestätigt oder nicht ausgeschlossen Lebensjahr durchgeführt werden. Bei
werden konnte, sollten ab dem 10. Lebens- unauffälligem Befund wird ein 3-Jahres-
jahr jährlich rekto-sigmoidoskopiert Intervall empfohlen. Das Intervall sollte
werden. Bei Nachweis von Adenomen soll in Abhängigkeit vom Schweregrad
eine komplette Koloskopie erfolgen und vorhandener Adenome auf bis zu einem
bis zur Proktokolektomie jährlich wieder- Jahr verkürzt werden. Bei Nachweis von
holt werden. Duodenal-/Papillenadenomen ist die Indi-
• Patienten mit klassischer FAP sollten kation zur endoskopischen Polypektomie zu
prophylaktisch und unabhängig vom überprüfen. Bei schwergradiger Duodenal-
Ergebnis der molekulargenetischen polyposis und invasivem nicht-fernmetas-
Testung – wann immer möglich kontinenz- tasiertem Karzinom besteht eine Indikation
erhaltend – proktokolektomiert werden, zur operativen Resektion.
wenn vertretbar erst nach Abschluss der • Eine jährliche Sonographie der Schild-
Pubertät. drüse kann ab dem 15. Lebensjahr bei
• Nach einer Proktokolektomie soll regel- weiblichen FAP-Patientinnen durchgeführt
mäßig eine Pouchoskopie erfolgen. Bei werden.
16LADR Themenheft Familiärer Darmkrebs
Geschwister eines Patienten mit MUTYH
assoziierter FAP (MAP) haben aufgrund des
autosomal-rezessiven Erbgangs ein Erkran-
kungsrisiko von 25 % und gelten als Risikoper-
sonen. Diesen sollte ab dem 18. – 20. Lebens-
jahr im Anschluss an eine humangenetische
Beratung eine prädiktive genetische Diagnostik
angeboten werden
Für Verwandte eines MAP-Patienten, bei
denen lediglich eine von zwei der beim Index-
patienten nachgewiesenen MUTYH-Mutationen
vorliegt (heterozygote Anlageträger) wie den
Eltern, werden Vorsorgeuntersuchungen wie
bei erstgradig Verwandten eines Patienten
mit sporadischem KRK empfohlen (d. h. erste
Ein Patient mit einer Attenuierten FAP (AFAP) Koloskopie spätestens mit 40 – 45 Jahren oder
sollte in Abhängigkeit von Alter, Polypenzahl 10 Jahre vor dem frühesten Manifestationsalter
und histologischem Befund therapiert werden. in der Familie bei einem Untersuchungsintervall
Bei endoskopisch nicht beherrschbarer Poly- von 10 Jahren), da davon ausgegangen werden
posis ist eine Kolektomie indiziert. Patienten, die kann, dass auch heterozygote Anlageträger ein
nicht kolektomiert sind, sollten zeitlebens jedes geringgradig erhöhtes Risiko für ein KRK im
Jahr koloskopiert werden. fortgeschrittenen Alter tragen.
Risikopersonen aus Familien mit AFAP Asymptomatische MUTYH-Mutationsträger
sollten im Rahmen der Vorsorgeuntersuchung (mit Mutationen in beiden Genkopien) sollten
im Alter von 15 Jahren erstmals koloskopiert im Alter von 18 – 20 Jahren erstmals kolosko-
werden. Finden sich keine Polypen, sollten diese piert werden. Finden sich keine Polypen, sollten
Personen ab dem 20. Lebensjahr jährlich kolos- diese Patienten weiterhin überwacht werden.
kopiert werden. Ein Patient mit einer MAP sollte in Abhängig-
Da die klinische Ausprägung stark variieren keit von Alter, Polypenzahl und histologischem
kann, ist die Therapieentscheidung individuell Befund therapiert werden. Bei endoskopisch
abzuwägen. Da extrakolonische Manifestationen nicht beherrschbarer Polyposis ist eine Kolek-
genau wie bei der klassischen FAP auftreten tomie indiziert. Patienten, die nicht kolektomiert
können, gelten diesbezüglich die Empfehlungen sind, sollen zeitlebens jedes Jahr kolosko-
für die klassische FAP. Bis zu welchem Alter piert werden. Eine ÖGD und Duodenoskopie
Vorsorgeuntersuchungen bei Risikopersonen (Seitblickoptik) mit besonderer Inspektion der
mit unauffälligem endoskopischem Befund fort- Papillenregion soll ab dem 25. – 30. Lebens-
geführt werden sollen, ist aufgrund der Daten- jahr mindestens alle drei Jahre durchgeführt
lage derzeit unklar. werden. Spezifische Vorsorgeuntersuchungen
für extraintestinale Manifestationen sind bei
MAP-Patienten nicht gerechtfertigt.
17LADR Themenheft Familiärer Darmkrebs
Nicht-adenomatöse Polyposis-Syndrome interdisziplinäre Zusammenwirken von Gastro-
Hierzu zählen u. a. die hamartomatösen Poly- enterologen, Chirurgen, Pathologen, Human-
posis-Syndrome: Peutz-Jeghers-Syndrom, genetikern, Radiologen und anderen klini-
Familiäre Juvenile Polyposis und Cowden- schen Fachdisziplinen (vor allem Gynäkologie,
Syndrom (Tab. 5). Urologie). Die Diagnose und klinische Betreuung
Die Differenzialdiagnose der nicht-adeno- der Patienten sollte daher in Abstimmung und
matösen Polyposis-Syndrome kann in Einzel- Zusammenarbeit mit Zentren erfolgen, die
fällen sehr schwierig sein und erfordert das Erfahrung mit diesen Syndromen haben.
Tab. 5: Polypen Lebens-
Syndrome mit über- Polypen-
Krankheit Gen Lokalisa- zeitrisiko weitere Symptome
zahl
wiegend hamarto- tion für KRK
matösen Polypen
(modifiziert nach Peutz-Jeghers- STK11 < 20 Dünndarm, 40 % Mukokutane/periorale Hyperpig-
Syndrom (LKB1) Dickdarm, mentierungen, Ovarialtumoren,
Aretz, 2010)
Magen Brustkrebs, Pancreas-Ca, Cervix-Ca
Familiäre juvenile SMAD4 5 bis Dünndarm, > 90 % ? Bei SMAD4-Mutationsträgern: here-
Polyposis BMPR1A Hunderte Dickdarm, ditäre, hämorrhagische Teleangi-
+PTEN Magen ektasien in ca. 20 %, Magenpolypen
und -karzinome
Cowden- PTEN Multiple Dünndarm, Gering Mukokutane Tumoren, Brustkrebs,
Syndrom Dickdarm, Endometrium-Ca, Schilddrüsen-Ca,
Magen andere hamartomatöse Tumoren
Die hamartomatösen Polypen von Patienten Das kumulative Lebenszeitrisiko für einen
mit Peutz-Jeghers-Syndrom (PJS) zeigen eine malignen Tumor wird mit 85 – 90 % angegeben.
charakteristische Histologie, wegweisend für die Für Tumore im gesamten Gastrointestinal-
Diagnose sind darüber hinaus die charakteris- trakt besteht ein kumulatives Lebenszeitrisiko
tischen perioralen Pigmentierungen, die meist von ca. 60 %, das KRK-Risiko allein beläuft sich
schon vor dem 5. Lebensjahr nachweisbar auf ca. 40 %. Tumore treten selten vor dem 30.
sind, im Laufe des Lebens aber oft abblassen. Lebensjahr auf, das Risiko steigt dabei nach
Das Manifestationsalter ist sehr variabel. Die dem 50. Lebensjahr rasch an. Das Lebenszeit-
Erkrankung kann bereits im frühen Kindesalter risiko für gynäkologische Tumore wird mit ca.
durch eine Invagination oder obstruktiven Ileus 15 % angegeben. Ovarialtumoren bei PJS werden
und eine chronisch gastrointestinale Blutung zum Teil bereits auch bei jungen Mädchen diag-
mit sekundärer Anämie imponieren. Das PJS nostiziert. Cervixkarzinome treten mit einem
disponiert zu einem breiten Spektrum gut- und Lebenszeitrisiko von fast 10 % auf und entspre-
bösartiger Tumore wie Brustkrebs, Pankreas- chen histologisch in mehr als drei Viertel der
Ca., Ovarialtumoren. Fälle einem Adenoma malignum.
18LADR Themenheft Familiärer Darmkrebs
Solitäre juvenile Polypen sind die Das Cowden-Syndrom wird heute
häufigsten Polypen des Kindes- und Jugend- zusammen mit weiteren Varianten als PTEN-
alters und sind in der Regel harmlos. Von der Hamartoma-Syndrom bezeichnet. Kolorektale
familiären juvenilen Polyposis sollte erst Polypen treten bei weniger als 10 % der Betrof-
gesprochen werden, wenn mehr als 5 Polypen fenen auf und sind hierbei kein Leitsymptom,
mit typischer Histologie im Kolorektum bzw. sie besitzen ein allenfalls geringes malignes
multiple Polypen im GI-Trakt oder ein oder Potenzial. Typisch ist eine Makrozephalie. Beim
mehrere Polypen bei positiver Familienanam- Cowden-Syndrom ist insbesondere das Risiko
nese nachgewiesen wurden. In einem nennens- für Mamma- und Schilddrüsenkarzinome
werten Teil der genetisch gesicherten Fälle wird erhöht. Weiterhin wurden erhöhte Risiken für
die juvenile Polyposis initial als Colitis ulcerosa Karzinome des Endometriums und der Nieren
oder hyperplastische Polyposis fehlgedeutet. sowie für Melanome beschrieben.
Typisch ist eine exsudative Enteropathie mit
begleitender Entwicklungsverzögerung. Vorsorgeprogramme
Das Lebenszeitrisiko für die Entwick- Spezielle Vorsorgeprogramme bzw. Empfeh-
lung eines KRK beträgt bis zu 70 %. Bei sehr lungen für die genannten Entitäten haben
schweren frühmanifesten Verläufen ist an die aufgrund der begrenzten Datenlage eine nur
seltene juvenile Polyposis des Kleinkindes- eingeschränkte Verbindlichkeit. Sie richten
alters zu denken. Die endoskopisch-histolo- sich nach dem jeweiligen Tumorspektrum und
gische Abgrenzung einer juvenilen Polyposis hinsichtlich der kolorektalen Tumore nach deren
vom auf PTEN-Mutationen beruhenden Cowden- Auftreten im Rahmen des Syndroms (s. Aretz,
Syndrom kann ebenfalls Probleme bereiten und 2010).
erfolgt in der Regel durch das im Vordergrund
stehende extraintestinale Tumorspektrum und
die Molekulargenetik.
19LADR Themenheft Familiärer Darmkrebs
Umfassende interdisziplinäre
Beratung betroffener Familien ist von
entscheidender Bedeutung
Umfassende humangenetische und fachärzt-
liche Beratungen sind für betroffene Familien Eine prädiktive genetische Analyse kann
und Risikopersonen von besonderer Bedeutung. bei einer Risikoperson nur dann zum
In der humangenetischen Beratung wird ermit- Ausschluss eines erhöhten Erkrankungs-
telt, ob überhaupt eine spezielle Risikosituation risikos führen, wenn bei einer betroffenen
besteht und eine genetische Testung sinnvoll Person der Familie die verantwortliche
sein kann. Der individuelle Sachverhalt wird den Mutation nachgewiesen wurde.
Ratsuchenden verständlich erklärt und danach
schriftlich zusammengefasst.
Die Diagnose erblicher Darmkrebs kann Bei dieser Fragestellung sollte daher in
für betroffene Familien wichtige Konsequenzen jedem Fall die molekulargenetische Diagnose
haben: sicherung bei einer betroffenen Person erfolgen.
In Familien, in denen eine betroffene Person
Wichtige Konsequen- nicht untersucht werden kann bzw. bereits
zen für betroffene • Anlageträger/-innen können Vorsorge- verstorben ist, sollte überlegt werden, ob eine
Familien und Therapieoptionen in Anspruch molekulargenetische Analyse aus Tumorma-
nehmen. terial einer betroffenen Person der Familie
• Nahe Verwandte betroffener Personen möglich ist.
(Risikopersonen) haben ein deutlich Vor der Entscheidung zur molekulargeneti-
erhöhtes Risiko, ebenfalls die krank- schen Untersuchung sollen Risikopersonen im
heitsverursachende Mutation zu tragen. Rahmen einer genetischen Beratung umfassend
Es beträgt für Kinder von Anlageträgern beraten werden. Die Entscheidung für oder
50 %, die selbst wiederum ein stark gegen eine molekulargenetische Testung ist
erhöhtes Erkrankungsrisiko tragen. immer individuell und persönlich.
Risikopersonen und gesicherten Anlage-
trägern wird empfohlen das spezielle
engmaschige Vorsorgeprogramm wahr-
zunehmen, das die Früherkennung
erleichtern und damit die Heilungs-
chancen deutlich verbessern kann.
20LADR Themenheft Familiärer Darmkrebs
Die in der AWMF-Leitlinie Mammakarzinom Nach Erhalt des molekulargenetischen
ausgeführten Überlegungen zur genetischen Befundes sollten in der Beratung vor dem
Testung gelten entsprechend auch für den Angebot präventiver Maßnahmen insbesondere
familiären Darmkrebs. Danach soll die human- folgende Inhalte vertieft werden:
genetische Beratung eine partizipative Entschei-
dungsfindung ermöglichen, die eine umfas-
sende Information in den Entscheidungsprozess • Erkrankungsrisiko in Abhängigkeit vom
voraussetzt. genetischen Befund, Alter und Begleit-
Bei der Risikoberatung vor der genetischen erkrankungen (natürlicher Verlauf)
Testung sollten insbesondere folgende Inhalte • Wahrscheinlichkeit für falsch positive und
berücksichtigt werden: falsch negative Testergebnisse der inten-
sivierten Früherkennung
• Nutzen der präventiven Optionen (intensi-
• Wahrscheinlichkeit für das Vorliegen vierte Früherkennung, prophylaktische
einer Mutation Operationen, medikamentöse Therapien)
• Erkrankungsrisiken bei positivem Befund • Risiken der präventiven Optionen
• Nutzen präventiver und therapeutischer einschließlich Langzeitfolgen
Optionen einschließlich der Option, nichts • Konkurrierende Risiken, Prognose
zu tun und Therapierbarkeit im Falle eines
• Wahrscheinlichkeit falsch negativer Krankheitseintrittes ohne präventive
Befunde Maßnahmen unter Berücksichtigung des
• Bedeutung der genetischen Testung für spezifischen Erscheinungsbildes des
die Familienangehörigen. genetisch definierten Tumorsubtyps
• Gegebenenfalls Risiken für assoziierte
Tumoren
• Psychoonkologische Beratungsangebote
21Wie wird eine genetische
Untersuchung veranlasst?
Bei einer erkrankten Person kann nach um Bei weitergehenden Fragen, z. B. zu konkre-
fassender Aufklärung und schriftlicher Einwil- ten Einzelfällen, stehen Ihnen die Mitarbeiter
ligung ein diagnostischer molekulargentischer des LADR Fachbereichs Humangenetik gern zur
Test von jedem Arzt veranlasst werden. Eine Verfügung (T: 02361 30 00-201). Hier können auch
vorhergehende genetische Beratung – die bei Termine zur genetischen Beratung vereinbart
uns erfolgen kann – ist unbedingt zu empfehlen werden.
(siehe Seite 12: Wem sollte eine molekularpa- Für Patienten und Risikopersonen steht ein
thologische bzw. genetische Mutationsanalyse LADR Informationsblatt „Erblicher Darmkrebs“
angeboten werden?). zur Verfügung (Best.-Nr. 116477).
Eine prädiktive Testung bei einer bisher Die Analyse erfordert eine 4 ml EDTA-Probe,
nicht betroffenen Risikoperson darf nach dem die zusammen mit einer Einwilligungserklärung
GenDG nur nach einer genetischen Beratung an das LADR Laborzentrum Recklinghausen,
veranlasst werden. Die Beratung darf nur durch Abteilung Humangenetik, eingesandt werden
Fachärzte für Humangenetik, Ärzte mit der kann.
Zusatzbezeichnung Medizinische Genetik oder
der Qualifikation zur fachgebundenen geneti-
schen Beratung erfolgen.
Fachbereich Humangenetik
LADR Laborzentrum Recklinghausen PD. Dr. med. Bianca Miterski
LADR MVZ Dres. Bachg, Haselhorst & Kollegen Fachärztin für Humangenetik;
Recklinghausen GbR Ärztliche Leitung Humangenetik
Fachbereich Humangenetik
Berghäuser Straße 295 PD. Dr. rer. nat. Larissa Arning
45659 Recklinghausen Fachhumangenetikerin
T: 02361 30 00 - 201
F: 02361 30 00 - 211 Dr. rer. nat. Beatrix Böckmann
humangenetik@LADR.de Dipl. Biologin, Molekulargenetik
www.LADR.de
Dipl. Biologin Anne Purczeld
Zytogenetik
Prof. Dr. med. Klaus Zerres
Facharzt für Humangenetik
22Fachliteratur
1. Leitlinienprogramm Onkologie (Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF):
S3-Leitlinie Kolorektales Karzinom, Langversion 2.1, 2019, AWMF Registrierungsnummer:
021/007OL, http://www.leitlinienprogramm-onkologie.de/leitlinien/kolorektales-karzinom/
[abgerufen am: 16.09.2019]
2. Kohlmann W, Gruber SB: Lynch Syndrome. GeneReviews® [Internet]. Seattle (WA): University of
Washington, Seattle; 1993 – 2019. 2004 Feb 5 [updated 2018 Apr 12]
3. Jasperson KW, Patel SG and Ahnen DJ, APC-Associated Polyposis Conditions. GeneReviews®
[Internet]. Seattle (WA): University of Washington, Seattle; 1993 – 2019. 2004 Feb 5 [updated
2018 Apr 12]
4. A retz S. Differenzialdiagnostik und Früherkennung hereditärer gastrointestinaler Polyposis-
Syndrome. Deutsches Ärzteblatt 2010;107:163 –173
Alle Rechte – auch der auszugsweisen Wiedergabe – vorbehalten.
© LADR Der Laborverbund Dr. Kramer & Kollegen GbR 2020,
Bildrechte bei den jeweiligen Fotografen und Bildarchiven.
Die Autoren haben das Werk mit großer Sorgfalt und nach ihrem aktuellen Wissensstand zusam-
mengestellt. Da die Medizin sich ständig weiterentwickelt, sollten bei Verwendung in Diagnostik und
Therapie alle Angaben immer den jeweiligen Beipackzetteln und Fachinformationen der Hersteller
entnommen werden. Sollten Sie auf Unstimmigkeiten stoßen oder Rückfragen haben, kontaktieren
Sie uns bitte.
23Im LADR Laborverbund Dr. Kramer & Kollegen
werden Sie gerne beraten.
LADR Laborzentrum Hormonzentrum LADR Laborzentrum Partner des Labor
Baden-Baden Münster Nord-West, Schüttorf verbundes:
T: 07221 21 17-0 T: 0251 871 13-23 T: 05923 98 87-100 LIS Labor im Sommershof,
Zweigpraxis Leer Köln
LADR Laborzentrum LADR Laborzentrum T: 0491 454 59-0 T: 0221 93 55 56-0
Berlin an den Immanuel Kliniken,
T: 030 30 11 87-0 Hennigsdorf LADR Laborzentrum LADR Der Laborverbund
T: 03302 20 60-100 Paderborn Dr. Kramer & Kollegen GbR
LADR Laborzentrum Zweigpraxis Bernau, T: 05251 28 81 87-0 Lauenburger Straße 67
Braunschweig Zweigpraxis Rüdersdorf 21502 Geesthacht
Best.-Nr. 116687 Stand 01/2020
T: 0531 310 76-100 LADR Laborzentrum T: 04152 803-0
LADR Laborzentrum Recklinghausen F: 04152 803-369
LADR Laborzentrum Neuruppin T: 02361 30 00-0 interesse@LADR.de
Bremen T: 03391 35 01-0
T: 0421 43 07-300 LADR Zentrallabor Diese GbR dient aus-
LADR Laborzentrum Dr. Kramer & Kollegen, schließlich der Präsen
LADR Laborzentrum Nord, Flintbek Geesthacht tation des LADR Labor-
Hannover T: 04347 90 80-100 T: 04152 803-0 verbundes unabhängiger
T: 0511 901 36-0 LADR Einzelgesellschaften.Sie können auch lesen

















































