Der Calciumantagonist Amlodipin inhibiert die SDF-1α induzierte Migration von humanen CD4+-Zellen
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Medizinische Fakultät der Universität Ulm
Bereich: Innere Medizin II
Ärztlicher Direktor: Prof. Dr. Wolfgang Rottbauer
Der Calciumantagonist Amlodipin inhibiert die
SDF-1α induzierte Migration von humanen CD4+-Zellen
Dissertation
zur Erlangung des Doktorgrades der Zahnmedizin
der Medizinischen Fakultät
der Universität Ulm
vorgelegt von
Dominik Alexander Bedau
geboren in Ochsenhausen
2016
Amtierender Dekan: Prof. Dr. Thomas Wirth
1. Berichterstatter: Prof. Dr. Daniel Walcher
2. Berichtersatter: Prof. Dr. Lars Bullinger
Tag der Promotion: 14.12.2017
I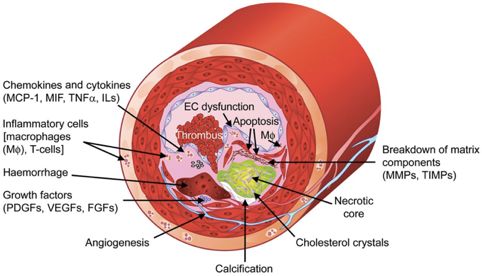
Meiner Familie gewidmet
II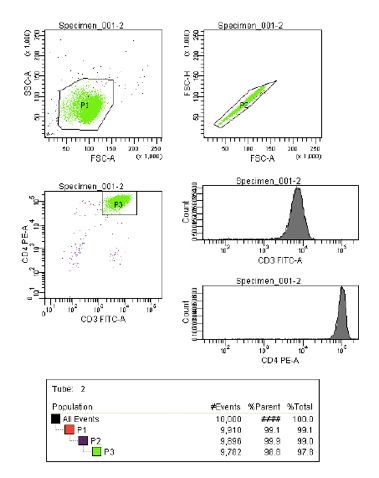
Inhaltsverzeichnis
Abkürzungsverzeichnis ............................................................................................................. V
1. Einleitung .............................................................................................................................. 1
1.1. Atherosklerose ................................................................................................................... 1
1.2. An der Atherogenese beteiligte Zelltypen und die Rolle der T-Zellen ............................. 4
1.3. Inflammation und Atherosklerose .................................................................................... 6
1.4. Medikamentöse Therapie bei Atherosklerose - ein Überblick ......................................... 8
1.5. Der Calciumantagonist Amlodipin................................................................................... 12
1.6. Zielsetzung dieser Arbeit und Fragestellungen............................................................... 14
2. Material und Methoden ..................................................................................................... 15
2.1. Chemikalien und Reagenzien .......................................................................................... 15
2.2. Lösungen und Puffer ....................................................................................................... 17
2.3. Antikörper ........................................................................................................................ 20
2.4. Geräte, Verbrauchsmaterialien und Computerprogramme........................................... 20
2.5. Isolation CD4-positiver Lymphozyten ............................................................................. 23
2.6. Migration CD4-positiver Lymphozyten ........................................................................... 26
2.7. PI3-Kinase Assay .............................................................................................................. 28
2.8. Western Blot .................................................................................................................... 31
3. Ergebnisse ........................................................................................................................... 35
3.1. Reinheit der isolierten CD4-positiven Lymphozyten (FACS-Messung) ........................... 35
3.2. Amlodipin inhibiert die Migration CD4-positiver Lymphozyten ..................................... 37
3.3. Amlodipin inhibiert konzentrationsabhängig die Aktivierung von p-SRC ...................... 39
3.4. Amlodipin inhibiert die PI-3-Kinase in humanen CD4-positiven Lymphozyten ............. 41
3.5. Amlodipin inhibiert konzentrationsabhängig die Aktivierung von p-AKT ...................... 43
3.6. Amlodipin inhibiert konzentrationsabhängig die Aktivierung von p-ERK ...................... 45
3.7. Amlodipin inhibiert konzentrationsabhängig die Aktivierung von p-MLC ..................... 47
4. Diskussion ........................................................................................................................... 49
4.1. Die Rolle der T-Zellen in der Atherosklerose .................................................................. 49
4.2. Bisherige Methoden zur Inhibition der Migration CD4-positiver Lymphozyten ............ 52
4.3. Der Calciumantagonist Amlodipin................................................................................... 54
III
5. Zusammenfassung .............................................................................................................. 61
6. Literaturverzeichnis ............................................................................................................ 63
Danksagung............................................................................................................................. 87
Lebenslauf ............................................................................................................................... 88
IVAbkürzungsverzeichnis
A Ampere
ACE Angiotensin-konvertierendes Enzym
ApoE Apolipoprotein E
APS Ammoniumpersulfat
Aqua bidest. zweifach destilliertes Wasser
ASS Acetylsalicylsäure
AT1 Angiotensin-II-Rezeptor-Subtyp-1
AV-Knoten Atrioventrikularknoten
BSA Bovines Serum Albumin
bzw. Beziehungsweise
C Celsius
ca. Circa
Ca2+ Calcium
CD Cluster of differentiation
cm Zentimeter
Co Kontrolle
CO2 Kohlenstoffdioxid
COX Cyclooxygenase
cPLA2 Cytosolic phospholipase A2
CRP C-reaktives Protein
CXCL Chemokine (C-X-C Motif) Ligand
CXCR3 CXC-Motiv-Chemokinrezeptor 3
DPBS Dulbecco’s Phosphate Buffered Saline
DMSO Dimethylsulfoxid
DNA Desoxyribonukleinsäure (deoxyribonucleic acid)
DR4 Direct-repeat-4
DTT Dithiothreitol
EC Endothelial cells
EDTA Ethylendiamintetraessigsäure
EGTA Ethyleneglycol-bis(aminoethylether)-tetraessigsäure
Vet al. Et alii (und andere)
FACS Fluorescence-activated cell sorting
FGF Fibroblast growth factor
FitC Fluorescin isothiocynanate
FSC Forward scatter
g Gramm
γ-ATP Gamma-Adenosintriphosphat
GAPDH Glycerinaldehyd-3-phosphat-Dehydrogenase
h Stunde
H2 O Dihydrogenmonoxid (Wasser)
HCl Chlorwasserstoff
HEPES 2-(4-(2-Hydroxyethyl)-1-piperazinyl)-ethansulfonsäure
HMG-CoA 3-Hydroxy-3-Methylglutaryl-Coenzym-A
HRP Horseradish peroxidase
huSerum Humanes Serum
HLA-DR Human leukocyte antigen - antigen D related
ICAM-1 Intercellular adhesion molecule 1
IgG Immunglobulin G
INF-γ Interferon-gamma
IL Interleukin
IP-10 Interferon gamma-induced protein 10
I-TAC Interferon-inducible t-cell alpha chemoattractant
l Liter
LDL Low density lipoprotein
LIMK LIM domain-containing kinases
LXR Liver-X-Rezeptor
M Molar
MACS Magnetic Cell Separation oder auch Magnetic Activated Cell Sorting
MAP-Kinase Mitogen-aktivierte Protein-Kinase
MCP-1 Monocyte chemoattractant protein-1
µg Mikrogramm
MgCl2 Magnesiumchlorid
VIMHC Major histocompatibility complex
MIF Migration inhibitory factor
MIG Monokine induced by gamma interferon
min Minute
Mio Millionen
ml Milliliter
µl Mikroliter
mm Millimeter
mM Millimolar
MMP Matrix-Metalloprotease
µM Mikromolar
MnCl2 Mangan(II)-chlorid
Mφ Makrophagen
n Fallzahl
NaCl Natriumchlorid
NaF Natriumfluorid
NaOH Natriumhydroxid
NaSO4 Natriumsulfat
ng Nanogramm
NO Stickstoffmonoxid
NP-40 Nonidet P-40 (Ethylphenyl-polyethylen glycol)
P Population
p p-Wert (Signifikanzwert)
p-AKT phospho-aktivierte Proteinkinase B
PDGF Platelet-derived growth factors
PDK1 3-phosphoinositide dependent protein kinase-1
PDK2 3-phosphoinositide dependent protein kinase-2
PE-A Phycoerythrin-A
p-ERK phospho-extracellular signal-regulated kinase
pH Potentia hydrogenii
PH-Domäne Pleckstrin-Homologie-Domäne
PI3-Kinase Phosphatidylinositol-3-Kinase
VIIPIP 1-Phosphatidylinositol-4-Phosphat
PIP3 1-Phosphatidylinositol-3,4,5-Trisphosphat
p-MLC phospho-Myosin light-chain
PMSF Phenylmethylsulfonylfluorid
PPAR Peroxisome proliferator-activated receptor
PS Penicillin/Streptomycin
p-SRC phospho-sarcoma/proto-oncogenic tyrosine Kinase
PvdF Polyvinylidenfluorid
RANTES Regulated upon activation normal t-cell expressed and secreted
rcf Relative centrifugal force
RNA Ribonukleinsäure (ribonucleic acid)
rpm Revolutions per minute
RPMI Roswell Park Memorial Institute
RT Raumtemperatur
RXR Retinsäure-X-Rezeptor
s Sekunde
SDF-1α Stromal cell derived factor 1alpha
SDS Sodiumdodecylsulfat
SH-Domäne Src-homology-Domäne
SOV Sodiumorthovanadat (Na3VO4)
SSC Side scatter
TBS Tris-gepufferte Saline
TbsT Tris-gepufferte Saline mit 0,05% Tween 20
TCFA Thin-Cap-Fibro-Atherome
TEMED N,N,N’,N'-Tetramethylethylendiamin
TGF-β Transformationswachstumsfaktor-beta
TH-0 Typ0-T-Helferzellen
TH-1 Typ1-T-Helferzellen
TH-2 Typ2-T-Helferzellen
TIMP Tissue inhibitors of metalloproteinases
TLC Thin layer chromatography
TNF-α Tumornekrosefaktor-alpha
VIIITregs Regulatorische T-Zellen
TRIZMA (TRIS) Tris(hydroxymethyl)-aminomethan
TWEEN 20 Polyoxyethylen(20)-sorbitan-monolaurat
V Volt
VCAM-1 Vascular cell adhesion protein 1 oder auch
Vascular cell adhesion molecule 1
VEGF Vascular endothelial growth factor
WHO World Health Organization
z. B. zum Beispiel
IX1. Einleitung
1.1. Atherosklerose
Die Atherosklerose gilt als die häufigste Systemerkrankung der Gefäßwände, typischerweise
der Intima und Media der kleinen bis mittelgroßen Arterien [144]. Charakterisiert wird sie
durch endotheliale Dysfunktion, Entzündung und Proliferation glatter Gefäßmuskelzellen
sowie Lipoproteineinlagerung und Schaumzellbildung [138]. Durch die WHO wird sie
darüber hinaus als eine Kombination von morphologischen Veränderungen der
Arterienintima und der herdförmigen Anhäufung von Lipiden, komplexen Kohlenhydraten,
Blut sowie Blutbestandteilen, der Bildung fibrösen Gewebes und Kalkablagerungen definiert
[171]. Die Atherogenese ist ein meist langsam fortschreitender, chronischer
Entzündungsprozess in der Gefäßwand mit Entzündungszellen wie Monozyten,
Makrophagen und CD4-positiven Lymphozyten [47, 178].
Die Ursachen der Atherosklerose sind vielseitig und bei der Entstehung handelt es sich um
einen oftmals über Jahrzehnte andauernden Prozess, dessen Verlauf auch von einer Reihe
möglicher Risikofaktoren beschleunigt werden kann [10]. Zu diesen gehören erhöhter
Blutdruck, Diabetes mellitus, freie Radikale durch Nikotinabusus, Störungen des
Fettstoffwechsels, mangelnde Bewegung, psychosozialer Stress und Adipositas [105,
137]. Weitere Faktoren sind neben dem männlichen Geschlecht, fortgeschrittenem Alter,
und genetischen Voraussetzungen auch eine bestehende Parodontitis, deren spezifische
Erreger in pathologisch veränderten Gefäßwänden nachgewiesen werden konnten [1, 10,
116].
Die Atherosklerose und die mit ihr verknüpfte atherosklerotische Intimaverkalkung führen
mit ihren Folgen wie Herz-Kreislauf-Erkrankungen, Apoplex oder peripheren arteriellen
Verschlusskrankheiten die Rangliste der häufigsten Todesursachen in den westlichen
Industriestaaten an [23, 89, 117]. Alleine in Deutschland sind laut Berechnungen des
Robert-Koch-Institutes derzeit ca. 45 % aller Todesfälle auf sie zurückzuführen, weltweit sind
es 16,7 Millionen Todesfälle pro Jahr [21, 90, 136].
1Es wird prognostiziert, dass die Atherosklerose im Jahr 2020 die häufigste Todesursache
weltweit sein wird [144].
Die Studien von Ross et al. führten zu der Erkenntnis, dass die Atherosklerose eine
chronische Entzündung der Gefäßwand ist [138]. Ursächlich hierfür sind Interaktionen
zwischen oxidativ veränderten Lipoproteinen mit Makrophagen, T-Zellen und weiteren
zellulären Bestandteilen wie z. B. glatten Muskelzellen. Dieser Prozess führt schließlich zur
Ausbildung von atherosklerotischen Läsionen und Plaques, die die Blutgefäße einengen.
Diese Plaques sind aus einer fibrösen Deckplatte und einem Lipidkern aufgebaut, der vor
allem aus Entzündungszellen und Cholesterinester besteht. Die primär stabilen Plaques
können zu einer Verringerung des Blutstroms führen, allerdings ist die Gefahr eines Infarkts
zunächst recht gering [24]. Im Gegensatz dazu gibt es aber auch sogenannte Thin-Cap-
Fibro-Atherome mit einer dünneren, fibrösen Deckplatte und einem größeren, weichen und
teils nekrotischen Lipidkern, der eine große Anzahl Entzündungszellen enthält. Die
Deckplatten sind dabei in der Regel dünner als 65 µm [65].
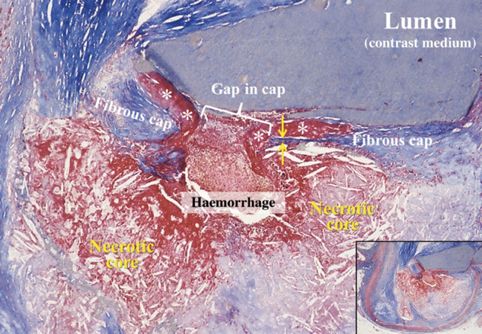
2Abbildung 1: Rupturierte Plaque in einem Herzkranzgefäß.
Die Abbildung zeigt einen Querschnitt der Herzkranzgefäße. Es ist die Ruptur eines Thin-Cap-Fibro-Atherome
zu sehen. Die fibröse Deckplatte in der Nähe der Bruchstelle ist sehr dünn (zwischen den Pfeilen) und ein
Thrombus (Sternchen) grenzt an die Lücke zu der gerissenen Deckplatte. Eine Blutung aus dem Lumen ist durch
den Spalt in den lipidreichen, nekrotischen Kern, in dem die charakteristischen Cholesterinspalten ersichtlich
sind, eingedrungen. Das Lumen enthält Kontrastmittel, welches postmortal injiziert wurde [175].
Ylä-Herttuala, Seppo; Bentzon, Jacob Fog, Stabilization of atherosclerotic plaques: an update, European Heart
Journal, 2013, volume 34, issue 42, 3251-3258, by permission of Oxford University Press.
Die Ruptur eines solchen Thin-Cap-Fibro-Atherome kann letztendlich einen Thrombus
auslösen, welcher zum Verschluss eines gesamten Gefäßes führen kann. Daraufhin kommt
es zu einer akuten Ischämie des distal liegenden Gewebes und somit z. B. zu einem
Myokardinfarkt oder einem Apoplex [81]. Die Mehrheit (ca. 75 %) der koronaren Thromben
werden durch eine Plaqueruptur ausgelöst [174].
In Abbildung 2 sind mögliche Faktoren zusammengefasst, die zur Bildung einer instabilen
Plaque beitragen können.
3Abbildung 2: Aufbau einer atherosklerotischen Läsion.
Die Abbildung zeigt den typischen Aufbau einer atherosklerotischen Läsion und mögliche Faktoren, die zur
Bildung von Plaques beitragen können [175].
Ylä-Herttuala, Seppo; Bentzon, Jacob Fog, Stabilization of atherosclerotic plaques: an update, European Heart
Journal, 2013, volume 34, issue 42, 3251-3258, by permission of Oxford University Press.
Abkürzungen:
EC (Endothelial cell), FGFs (Fibroblast growth factors), ILs (Interleukine), Mφ (Makrophagen), MCP-1
(Monozyten-chemotaktisches Protein-1), MIF (Migration inhibitory Factor), MMPs
(Matrixmetalloproteinasen), PDGFs (Platelet-derived growth factors), TIMPs (Tissue inhibitors of
metalloproteinases), TNFα (Tumornekrosefaktor-α), VEGF (Vascular endothelial growth factors)
1.2. An der Atherogenese beteiligte Zelltypen und die Rolle der T-Zellen
Instabile Plaques enthalten Monozyten, Makrophagen und T-Zellen. Die wichtigsten
Vertreter der T-Zellen sind dabei die CD4-positiven T-Lymphozyten [49].
In atherosklerotischen Läsionen kommen vorwiegend zwei Makrophagenphänotypen vor:
Einerseits der proinflammatorische Typ M1 Makrophage (IFN-γ-induziert) und andererseits
der entzündungshemmende oder regulatorische Typ M2 Makrophage (IL-4- und
IL-13-induziert) [98].
4T-Zellen mit Helferfunktion sezernieren unterschiedliche Zytokine und können danach
eingeteilt werden, ob diese Botenstoffe an der zellvermittelten Immunantwort beteiligt sind
oder ob die humorale Immunantwort der B-Lymphozyten stimuliert wird. Die Einteilung
erfolgt dabei in einen TH1- oder einen TH2-Phänotyp. So induziert die Anwesenheit von
IL-12 und IFN-γ die Differenzierung zur TH1-Zelle, während IL-4, IL-10 und IL-13 eine
Differenzierung zur TH2-Zelle fördern. Beispielsweise gehören unter den CD4-positiven
Lymphozyten solche zur ersten Gruppe (Typ 1), die IFN-γ, IL-2 und TNF-α sezernieren.
CD4-positive Lymphozyten, die die Zytokine IL-4, IL-5, IL-6, IL-10 und IL-13 erzeugen, werden
dem Typ 2 zugerechnet [114, 175].
T-Zellen fördern die Anfälligkeit der Plaques durch ihre Wirkung auf Makrophagen.
Die Migration CD4-positiver Lymphozyten in die Gefäßwand mit anschließender Aktivierung
anderer Gefäßzellen stellt einen wichtigen Schritt in der frühen Atherogenese dar [47]. Dies
geschieht durch die Aktivierung der T-Lymphozyten und deren anschließende Bildung
proinflammatorischer Zytokine wie IFN-γ, TNF-α und IL-2 [31, 102, 180].
Tatsächlich zeigen Patienten mit Atherosklerose und akuten Koronarsyndromen eine
erhöhte T-Zell-Aktivierung und einen erhöhten IFN-γ Spiegel im Serum [88, 119].
Die CD4-positiven Lymphozyten werden durch chemotaktische Proteine wie RANTES und
SDF-1α angezogen und betreten die Gefäßwand als TH0-Zellen. Im Subendothel treffen sie
dann auf Antigene, wie beispielsweise oxidiertes LDL, wodurch es zu einer Differenzierung
zu TH1-Zellen mit anschließender Freisetzung der oben genannten proinflammatorischen
Mediatoren kommt [46]. Diese Zytokine bewirken daraufhin die entzündliche Reaktion in
der Gefäßwand durch Aktivierung und Modulation anderer Zellen wie Endothelzellen,
Makrophagen und glatter Gefäßmuskelzellen [46, 78]. Damit fördern sie den entzündlichen
Prozess der Atherogenese und tragen zur Bildung von Plaque bei [83].
Die zelluläre Zusammensetzung atherosklerotischer Plaques wurde von Hansson et al. durch
Immunzytochemie unter Verwendung von zelltypspezifischen, monoklonalen Antikörpern
analysiert. T-Lymphozyten aber auch Monozyten und Makrophagen wurden sowohl in
frühen, fibrösen Plaques als auch in fortgeschrittenen, komplizierten Plaques nachgewiesen.
Darüber hinaus konnte in glatten Muskelzellen dieser Plaques das
5Klasse-II-MHC-Antigen HLA-DR nachgewiesen werden. Da dieses Antigen durch
T-Zell-Produkte induziert wird, legen die Ergebnisse nahe, dass Interaktionen zwischen
T-Zellen und der glatten Muskulatur während der Atherogenese auftreten [45].
1.3. Inflammation und Atherosklerose
Im derzeitigen pathophysiologischen Verständnis der Atherosklerose spielt der Begriff der
Entzündung eine zentrale Rolle und stellt eine gemeinsame Verbindung zwischen
Risikofaktoren und den zellulären und molekularen Veränderungen dar [85, 86]. Bei diesem
Konzept wird die Atherosklerose als eine von Lipiden angetriebene entzündliche Erkrankung
gesehen, die durch die Ansammlung von Makrophagen und Schaumzellen in der
Arterienwand charakterisiert ist und von einer Kaskade proinflammatorischer Zytokine und
Chemokine begleitet wird. Die vaskuläre Entzündung führt zur Initiation, Progression und
schlussendlich zu den Komplikationen von atherosklerotischen Läsionen [85, 135].
Aktuelle klinische Strategien gegen Atherosklerose konzentrieren sich meist noch auf die
Reduzierung von Risikofaktoren wie Bluthochdruck, Hyperlipidämie oder auf die
Vermeidung von thromboembolischen Komplikationen und nicht direkt auf die Bekämpfung
der Entzündungsmechanismen bei der Progression einer Atherogenese [169]. Mit
zunehmender Anerkennung der Rolle der Entzündung bei der Atherosklerose sind nun aber
auch entzündungshemmende Strategien bei der Behandlung in den Fokus gerückt und
bieten neue Therapiemöglichkeiten [47, 49, 169].
Akute Inflammationsreaktion
Inflammation wird als eine sehr komplexe Reaktion des angeborenen Immunsystems in
vaskularisierten Geweben beschrieben. Sie bewirkt eine Anhäufung und Aktivierung von
Plasmaproteinen und Leukozyten. Bei einer Entzündungsreaktion kommt es neben den
gewünschten protektiven und inflammatorischen Effekten, die Infektionen bekämpfen und
Gewebe reparieren, auch zu gewebeschädigenden, destruktiven Veränderungen [141].
Die Hauptproduzenten der proinflammatorischen Zytokine wie z. B. TNF-α, IL-1, IL-6 und
6IL-8 sind dabei Makrophagen und Monozyten. Der menschliche Organismus reagiert im
Ablauf einer akuten Inflammationsreaktion je nach Intensität und Art des Auslösers mit
Fieber, Leukozytose und Stress. Zusätzlich kommt es zu einer veränderten
Zusammensetzung der Plasmaproteine. Vor allem das IL-6 stimuliert dabei in den
Hepatozyten die Produktion von Proteinen wie dem CRP, Haptoglobin, Serumamyloid A,
Tranferrin, Fibrinogen und Albumin [33, 71].
Chronische Mikroinflammation (Metainflammation)
Die akute Entzündung stellt mit all ihren Reaktionen einen überlebensnotwendigen Teil der
Immunantwort dar. Demgegenüber steht die chronische Entzündung mit ihren negativen
Begleiterscheinungen wie der dauerhaften Gewebeschädigung, dem Auslösen von
Autoimmunkrankheiten, kardiovaskulären oder auch neurodegenerativen Krankheiten. Bei
der chronischen Entzündungsreaktion sind T-Lymphozyten, Endothel- und glatte
Muskelzellen, Lipoproteine, Monozyten sowie die extrazelluläre Matrix der Arterienwand
involviert [115].
Als Folge der chronischen Entzündung sind deshalb zahlreiche Mediatoren der
inflammatorischen Immunantwort exprimiert und können in erhöhten Plasmaaktivitäten
bzw. -konzentrationen gemessen werden. Dazu zählen z. B. Adhäsionsmoleküle oder
Zytokine, welche sich somit zur Beurteilung des Entzündungsstatus und kardiovaskulärer
Ereignisse eignen [4].
Der Übergang von einer akuten in eine chronische Entzündungsreaktion ist gekennzeichnet
durch eine Verschiebung der rekrutierten granulozytären Zellen zugunsten der Monozyten.
Auch hier nimmt das IL-6 eine Schlüsselrolle ein. Ein Rezeptor wird von Neutrophilen
Granulozyten abgesondert, welcher freies IL-6 bindet. Dies führt zur Transkription von
MCP-1 und einer erhöhten Produktion von IL-6 im Endothel. MCP-1 ist ein Signalprotein,
welches zur Chemotaxis von Monozyten führt [71].
71.4. Medikamentöse Therapie bei Atherosklerose - ein Überblick
Statine
Ein zu hoher Cholesterinspiegel gilt als ein wichtiger Risikofaktor für koronare
Herzerkrankungen. Daher reduziert eine Senkung des Cholesterinspiegels das Risiko von
Herzinfarkt, Schlaganfall und allen Formen atherosklerotischer Gefäßerkrankungen [144].
Die HMG-CoA-Reduktase-Inhibitoren, die auch als Statine bekannt sind, sind die wirksamste
Klasse von Arzneimitteln zur Senkung der Serumkonzentrationen von LDL und Cholesterin
[148]. Statine spielen nachgewiesenermaßen eine wichtige Rolle bei der Verringerung der
Rate der kardiovaskulären Morbidität und Mortalität und sie senken das Risiko für ein
kardiovaskuläres Ereignis bei allen Patientengruppen um etwa 30 % [37, 38]. Seit ihrer
Einführung ging die Inzidenz von Myokardinfarkten um rund ein Drittel zurück [110].
Die Wirkung der Statine beruht auf der kompetitiven Hemmung des Enzyms HMG-CoA-
Reduktase in der Leber. Da HMG-CoA vom Körper zur Synthese von Cholesterin benötigt
wird, hemmen Statine dadurch die Cholesterinsynthese spezifisch [76]. Dies hat einen
Mangel an Cholesterin in den Zellen zur Folge, es kommt zu einer verstärkten Expression
von LDL-Rezeptoren und damit zu einer vermehrten Aufnahme von LDL aus dem Blut durch
Endozytose. Der LDL-Serumspiegel und die Cholesterinmenge werden somit minimiert und
damit auch ihre negativen Auswirkungen auf atherosklerotische Läsionen [67, 148].
Statine führen dazu, dass der große, lipidreiche Kern in instabilen Plaques in seiner Größe
reduziert und der Kollagengehalt erhöht wird. Dies bewirkt eine Stabilisierung der Plaques
und eine Abnahme der Entzündungsreaktion. Dabei scheint oxidiertes LDL einen wichtigen
Mediator darzustellen, dessen Einlagerung von den Statinen verhindert wird. LDL lockt
Monozyten und Lymphozyten an, aktiviert diese und stimuliert somit die Bildung von
Metalloproteasen. Diese Enzyme bauen vor allem das Strukturprotein Kollagen ab, welches
den Plaques eine gewisse mechanische Festigkeit verleiht [79, 152]. Das so vermehrt
vorhandene Kollagen führt also zu einer Beruhigung der Plaques, allerdings geht die Dicke
der Ablagerungen und somit die Einengung des Gefäßes nicht oder nur sehr wenig zurück
[16, 80]. Darüber hinaus führen Statine zu einer Verbesserung der Funktion des Endothels.
Dies beruht auf einer Erhöhung der in atherosklerotisch veränderten Gefäßen verminderten
Expression der NO-Synthase und führt zu einer Abnahme der pathologischen
Vasokonstriktion, einer Verringerung der Thrombogenizität und letztendlich zu einem
8geringeren Risiko der Ruptur einer Plaque [16, 17].
ACE-Hemmer
ACE-Hemmer werden in erster Linie zur Therapie des arteriellen Bluthochdrucks eingesetzt
und gehören dabei sowohl als Monotherapie als auch in Kombination mit anderen
blutdrucksenkenden Medikamenten zu den am häufigsten verordneten
blutdrucksenkenden Medikamenten [106]. Es konnte bewiesen werden, dass ACE-Hemmer
die Überlebensrate sowie die Symptome von Patienten mit Herzinsuffizienz signifikant
verbessern konnten [153]. Die Wirkung der ACE-Hemmer beruht auf der Hemmung des
Angiotensin-konvertierenden Enzyms, welches für die Synthese des gefäßverengend
wirkenden Angiotensin-II aus seiner Vorstufe, dem Angiotensin-I, essentiell ist. Dadurch
bewirkt es eine Senkung des Blutgefäßtonus und führt zu einer Abnahme des arteriellen
Blutdrucks [42, 132].
Darüber hinaus reduzieren ACE-Hemmer den Abbau des Kinins Bradykinin [177]. Bradykinin
wirkt über seine Bindung an ß2-Rezeptoren, vermittelt darüber seine vasodilatatorische
Wirkung und sorgt für antiproliferative Effekte [43, 87]. Die ansteigende Konzentration des
Bradykinin hat außerdem eine vermehrte Freisetzung von Stickoxid, Prostaglandin E2 und
Prostazyklin I2 zur Folge, welche eine zusätzliche Vasodilatation vermitteln. Zudem nehmen
die Prostaglandine positiven Einfluss auf die Mitogenese und die Proliferation glatter
Gefäßmuskelzellen [172].
Betablocker
Auch Betablocker spielen eine wichtige Rolle in der Behandlung von kardiovaskulären
Erkrankungen [91].
Zu ihnen gehören eine Reihe ähnlich wirkender Medikamente, die eine Blockade der
β-Adrenozeptoren vermitteln. Damit hemmen sie die aktivierende Wirkung von Adrenalin
und Noradrenalin auf diese Rezeptoren und reduzieren die negativen Effekte einer
Überaktivität des sympathischen Nervensystems [8, 164]. Dazu gehört die Senkung der
Herzfrequenz in Ruhe und des Blutdrucks [14]. Der Wirkmechanismus beruht dabei darauf,
dass Betablocker in ihrer molekularen Struktur diesen aktivierenden, neuroendokrinen
9Botenstoffen ähneln. Sie besetzen die Bindungsstelle an den Rezeptoren und wirken somit
als kompetitiver Antagonist [14, 91].
Betablocker tragen deutlich zur Verbesserung der Symptome der Herzinsuffizienz bei und
verringern das Risiko für kardiovaskuläre Ereignisse [74]. Die Gabe von Betablockern
minimiert das Risiko an Herzinsuffizienz oder an einem plötzlichen Herztod zu versterben
und führt zu einer Verbesserung der Hospitalisierungsrate [153].
Weitere Vorteile von Betablockern bei Patienten mit akutem Myokardinfarkt sind ihre anti-
ischämischen, antihypertensiven, antiarrhythmischen und antithrombotischen Effekte [91].
Acetylsalicylsäure
Bereits 1899 wurden die antipyretischen, analgetischen und auch die antiinflamma-
torischen Eigenschaften der Acetylsalicylsäure entdeckt [25]. Mitte des 20. Jahrhunderts
wurde erstmals die hemmende Wirkung durch ASS auf die Thrombozytenaggregation
beschrieben, die bis heute eine breite Anwendung bei der Vorbeugung des
Myokardinfarktes, Apoplex und weiterer Formen von pathologischen Gefäßveränderungen
thromboembolischen Ursprungs findet [18].
Ranke et al. konnten in einer klinischen Studie zeigen, dass ASS die Progression
atherosklerotischer Veränderungen verlangsamt [131]. In verschiedenen Studien wurde
beschrieben, dass ASS Modifikationen von LDL und Fibrinogen verhindern kann, die zur
Entstehung einer Atherosklerose beitragen [151, 159, 165]. Des weiteren verbessert ASS die
Acetylcholin-induzierte Vasodilatation, die als Maß für die Funktion des Endothels gilt,
sowohl bei Patienten mit Atherosklerose als auch bei Patienten mit Bluthochdruck [55, 111].
Die Funktionsweise von ASS beruht auf der Hemmung der Prostaglandinsynthese [161].
Dies geschieht durch die irreversible Hemmung der Prostaglandin-H2-Synthase, genauer der
beiden Enzyme COX-1 und COX-2. Diese katalysieren die Synthese von inflammatorisch
wirkenden Prostaglandinen sowie z. B. des in Thrombozyten vorkommenden Thromboxan
A2, welches thrombozytenaktivierend wirkt. ASS überträgt einen Acetylrest auf einen
Aminosäurerest, welcher sich kurz vor dem katalytischen Zentrum befindet. Als Folge dessen
kann die Arachidonsäure als Substrat des Enzyms das aktive Zentrum nicht mehr erreichen
und das Enzym wird dauerhaft inaktiviert [66]. Da es sich um eine irreversible Acetylierung
handelt, Thrombozyten keinen Zellkern besitzen und somit keine Enzyme nachproduzieren
10können, ist die gerinnungshemmende Wirkungsdauer dieselbe wie die Überlebenszeit der
Thrombozyten von etwa 7 bis 11 Tagen [139]. ASS hemmt zwar darüber hinaus auch die
Prostacyclinsynthese, allerdings wird der antithrombotische Effekt der Substanz dadurch
nicht merklich beeinflusst, da Prostacyclin in den kernhaltigen Endothelzellen schnell
nachproduziert werden kann. Daher kommt es durch die Gabe von ASS in erster Linie zur
Thrombozytenaggregationshemmung [134].
Calciumantagonisten
Calciumionen spielen eine große Rolle bei der kardiovaskulären Funktion. Äußere Reize
führen zu einer Öffnung von Calciumkanälen, was den Einstrom von Calciumionen in die
Zelle bewirkt. Das veränderte Membranpotential der Muskelzelle bewirkt eine
Muskelkontraktion, welche zu einer Vasokontriktion und somit zu einem erhöhten Druck in
Gefäßen führt [142]. Somit spielt eine Änderung der Calcium-Homöostase eine
Schlüsselrolle für mögliche pharmakologische Interventionen. Dabei werden
Calciumantagonisten oder im engeren Sinne Calciumkanalblocker in erster Linie zur
Therapie von Hypertonie, koronarer Herzkrankheit und Herzrhythmusstörungen eingesetzt
[123]. Sie hemmen spannungsabhängige L-Typ-Calciumkanäle, blockieren damit den
Calciumeinstrom ins Innere der Muskelzellen und sorgen so für eine Abnahme der
intrazellulären Calciumkonzentration [123]. Dies führt zu einer Entspannung der glatten
Muskulatur mit Verringerung des peripheren Widerstandes und somit zu einer Senkung des
Blutdruckes. Darüber hinaus sorgen sie für negative inotrope und chronotrope Effekte und
haben eine antiproliferative Wirkung [142].
Die Calciumantagonisten werden entsprechend ihrer spezifischen Bindungsstellen an den
Calciumkanälen in drei Typen eingeteilt [27, 34]:
1. Dihydropyridine (Nifedipin-Typ)
2. Phenylalkylamine (Verapamil-Typ)
3. Benzothiazepine (Diltiazem-Typ)
Sie unterscheiden sich zudem in ihrer molekularen Struktur, ihrem Ort und ihrer Art der
Wirkungsweise und in ihren Effekten auf verschiedene andere Herz-Kreislauf-Funktionen.
11Alle drei haben gemeinsam, dass sie den Blutdruck senken, wobei dies auf unterschiedliche
Art und Weise geschieht: bei den Dihydropyridinen durch eine Vasodilatation, bei den
Phenylalkylaminen durch eine Verlangsamung und gleichzeitige Abschwächung des
Herzschlags, bei den Benzothiazepinen durch eine Kombination aus beidem [52, 125].
Calciumantagonisten, insbesondere die neueren, länger wirkenden Vertreter, sind eindeutig
wirksam gegen Bluthochdruck und werden daher intensiv genutzt [121]. Klinische
Langzeitstudien über Calciumantagonisten konnten außerdem nachweisen, dass diese
Medikamente das Fortschreiten der Atherosklerose bei Patienten mit Hypertonie und
koronarer Herzkrankheit reduzieren konnten [52].
1.5. Der Calciumantagonist Amlodipin
Bereits 1988, als Amlodipin noch in der klinischen Entwicklungsphase steckte, stellten Reid
et al. fest, dass es ein völlig anderes pharmakokinetisches Profil als die bisher bekannten
Calciumantagonisten besitzt. Es ist wasserlöslich, photostabil und hat eine lange
Halbwertszeit von 35 bis 50 Stunden. Amlodipin wird langsam absorbiert, seine absolute
Bioverfügbarkeit ist hoch und es wird weitgehend in der Leber metabolisiert. Die lange
Halbwertszeit ist mit einer längeren (mehr als 24 Stunden) Dauer der
pharmakodynamischen Wirkung verbunden und kann somit praktische Vorteile gegenüber
bestehenden Calciumantagonisten bei der langfristigen Behandlung von kardiovaskulären
Erkrankungen bieten [133].
Als blutdrucksenkendes Medikament ist Amlodipin einer der am häufigsten verwendeten
1,4-Dihydropyridin-Calciumkanal-Antagonisten [75]. Es hemmt den Einstrom von Calcium-
ionen durch L-Typ-Calciumkanäle, die hauptsächlich in vaskulären glatten Muskelzellen
vorkommen, was zu einer verringerten Kontraktilität der vaskulären glatten Muskelzellen
und zu einer Vasodilatation führt [73]. Als eine der häufigsten Herz-Kreislauf-Erkrankungen
ist Bluthochdruck ein signifikanter Risikofaktor für verschiedene Herz-Kreislauf- und
zerebrovaskuläre Erkrankungen. Hierzu zählen zum Beispiel Atherosklerose, Schlaganfall,
12Herzinfarkt oder Herzinsuffizienz [126]. In einigen Studien konnte gezeigt werden, dass
Bluthochdruck regulierbar und gut einstellbar ist [72, 77, 113]. Die Verringerung des
Blutdrucks bei hypertensiven Patienten verringert das Auftreten von Schlaganfällen und
Herzkrankheiten und erhöht deren Lebensdauer [126].
Darüber hinaus bietet Amlodipin als eine neue Art von lang wirkenden Calciumkanalblockern
in der Kategorie der Dihydropyridine mehrere Vorteile. Dazu gehören die rasche und
verbesserte Wirkung, weniger Nebenwirkungen und langfristige antiatherosklerotische
Effekte. Es besitzt jedoch keine Wirkung auf den Blutzucker- und den Lipidstoffwechsel und
ist somit zwischenzeitlich für die Behandlung von Bluthochdruck weit verbreitet [39].
Zahlreiche internationale, multizentrische und klinische Studien haben gezeigt, dass die
Gabe von Amlodipin als Monotherapie bei der Behandlung von leichter bis mäßiger primärer
Hypertonie einen Wirkungsgrad von bis zu 70 % erreichte [28, 128]. Fogari et al. zeigten,
dass die effektive therapeutische Rate 60 % betrug [28]. Neben der blutdrucksenkenden
Wirkung wurden auch eine entzündungshemmende Wirkung bei der Atherosklerose und
eine Verringerung der Größe atherosklerotischer Läsionen nachgewiesen [146].
131.6. Zielsetzung dieser Arbeit und Fragestellungen
Die entscheidende Rolle der CD4-positiven T-Lymphozyten bei Entstehung und Progression
der Atherosklerose und die Annahme, dass der Calciumantagonist Amlodipin auch
antiinflammatorische Wirkungen besitzt, führen zu folgenden Fragen:
Inhibiert Amlodipin die SDF-1α induzierte Migration humaner CD4-positiver Lymphozyten,
wirkt Amlodipin antiinflammatorisch und welche Signalkaskademoleküle sind an diesen
Vorgängen beteiligt?
Im Einzelnen wurden in der vorliegenden Arbeit folgende Fragestellungen bearbeitet:
1. Welchen Effekt hat Amlodipin auf das Migrationsverhalten CD4-positiver
Lymphozyten?
2. Hat Amlodipin einen Einfluss auf die Vitalität der CD4-positiven Lymphozyten?
3. Welchen Effekt hat Amlodipin auf die PI3-Kinase in CD4-positiven Lymphozyten?
4. Welche Wirkung hat Amlodipin auf die Phosphorylierung von AKT, ERK, SRC und
MLC?
142. Material und Methoden
2.1. Chemikalien und Reagenzien
Acrylamide (30 %) Bio-Rad Laboratories, Hercules, USA
Amlodipine besylate Sigma-Aldrich, St. Louis, USA
Ammoniak Lösung 25 % Merck, Darmstadt, Deutschland
α-Phosphatidylinositol Sigma-Aldrich, St. Louis, USA
APS Sigma-Aldrich, St. Louis, USA
Ampuwa Spüllösung, Plastipur Fresenius Kabi, Bad Homburg, Deutschland
BenchMark Pre-Stained Protein Ladder Life Technologies, Carlsbad, USA
β-Glycerophosphat Sigma-Aldrich, St. Louis, USA
β-Mercaptoethanol Sigma-Aldrich, St. Louis, USA
Bromphenolblau Serva Electrophoresis, Heidelberg, Deutschland
BSA Fraction V, ph 7,0 GE Healthcare, Chalfont St Giles, England
Chloroform Sigma-Aldrich, St. Louis, USA
Collagen from calf skin Sigma-Aldrich, St. Louis, USA
Diff-Quik I und II Medion Diagnostics, Düdingen, Schweiz
DPBS 1-fach Life Technologies, Carlsbad, USA
DTT Sigma-Aldrich, St. Louis, USA
EDTA, UltraPure 0,5 M, pH 8,0 Life Technologies, Carlsbad, USA
EGTA Sigma-Aldrich, St. Louis, USA
Essigsäure Sigma-Aldrich, St. Louis, USA
Eukitt O. Kindler, Freiburg, Deutschland
γ-ATP Hartmann Analytics, Braunschweig, Deutschland
Glycerin Carl Roth, Karlsruhe, Deutschland
Glycine Sigma-Aldrich, St. Louis, USA
HCl Merck, Darmstadt, Deutschland
HEPES Sigma-Aldrich, St. Louis, USA
Histopaque-1077 Sigma-Aldrich, St. Louis, USA
Human SDF-1-α PeproTech, Hamburg, Deutschland
15huSerum „off the clot“ Type AB PAA, Cambridge, England
Hybri-Max DMSO Sigma-Aldrich, St. Louis, USA
MACS autoMACS Running Buffer Miltenyi Biotec, Bergisch Gladbach, Deutschland
MACS CD4+ T Cell Isolation Kit Miltenyi Biotec, Bergisch Gladbach, Deutschland
Methanol Sigma-Aldrich, St. Louis, USA
Micro BCA Reagent A (MA) Thermo Fisher Scientific, Waltham, USA
Micro BCA Reagent B (MB) Thermo Fisher Scientific, Waltham, USA
Micro BCA Reagent C (MC) Thermo Fisher Scientific, Waltham, USA
NaCl Sigma-Aldrich, St. Louis, USA
NaOH (steril) Sigma-Aldrich, St. Louis, USA
NP-40 USB, Cleveland, USA
p85α Antikörper Santa Cruz Biotechnology, Dallas, USA
Penicillin/Streptomycin Biochrom, Berlin, Deutschland
Protease inhibitors Sigma-Aldrich, St. Louis, USA
Protein A/G PLUS-Agarose Beads Santa Cruz Biotechnology, Dallas, USA
Protein Essay Dye Regent Concentrate Bio-Rad Laboratories, Hercules, USA
(„Bradford Lösung“)
Restore Western Blot Stripping Buffer Thermo Fisher Scientific, Waltham, USA
Roentoroll HC Entwickler Tetenal, Norderstedt, Deutschland
RPMI 1640 with L-Glutamine Life Technologies, Carlsbad, USA
SDS Carl Roth, Karlsruhe, Deutschland
Skim Milk Powder Sigma-Aldrich, St. Louis, USA
SOV Sigma-Aldrich, St. Louis, USA
Superfix MRP Fixierer Tetenal, Norderstedt, Deutschland
Super Signal West Pico Stable Thermo Fisher Scientific Waltham, USA
Peroxide Solution
Super Signal West Pico Luminol/ Thermo Fisher Scientific Waltham, USA
Enhancer Solution
Super Signal West Femto Stable Thermo Fisher Scientific Waltham, USA
Peroxide Solution
Super Signal West Femto Luminol/ Thermo Fisher Scientific Waltham, USA
Enhancer Solution
16TEMED Sigma-Aldrich, St. Louis, USA
TRIZMA (TRIS Base) Sigma-Aldrich, St. Louis, USA
Trypan Blue Solution (0,5 %) Serva Electrophoresis, Heidelberg, Deutschland
TWEEN 20 Merck, Darmstadt, Deutschland
2.2. Lösungen und Puffer
Amlodipin (10 mM) 10 mg Amlodipine besylate
5 ml DMSO
APS (10 %) 1 g APS
10 ml Aqua bidest.
Collagen Lösung 50 µl Collagen Stock
50 µl 1M NaOH (steril)
9,9 ml DPBS Collagen Stock
10 mg Collagen from calf skin
3,33 ml 0,5 M Essigsäure (steril)
LIMK-Lysepuffer 2,5 ml 7,4 mM Hepes, pH 7,4
1,5 ml 150 mM NaCl
0,5 ml 1 % NP40
2,5 ml 5 % Glycerol
50 µl 1 mM MgCl2
50 µl 1 mM MnCl2
500 µl 10 mM NaF
1 mM NaSO4
1 mM PMSF
1:200 Protease inhibitor
17Lipid Kinase Puffer (3-fach) 600 µl 60 mM TRIZMA Base, pH 7,4
120 µl 12 mM MgCl2
600 µl 100 mM NaCl
8,68 ml Aqua bidest.
Loading Buffer Western Blot (5-fach) 5 ml 0,5 M Trizma Base, pH 6,8
4 ml Glycerin
0,8 g SDS
0,5 ml β-Mercaptoethanol
25 mg Bromphenolblau
10 ml Aqua bidest.
Lymphozyten-Kulturmedium RPMI 1640 with L-Glutamine
1 % PS
0,5 % huSerum
Master-Mix 22,5 µl 3-fach Lipid Kinase Puffer
16,5 µl Aqua bidest.
12,5 µl α-Phosphatydiliositol
1 µl γ-ATP
Milch (5 %) 5 g Skim Milk Powder
100 ml TbsT
PI3-Kinase Lysepuffer (50 ml) 1 ml 1 M HEPES, pH 7,5
5 ml 100 mM EGTA
500 µl 100 % NP-40
125 µl 1 M MgCl2
500 µl 200 mM SOV
2 ml 1 M β-Glycerophosphat
50 µl 1 M DTT
250 µl Protease inhibitors
40 ml Aqua bidest.
Running-Buffer (1-fach) 100 ml 10-fach Running-Buffer
900 ml Aqua bidest.
18Samplegel (12 %) 72 ml 30 % Acrylamide
59,04 ml Aqua bidest.
45 ml 1,5 M TRIZMA Base
1,8 ml 10 % SDS
Stackgel (5 %) 15 ml 30 % Acrylamide
61,8 ml Aqua dest.
11,25 ml 1,0 M TRIZMA Base
0,9 ml 10 % SDS
TBS (10-fach) 24,2 g TRIZMA Base
80 g NaCl
1 l Aqua bidest.
auf pH 7,6 einstellen
TbsT 100 ml 10-fach TBS
900 ml Aqua bidest.
1 ml TWEEN 20
TLC Puffer 93 ml Methanol
112 ml Chloroform
16 ml Ammoniak (25 %)
10,5 ml Aqua bidest.
Transferpuffer (1-fach) 100 ml 10-fach Transferpuffer
100 ml Methanol
800 ml Aqua bidest. Transferpuffer (10-fach)
30,3 g TRIZMA Base
112,6 g Glycine
1 l Aqua bidest.
auf pH 8,3 einstellen
192.3. Antikörper
GAPDH (V-18), goat polyclonal IgG Santa Cruz Biotechnology, Dallas, USA
p-AKT Rabbit Antibody Cell Signaling Technologie, Boston, USA
p-ERK Rabbit Antibody Cell Signaling Technologie, Boston, USA
p-MLC Rabbit Antibody Cell Signaling Technologie, Boston, USA
Polyclonal Swine Anti-Rabbit Dako, Hamburg, Deutschland
Immunoglobulins/HRP
Polyclonal Rabbit Anti-Goat Dako, Hamburg, Deutschland
Immunoglobulins/HRP
2.4. Geräte, Verbrauchsmaterialien und Computerprogramme
Amersham Hyperfilm ECL, High performance GE Healthcare, Chalfont St Giles, England
chemiluminescence film
Biofuge fresco Heraeus, Hanau, Deutschland
Biofuge pico Heraeus, Hanau, Deutschland
C-Chip Disposable Hemocytometer NanoEnTek, Seoul, Korea
Cell Scraper (klein) Falcon, New York, USA
Cell Scraper, Disposable (groß) Sarstedt, Nümbrecht, Deutschland
Cellstar Cell Culture Flasks 250 ml Greiner Bio-One, Kremsmünster, Österreich
Deckgläser, 24 x 60 mm Menzel-Gläser, Braunschweig, Deutschland
DUOMAX 1030 (Wippe) Heidolph Instruments, Schwabach,
Deutschland
Extend (Waage) Sartorius, Göttingen, Deutschland
20Extra thick blot paper, Filter paper Bio-Rad Laboratories, Hercules, USA
FACScalibur Flow Cytometer Becton Dickinson, Franklin Lakes, USA
Folded Filters Schleicher & Schuell, Dassel, Deutschland
Herasafe (Sicherheitswerkbank) Heraeus, Hanau, Deutschland
ImageJ (Bildbearbeitungsprogramm) Rasband, W.S., U.S. National Institutes of
Health, Bethesda, USA
KC4 Kineticalc for Windows Bio-Tek Instruments, Winooski, USA
Lambda K (Fotometer) MWG-Biotech, Ebersberg, Deutschland
LB122 (Geigerzähler) Berthold, Bad Wildbad, Deutschland
Leucosep Tube, Porous Barrier, 50 ml Greiner Bio-One, Kremsmünster, Österreich
MACS MidiMACS Separator Miltenyi Biotec, Bergisch Gladbach,
Deutschland
MACS MultiStand Miltenyi Biotec, Bergisch Gladbach,
Deutschland
MACS Pre-Separation Filter, 30 µm Miltenyi Biotec, Bergisch Gladbach,
Deutschland
MACS Separation LS Columns Miltenyi Biotec, Bergisch Gladbach,
Deutschland
Micro Chemotaxis Chamber, 48-Well Neuro Probe, Gaithersburg, USA
Microplate, 96 Well, Clear, F-Bottom Greiner Bio-One, Kremsmünster, Österreich
Mini Trans-Blot Cell Bio-Rad Laboratories, Hercules, USA
MR 3002 (Magnetrührer) Heidolph Instruments, Schwabach,
Deutschland
Multipette plus Eppendorf, Hamburg, Deutschland
Nuclepore Track-Etch Membrane GE Healthcare, Chalfont St Giles, England
Objektträger, 76 x 26 mm Carl Roth, Karlsruhe, Deutschland
OV5 Inkubator Biometra, Göttingen, Deutschland
pH 538 (pH Meter) WTW, Weilheim, Deutschland
Pipetman Classic Gilson, Middleton, USA
Pipetus Hirschmann Laborgeräte, Eberstadt,
Deutschland
Polypropylene Conical Tube, 50 ml Falcon, New York, USA
21Polypropylene Conical Tube, Falcon, New York, USA
High-Clarity 15 ml
PvdF Transfer Membrane, 0,45 µm Thermo Fisher Scientific, Waltham, USA
REAX2 (Rotating-Maschine) Heidolph Instruments, Schwabach,
Deutschland
Replacement Gaskets Neuro Probe, Gaithersburg, USA
Scanjet 5530 photosmart scanner Hewlett-Packard, Palo Alto, USA
Scout Pro (Waage) Ohaus, Parsippany, USA
Serological Pipet, 10 ml Falcon, New York, USA
Standard Power Pack P25 Biometra, Göttingen, Deutschland
Stripette Serological Pipet, 25 ml und 50 ml Sigma-Aldrich, St. Louis, USA
Thermomixer compact Eppendorf, Hamburg, Deutschland
TLC Silica gel 60 F254 Merck, Darmstadt, Deutschland
Vortex-Genie 2 Scientific Industries, Bohemia, USA
Wasserbad mit Shaker Köttermann, Uetze/Hänigsen, Deutschland
Minigel - Twin (Westernblotapparatur) Biometra, Göttingen, Deutschland
X-Ray Cassette Rego X-Ray, Augsburg, Deutschland
Zellkulturschalen, 100 x 20 mm Falcon, New York, USA
222.5. Isolation CD4-positiver Lymphozyten
Herkunft der Blutpräparate
Für die Isolierung CD4-positiver Lymphozyten wurden sogenannte Buffy Coats verwendet.
Der zum Großteil aus Thrombozyten und Leukozyten bestehende Zellüberschuss entsteht
bei der Gewinnung von Erythrozytenkonzentraten. Gemäß aktueller Richtlinien wurden die
Buffy Coats über Nacht in der Blutspendezentrale des Instituts für klinische Transfusions-
medizin und Immungenetik Ulm gekühlt und erst am nächsten Morgen verarbeitet.
Eine Genehmigung durch die Ethikkommission liegt unter der Antragsnummer 180/12 vor.
Isolierung von mononuklearen Zellen
Zunächst wurde eine Puffermischung aus 500 ml DPBS mit einem pH-Wert von 7,2 und 2 ml
0,5 molarem EDTA hergestellt und bei Raumtemperatur gelagert. Für die beiden Buffy Coats
wurden anschließend jeweils zwei Falcon Leucosep-Isolationsröhrchen, welche mittig einen
Filter besitzen, mit 16 ml des Histopaque Isolationsmediums (Ficoll-Paque) gefüllt und für
30 s mit 1000 rcf bei Raumtemperatur zentrifugiert. Dadurch gelangte das
Leukozytenseparationsmedium unter den Filtereinsatz.
Die Buffy Coats wurden derweil mit der vorbereiteten Puffermischung aus DPBS und EDTA
auf 80 ml um ein Vierfaches verdünnt. In die vier Leucosep-Röhrchen wurden nun je 20 ml
der Blutverdünnung gefüllt und mit DPBS und EDTA auf 50 ml aufgefüllt. Durch
Zentrifugieren in einer Zentrifuge ohne Bremse bei 1000 rcf für 15 min wurden alle
Bestandteile anhand ihres Dichtegradienten aufgetrennt.
Anschließend wurden alle Serumreste bis ca. 1 cm oberhalb der sichtbaren Bande vorsichtig
abgesaugt. Die abgesaugten Reste wurden in einem separaten Behältnis aufgefangen und
mit dem restlichen, potentiell infektiösen Müll entsorgt.
Die Interphasen der mononuklearen Zellen aus je zwei zusammengehörigen Leucosep-
Röhrchen wurden nun in je ein neues Falcon-Röhrchen zusammengeführt. Diese Röhrchen
besitzen keinen Filtereinsatz und wurden zusätzlich mit der DPBS und EDTA Mischung auf
50 ml aufgefüllt, gut gemischt und dann mit 300 rcf für 10 min bei Raumtemperatur
zentrifugiert. Die Zellen waren nun als Pellet am Boden zu erkennen, und der Überstand mit
den Resten des verbliebenen Serums wurde vorsichtig abgesaugt. Mit 1 ml DPBS und EDTA
23wurden die Pellets resuspendiert und anschließend wieder mit DPBS und EDTA auf 50 ml
aufgefüllt. Erneut erfolgte eine Zentrifugation mit 200 rcf für 10 bis 15 min. Alle Zellen vom
gleichen Spender wurden nun in ein gemeinsames Falcon-Röhrchen gegeben, erneut mit
DPBS und EDTA resuspendiert, auf 50 ml aufgefüllt und nochmals zentrifugiert.
Magnetic labeling
Anschließend wurde erneut der Überstand abgesaugt, das Pellet zunächst mit 1 ml DPBS
und EDTA resuspendiert und schließlich auf 10 ml aufgefüllt. Nun wurden die entstandenen
Zellsuspensionen durch jeweils einen MACS-Filter geschickt. Das Filtern verhindert, dass die
spätere Magnetmarkierung und Trennung der Zellen durch größere Partikel behindert wird.
Der Filter besitzt eine Porengröße von 30 µm. Aus der nun erhaltenen Suspension wurde
eine Verdünnung zur genauen Zellzählung hergestellt. Dafür wurden 10 µl der gefilterten
Suspension entnommen und mit 990 µl des DPBS und EDTA Puffers aufgefüllt. Jeweils 10 µl
der beiden Suspensionen wurden in eine der beiden Kammern A und B eines
Hemozytometers gefüllt. Unter dem Mikroskop konnten die Zellen als leuchtende Punkte in
den 16 Quadraten gezählt werden. Die gewünschte Anzahl der Zellen wurde ermittelt,
durch Verwerfen der übrigen Suspension hergestellt und anschließend bei 300 rcf für 10 min
zentrifugiert. Erneut wurde der Überstand abgesaugt. Das entstandene Zellpellet wurde in
40 µl kaltem MACS-Puffer pro 107 Zellen resuspendiert. Dann wurden aus dem MACS CD4+
T Cell Isolation Kit 10 µl des Biotin-Antibody Cocktail pro 107 Gesamtzellen hinzugegeben.
Nach gutem Durchmischen wurde die Suspension für 5 Minuten bei 2-8 °C im Kühlschrank
inkubiert. Danach wurden erneut 30 µl des MACS-Puffers und schließlich 20 µl des
MicroBead Cocktails pro 107 Zellen zugefügt und abschließend für weitere 10 min bei
2 bis 8 °C inkubiert.
Magnetic separation
Die biotinylierten Antikörper richten sich gegen nicht CD4-positive Zellen, indem sie an CD8,
CD14, CD16, CD19, CD36, CD56, CD123, T-Zell-Rezeptor γ/δ und Glycophorin A binden und
so z. B. CD8-positive Zellen, γ/δ T-Zellen, natürliche Killerzellen, dentritische Zellen,
Monozyten, Granulozyten und Erythrozyten markieren. Der zweite Antikörper ist gegen
24Biotin gerichtet und an eisentragende Micro Beads gekoppelt, so dass nicht-CD4-tragende
Zellen magnetisch markiert werden, während CD4-positive Zellen unmarkiert bleiben.
Zunächst wurden im MACS Multistand zwei MACS-Separationssäulen pro Buffy ohne
Stempel aufgebaut. Die Zellen sollten ohne den Stempeldruck durch die Säule sickern. Unter
die Säulen wurde je ein Falcon Reagenzglas positioniert und zunächst durch jede Säule zur
Vorbereitung 3 ml des MACS-Puffers laufen gelassen. Anschließend wurden die
Zellsuspensionen jeweils zur Hälfte in die MACS-Säulen gegeben und vollständig hindurch
laufen gelassen. Abschließend wurde erneut mit 3 ml des Puffers nachgespült.
Bei der magnetischen Zellseparation handelt es sich um eine sogenannte negative Selektion.
Nur die nicht mit Antikörpern markierten CD4-positiven Zellen werden wieder
ausgewaschen, während die nicht CD4-positiven Zellen in der Säule verbleiben.
Die nun verbliebene Suspension mit den CD4-positiven Zellen wurde nochmals bei 300 rcf
für 12 min zentrifugiert und der Überstand bis auf das Pellet abgesaugt. Die beiden Pellets
einer Probe wurden jeweils mit 1 ml Kulturmedium resuspendiert, anschließend
zusammengeführt und auf 10 ml mit dem Kulturmedium aufgefüllt. Bei dem Kulturmedium
handelt es sich um RPMI mit 0,5 % huSerum und 1 % PS, das die Lebenserhaltung der Zellen
garantiert, ihren Stoffwechsel aber herabsetzt.
Für die erneute Zellzählung wurde aus den beiden 10 ml Suspensionen eine neue
Verdünnung hergestellt. Hierfür wurden 100 µl der Zellsuspension mit 900 µl des RPMI
Kulturmediums vermischt und dann wie oben beschrieben jeweils 10 µl im Hemocytometer
gemessen.
Die Zellen wurden nun über Nacht bei 37 °C und 5 % CO2 im Brutschrank inkubiert.
FACS-Messung
Nachdem die CD4-positiven Lymphozyten mittels MACS isoliert waren, wurden sie für die
Durchflusszytometrie vorbereitet. Dafür wurden sie fluoreszenzmarkiert und mit einem
speziellen FACS-Gerät zur Reinheitsmessung sortiert.
2 Millionen Zellen wurden zuvor bei 3200 rpm für 5 min zentrifugiert, in 100 µl PBS
resuspendiert und 10 µl AK CD3 FitC und 10 µl AK CD4 PE zugegeben. Nach 10 min bei 4 °C
in Dunkelheit wurden die Zellen anschließend zweimal mit 1000 µl PBS gewaschen und
erneut bei 3200 rpm für 5 min zentrifugiert. Danach wurde die Mischung in 100 µl 1 %
25Formalin in PBS aufgenommen und im Dunkeln bei 4 °C über Nacht stehen gelassen. Vor der
FACS Messung wurde diese noch einmal mit 1000 µl PBS gewaschen und in 300 µl PBS
resuspendiert. Die Proben wurden wie vorgeschrieben bis zur FACS Messung dunkel
gelagert.
Die Messung der Zellen wurde in einem FACS-Durchflusszytometer durchgeführt. Zur
Auswertung wurde jeweils der Prozentsatz CD4-positiver Zellen an der Gesamtpopulation
bestimmt, wobei sich die Stärke der CD4-Expression aus der Stärke der mittleren
Fluoreszenz ergab.
2.6. Migration CD4-positiver Lymphozyten
Migration von CD4-positiven Lymphozyten
Um die Migration der CD4-positiven Zellen untersuchen zu können, wurde eine Micro 48-
Well Chemotaxis Chamber verwendet. Diese besitzt 48 Vertiefungen (= Wells) und wird mit
einer Grund-, einer Deckplatte und der dazwischen gehörigen Gummidichtung geliefert. In
allen drei Teilen sind die 48 Wells identisch angeordnet und somit von oben nach unten
durchgängig. Zwischen die Grundplatte und die Gummidichtung wurde eine
Polycarbonatmembran mit einer Porengröße von 5,0 µm mit der rauen Seite nach unten
eingelegt. Schließlich wurde die Kammer mit 6 Schrauben verschlossen.
Bereits am Vortag wurden die Kammern über Nacht bei 37 °C in den Brutschrank gestellt,
um am nächsten Tag vorgewärmt zu sein. Die Membranen wurden für ca. 4 h bei
Raumtemperatur in einer Petrischale in Collagenlösung eingelegt. Dabei war auf eine
vollständige Benetzung der Membran zu achten. Anschließend wurden die Membranen zum
Trocknen für etwa 30 min bei Raumtemperatur aufgehängt. Währenddessen wurde RPMI
im Wasserbad auf 37 °C aufgewärmt. Nun wurden die Membranen im erwärmten RPMI für
30 min im Brutschrank eingelegt.
Die mittels MACS isolierten Zellen, die über Nacht im Nährmedium im Brutschrank gelagert
waren, wurden bei 300 rcf für 10 min bei Raumtemperatur zentrifugiert. Der Überstand
wurde abgesaugt und das entstandene Pellet mit 1 ml RPMI resuspendiert und auf 10 ml
mit RPMI aufgefüllt. Davon wurden 100 µl zu 900 µl RPMI gegeben um eine 1:10
26Verdünnung zu erhalten, wovon 10 μl zur Zellzahlbestimmung in einem Hemocytometer
mikroskopiert wurden. Die Zellzahl wurde durch Verdünnung mit RPMI auf 5 Mio/ml
eingestellt. Jeweils 1 ml dieser Zellsuspension wurde in 1,5 ml Reaktionsgefäße verteilt. Dazu
wurden außer bei der Kontrolle je 1 μl der Amlodipinkonzentration (1 mM, 5 mM, 10 mM)
pipettiert, um die gewünschte Amlodipin-Endkonzentration von 1 µM, 5 µM bzw. 10 µM zu
erhalten. Die Reaktionsgefäße wurden in einem Vortexmischer gründlich durchmischt und
für 30 min im Brutschrank inkubiert. Nun wurde die Grundplatte mit je 28,5 μl pro Well
gefüllt. Die Vertiefung der späteren Kontrolle und die Vertiefung mit 10 µM Amlodipin
alleine ohne SDF wurden mit RPMI gefüllt. Die chemotaktische Substanz SDF-1α wurde mit
RPMI verdünnt (1 ml RPMI zu 1 μl SDF-1α 100 µg/ml), so dass die Endkonzentration von
100 ng/ml entstand, und in die weiteren Wells gefüllt.
Auf die fertig befüllte Grundplatte wurde die Membran mit der rauen Seite nach unten
aufgelegt, die Gummidichtung darauf fixiert und schließlich die Deckplatte darauf
festgeschraubt. In die Wells der Deckplatte wurden je 45 μl der präinkubierten
Zellsuspensionen gegeben. Um eine reibungslose Migration zu ermöglichen, ist darauf zu
achten die Suspensionen möglichst blasenfrei zu pipettieren. Die Kammer wurde nun für
3 h im Brutschrank bei 37 °C inkubiert. In dieser Zeit erfolgte die Migration, wobei die Zellen
entlang des chemotaktischen Gradienten nach unten durch die Membran wanderten. Die
raue Seite erleichterte hierbei den Zellen das Anheften an die Membran.
Nach der dreistündigen Migrationsphase wurde die Kammer aufgeschraubt, die Membran
entnommen und für 10 min in Methanol fixiert. Anschließend wurde die Membran für
1,5 min in Eosin (Diff-Quick I) gefärbt und dann für 3 min in Thiazin (Diff-Quick II) eingelegt.
Sowohl Diff-Quick I als auch II musste zuvor durch einen Filter geschickt werden, da es eine
ölige Oberfläche bildet, die entfernt werden musste. Nach erfolgter Färbung wurde die
Membran in Aqua bidest. gewaschen. Mit der rauen Seite nach unten wurde die Membran
nun auf einem mit 70 % Ethanol gereinigten Objektträger positioniert. Die Oberseite wurde
mit einem fusselfreien Tuch vorsichtig abgetrocknet und gereinigt, um die Zellen
abzuwischen, die zwar bis zur Membran gewandert, aber nicht durch sie hindurch migriert
waren. Abschließend wurde ein Deckglas mittels Eukitt aufgeklebt und über Nacht bei
Raumtemperatur trocknen gelassen. Am darauf folgenden Tag wurde die Migration bei
40-facher Vergrößerung unter dem Mikroskop ausgewertet. Pro Dreierreihe wurde die
Vertiefung mit der gleichmäßigsten Zellverteilung ausgewählt und davon wurden fünf
27Sie können auch lesen

















































