DISSERTATION - SciDok ...
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Aus dem Bereich Innere Medizin IV
Klinische und Experimentelle Medizin
der Medizinischen Fakultät der Universität des Saarlandes, Homburg/Saar
Direktor: Univ.-Prof. Dr. Danilo Fliser
LDL-Carbamylierung als Mechanismus endothelialer
Dysfunktion bei Patienten mit chronischer
Nierenerkrankung
DISSERTATION
zur Erlangung des Grades eines Doktors der Medizin
der Medizinischen Fakultät
der UNIVERSITÄT DES SAARLANDES
2017
vorgelegt von:
Felix Thomas Lothar Frenzel
geboren am 14.02.1990 in Neunkirchen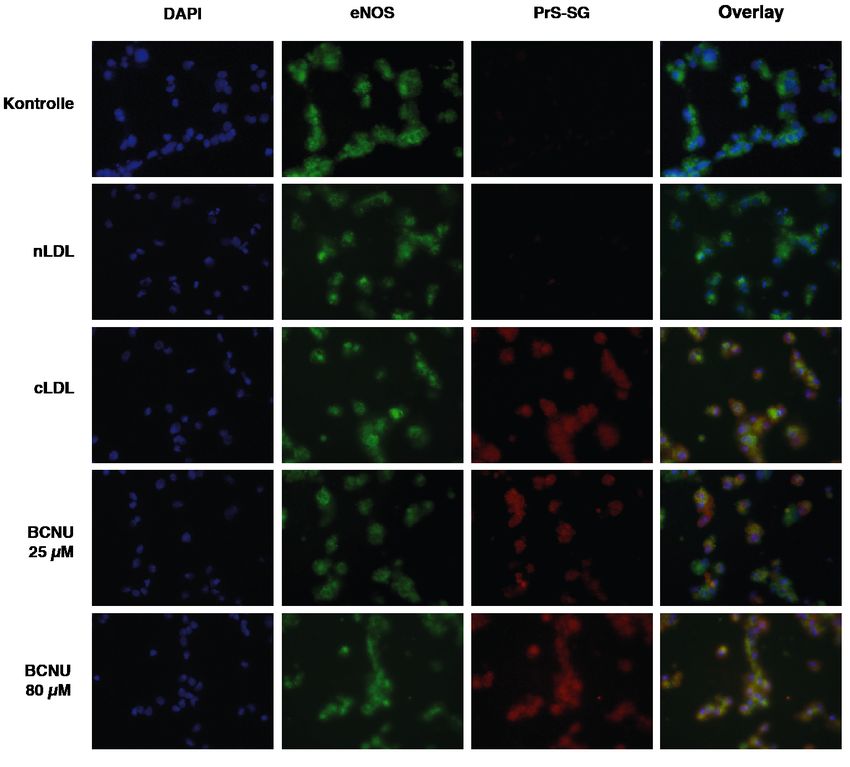
Tag der Promotion: Dekan: Univ.-Prof. Dr. M. Menger 1. Berichterstatter: Univ.-Prof. Dr. D. Fliser 2. Berichterstatter: 1
Inhaltsverzeichnis 1. Zusammenfassung 7 1.1 cLDL induziert endotheliale Dysfunktion in klinisch relevantem Ausmaß 7 1.2 cLDL induces endothelial dysfunction in a clinically relevant dimension 8 2. Einleitung 10 2.1 Kardiovaskuläre Erkrankungen 10 2.1.1 Klassische kardiovaskuläre Risikofaktoren 10 2.1.2 Chronische Nierenerkrankung (CKD) als kardiovaskulärer Risikofaktor 10 2.2 Atherogenetische Grundprinzipien 11 2.2.1 Pathophysiologie der Atherosklerose 11 2.2.2 Endotheliale Dysfunktion initiiert Atherogenese 13 2.3 Lipoproteine 14 2.3.1 Lipoproteine als Regulatoren der Endothelfunktion 14 2.3.2 Carbamylierung von Lipoproteinen 16 2.4 Voruntersuchungen der AG Dr. Dr. Speer: cLDL induziert endotheliale Dysfunktion in Aortenringen ex vivo 17 2.5 Fragestellung und Zielsetzung der Arbeit 20 3. Material 21 3.1 Antikörper 21 3.1.1 Primärantikörper 21 3.1.2 Sekundärantikörper 21 3.2 Kits und siRNA 22 3.3 Medien, Lösungen, Puffer 22 3.4 Geräte 24 3.5 Verbrauchsmaterialen 25 3.6 Chemikalien und sonstige Reagenzien 26 3.7 Analyse- und Statistikprogramme 28 4. Methoden 29 4.1 LDL-Isolation, -Analyse und -Modifikation 29 4.1.1 LDL-Isolation mittels Ultrazentrifugation 29 4.1.1.1 Langzeitprotokoll 29 4.1.1.2 Kurzzeitprotokoll 31 4.1.2 LDL-Konzentration und -Sterilfiltration 33 4.1.3 LDL-Konzentrationsbestimmung 33 4.1.4 LDL-Carbamylierung ex vivo 33 4.1.5 TBARS Assay 34 4.1.6 UHPLC-Aminosäureanalyse 34 4.2 HAEC-Zellkultur 35 4.3 Small interfering RNA-Transfektion 36 4.4 Tierversuche an C57BL/6J-Mäusen 37 4.4.1 Verwendete Mausstämme 37 4.4.2 Überexpression von Lox1 38 4.4.3 nLDL-/cLDL-Stimulation in vivo 38 4.4.4 Blut- und Organentnahme 38 4.4.5 nLDL-/cLDL-Stimulation ex vivo 39 2
4.5 Elektronenspinresonanz-Spektroskopie 39 4.5.1 Detektion reaktiver Sauerstoffspezies (ROS) 39 4.5.1.1 ROS-Bestimmung in HAEC 40 4.5.1.2 ROS-Bestimmung in murinen Aortenringen 41 4.5.1.3 ROS-Bestimmung in murinem Vollblut 41 4.5.1.4 Messung 41 4.5.2 Detektion von Stickstoffmonoxid (NO) 42 4.5.2.1 NO-Bestimmung in HAEC 42 4.5.2.2 Messung und Normalisierung 43 4.6 Western Blot 44 4.6.1 Herstellung von Zelllysaten 44 4.6.2 Bestimmung der Proteinkonzentration nach der Bradford-Methode 45 4.6.3 SDS-PAGE 45 4.6.4 Protein-Transfer (Blot) 46 4.6.5 Blocken 47 4.6.6 Antikörperinkubation 47 4.6.7 Auswertung 47 4.7 Immunfluoreszenz-Färbung der eNOS S-Glutathionylierung 48 4.8 MALDI-ToF-Massenspektrometrie 49 4.9 Klinische Studie 49 4.10 Carbamyl-Lysin FLISA 50 4.11 Statistische Analyse 50 5. Ergebnisse 52 5.1 Ex vivo Carbamylierung von LDL 52 5.2 Effekte von cLDL auf die endotheliale ROS Produktion 53 5.3 Effekte von cLDL auf die endotheliale NO-Produktion 55 5.4 Effekte von cLDL auf die endotheliale NO-Synthase (eNOS) 55 5.4.1 eNOS-Phosphorylierung 55 5.4.2 eNOS-Entkopplung 56 5.5 Rolle von LOX-1 als Rezeptor für cLDL 59 5.6 Zelluläre Mechanismen der cLDL-induzierten ROS-Produktion 61 5.7 Potentielle therapeutische Beeinflussbarkeit der LDL-Carbamylierung 63 5.8 Klinische Relevanz der Forschungsergebnisse bei Patienten mit chronischer Nierenerkrankung 64 6. Diskussion 70 6.1 Proteincarbamylierung und deren funktionelle Konsequenzen 70 6.2 Carbamylierung bei CKD 72 6.3 Endotheliale Effekte von cLDL 74 6.4 Rolle des Rezeptors LOX-1 79 6.6 Therapeutische Konsequenzen 82 6.7 Zusammenfassung 83 7. Literaturverzeichnis 85 8. Publikation 93 9. Danksagung 94 3

Vorbemerkung Die in dieser Arbeit erhobenen Daten wurden in Form einer Originalarbeit publiziert (Speer et al. Eur Heart J 2014). Bei der Durchführung der Arbeit und bei der Auswertung der Daten wurde ich von Herrn Dr. med. Dr. sc. nat. Timo Speer betreut. Der Text und die Abbildungen dieser Dissertationsschrift sind daher in weiten Teilen eine deutsche Reproduktion der Originalarbeiten. 4
Verzeichnis der verwendeten Abkürzungen
APS Ammonium-Persulfat
Aqua dest. Aqua destillata, destilliertes Wasser
AU arbitrary unit (willkürliche Einheit)
AUC Area under the curve (Fläche unter der Kurve)
BCNU 1,3-Bis(2-Chlorethyl)-1-Nitrosourea
BMI Body Mass Index
BSA Bovines Serumalbumin
CKD Chronic kidney disease (chronische Nierenerkrankung)
cLDL carbamyliertes LDL
CMH 1-Hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine
CRP C-reaktives Protein
DAPI 4',6-Diamidin-2-phenylindol
DETC Diethyldithiocarbaminsäure
DFO Deferoxamin
DMEM Dulbecco's Modified Eagle Medium
DMSO Dimethylsulfoxid
DPI Diphenylen-Iodonium
ECL Enhanced chemoluminescence (erweiterte Chemolumineszenz)
EDTA Ethylendiamintetraacetat
eGFR estimated GFR (geschätzte glomeruläre Filtrationsrate)
ESR Elektronenspinresonanz
FLISA Fluorescence-linked Immunosorbent Assay
GAPDH Glycerinaldehyd-3-phosphat-Dehydrogenase
HAEC Human aortic endothelial cells (humane aortale Endothelzellen)
HbA1C Glykosyliertes Hämoglobin
HBSS Hank's Balanced Salt Solution
HDL High Density Lipoprotein
HRP Horseradish Peroxidase
hsCRP high sensitivity CRP
IF Immunfluoreszenz
KDIGO Kidney Disease: Improving Global Outcomes
KHB Krebs-HEPES-Puffer
KHK koronare Herzkrankheit
L-NAME L-Nitro-Arginin-Methyl-Ester
LDL Low Density Lipoprotein
LOX-1 Lectin-like oxLDL Rezeptor-1
MALDI-ToF-MS Matrix-unterstützte Laser-Desorptions/Ionisations-Massenspektrometrie mit
Flugzeitanalysator
MDRD Modifikation of Diet in Renal Disease
milk Magermilchpulver
mind. mindestens
5MS Massenspektrometrie
MW Molekulargewicht
MW ± SE Mittelwert ± Standardfehler
NFS Nanoparticle Formation Solution
nLDL natives LDL
NO Stickstoffmonoxid
PBS Phosphate buffered saline (Phosphatgepufferte Salzlösung)
PEG-SOD Polyethylenglykol-Superoxid Dismutase
PFA Paraformaldehyd
PMSF Phenylmethylsulfonylfluorid
PVDF Polyvinylidenfluorid
ROS Reactive oxygen species (reaktive Sauerstoffspezies)
RPM Rounds per minute
RT Raumtemperatur
SDS-Page sodium dodecyl sulfate polyacrylamide gel electrophoresis,
(Natriumdodecylsulfat-Polyacrylamidgelelektrophorese)
sog. sogenannt
TBS Tris buffered saline (Trisgepufferte Salzlösung)
TEMED Tetramethylethylendiamin
ToF time of flight (Flugzeitberechnung)
ü.N. über Nacht
WB Western Blot
WT Wildtyp
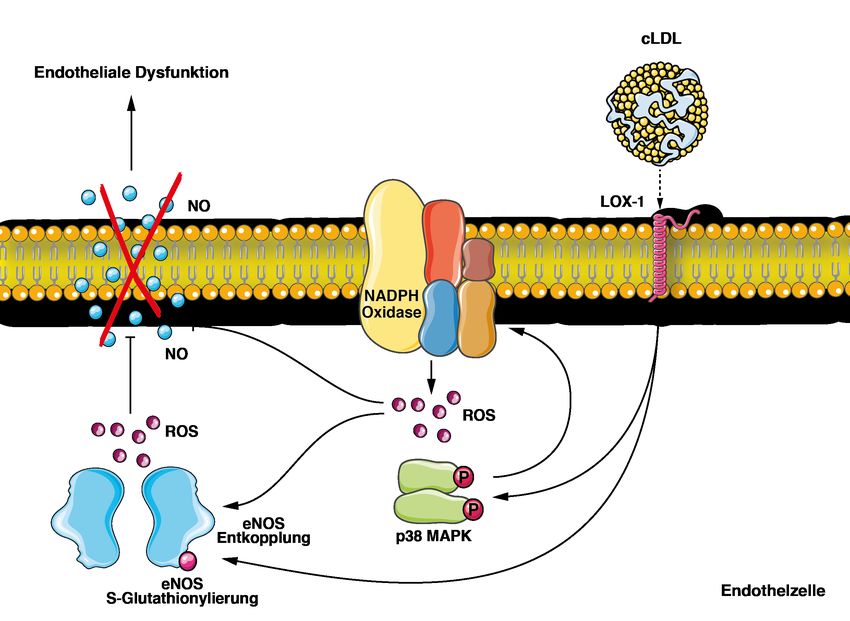
61. Zusammenfassung 1.1 cLDL induziert endotheliale Dysfunktion bei Patienten mit chronischer Nierenerkrankung (CKD) Erhöhte LDL-Cholesterinspiegel sind insbesondere auch bei Patienten mit chronischer Nierenerkrankung wesentlich mit Atherosklerose und dem Auftreten von Gefäßkomplikationen vergesellschaftet. Vermehrt werden bei diesen Patienten Serumproteine posttranslational carbamyliert. Die Carbamylierung von Lipoproteinen kann die Schaumzellenbildung und endotheliale Dysfunktion begünstigen, eine mechanistische Bestätigung dieser Hypothese wurde bislang nicht erbracht. In dieser Studie wurden die endothelialen Effekte carbamylierten LDLs untersucht. Zunächst wurde LDL aus dem Serum gesunder Probanden isoliert und anschließend ex vivo carbamyliert. ESR-spektroskopisch zeigte sich eine gesteigerte ROS- und verminderte NO-Produktion nach Inkubation von murinen Aortenringen sowie in humanen aortalen Endothelzellen (HAEC) nach Inkubation mit carbamyliertem LDL (cLDL). Western Blot-Analysen ergaben einen durch cLDL hervorgerufenen aktivitätsmindernden Phosphorylierungszustand der endothelialen NO-Synthase (eNOS). Der als Entkopplung bezeichnete Zerfall der dimeren eNOS in Monomere, welche folglich ROS statt NO bilden, wurde ergänzend immunfluoreszenzmikro- skopisch durch cLDL-abhängige S-Glutathionylierung aufgezeigt. Zudem konnte LOX-1 als Rezeptor identifiziert werden, der diese Effekte von cLDL auf HAEC vermittelt. C57BL/6J-Wildtypmäuse wiesen nach Injektion mit cLDL erhöhte ROS- Spiegel in Vollblut und explantierten Aortenringen auf, wobei sich in Lox1 transgenen C57BL/6J- Mäusen mit endothelialer Überexpression von LOX-1 eine weitere deutliche Zunahme der Effektstärke objektivieren lies. Durch Inkubation mit spezifischen Inhibitoren wurden die Beteiligung der NADPH-Oxidase (Inhibition durch Captopril/ DPI) sowie p38 MAPK (Inhibition durch SB202190) an der LOX-1 Rezeptoraktivierung nachgeschalteten zellulären Signalkaskade bewiesen. Die klinische Relevanz der LDL-Carbamylierung verdeutlichte der Vergleich in vivo carbamylierter Lysin-Reste im LDL gesunder Probanden (kein Nachweis) und chronisch Nierenkranker (54 ± 4) mittels MALDI-ToF-Massenspektroskopie. Analog der ESR-spektroskopischen Studien mit ex vivo carbamyliertem LDL führte das aus dem Plasma von CKD-Patienten isolierte LDL zu einer signifikanten Reduktion der NO-Produktion in Zellkulturversuchen an HAEC. 7
In einer klinischen Studie an Patienten mit CKD zeigte sich, dass hohe LDL- Carbamyl-Lysinkonzentrationen mit höherer Mortalität und Inzidenz kardiovaskulärer Ereignisse assoziiert sind. Zusammenfassend zeigt diese Studie, dass cLDL bei chronisch Nierenkranken in klinisch relevantem Ausmaß endotheliale Dysfunktion über eine gesteigerte ROS- Produktion und eine verminderte NO-Bioverfügbarkeit induziert. Auf molekularer Ebene werden hier nach LOX-1 Rezeptorbindung p38 MAPK und NADPH-Oxidase aktiviert sowie eNOS gehemmt und via S-Glutathionylierung entkoppelt. Diese Ergebnisse weisen auf einen wichtigen neuen Pathomechanismus für die Entstehung kardiovaskulärer Ereignisse bei Patienten mit Niereninsuffizienz hin. Zudem konnte cLDL als neuer Biomarker sowie therapeutischer Angriffspunkt identifiziert werden. 1.2 cLDL induces endothelial dysfunction in patients with chronic kidney disease (CKD) Elevated levels of LDL-cholesterol are crucially involved in the pathogenesis of atherosclerosis and subsequent cardiovascular complications. This also applies to CKD-patients, whose serum proteins are increasingly subject to posttranslational carbamylation. Lipoprotein carbamylation is thought to promote foam cell formation and endothelial dysfunction but the underlying molecular mechanisms remained unclear. Here, we investigated the endothelial effects of carbamylated LDL. LDL was isolated from healthy donors’ serum and carbamylated ex vivo. ESR-spectroscopy showed an increased production of ROS and diminished release of NO in murine aortic rings and human aortic endothelial cells (HAEC) after incubation with cLDL. Western blot analyses revealed a cLDL-induced, activity-diminished phosphorylation state of eNOS, a key regulator of cellular redox state. In addition, uncoupling called disaggregation of dimeric eNOS into monomer units, which promotes the production of ROS instead of NO, was shown to be due to S-glutathionylation of eNOS. Moreover, LOX-1 was identified as the receptor mediating these adverse endothelial effects of cLDL. While C57BL/6J- wild type mice already featured increased levels of ROS in whole blood and explanted aortic rings after injection with cLDL, an even greater increase was observed in Lox1 transgenic C57BL/6J- mice exhibiting endothelial overexpression of LOX-1. Using specific inhibitors, further experiments demonstrated an involvement of NAPH-oxidase (inhibition by captopril/ DPI) and p38 8
MAPK (inhibition by SB202190) in the cellular downstream effects of LOX-1 receptor stimulation by cLDL. The clinical relevance of LDL carbamylation was proven by comparison of carbamylated lysine residues in LDL of healthy donors (none detectable) and CKD patients (54 ± 4) using MALDI-ToF mass spectrometry. Similar to LDL carbamylated ex vivo, incubation with LDL isolated from CKD patients’ plasma led to a significant reduction of basal NO-production in HAEC culture experiments. Notably, in a prospective clinical study of CKD patients, we could prove the clinical relevance of these experimental findings. Carbamyl-lysine levels in LDL were associated with increased all-cause mortality and cardiovascular event rate. In summary, cLDL induces endothelial dysfunction through increased production of ROS and diminished bioavailability of NO, in a clinically relevant dimension in CKD patients. LOX-1 receptor binding leads to activation of p38 MAPK and NADPH- oxidase such as inhibition and uncoupling of eNOS by S-glutathionylation. These findings highlight on carbamylation of LDL as an important pathomechanism for CKD-associated accelerated cardiovascular disease. Moreover, cLDL was identified as a novel biomarker and potential therapeutic target. 9
2. Einleitung 2.1 Kardiovaskuläre Erkrankungen Kardiovaskuläre Erkrankungen stellen die Haupttodesursache in westlichen Ländern dar. So waren kardiovaskuläre Ereignisse wie der akute Myokardinfarkt oder Schlaganfall entsprechend der Global Burden of Disease-Studie in 2013 für 247,9 Todesfälle/100.000 Personen verantwortlich. Demnach waren 28,2 % aller Todesfälle weltweit kardiovaskulären Erkrankungen geschuldet [1]. Ähnliche Zahlen publiziert die Todesursachenstatistik für Deutschland. Laut Daten des statistischen Bundesamts sind 38,9% der gesamten Todesursachen im Jahr 2014 Erkrankungen des Kreislaufsystems zuzuschreiben [2]. 2.1.1 Klassische kardiovaskuläre Risikofaktoren Seit Start der Framingham-Herz-Studie im Jahr 1948 sind mehrere unabhängige kardiovaskuläre Risikofaktoren identifiziert worden [3], die das globale Risiko für das Auftreten kardiovaskulärer Ereignisse bestimmen und seitdem in zahlreichen Folgestudien validiert wurden. So stratifizierte die deutsche „Prospektive Cardiovaskuläre Münster (PROCAM)" Studie hinsichtlich des Risikos für einen Myokardinfarkt neben dem Geschlecht als bedeutsamste Risikofaktoren das Lebensalter, dicht gefolgt von LDL-Cholesterin, Nikotinkonsum, HDL-Cholesterin, systolischem Blutdruck, positiver Familienanamnese mit frühzeitigen Myokardinfarkten, Diabetes mellitus und Triglyzeriden [4]. 2.1.2 Chronische Nierenerkrankung (CKD) als kardiovaskulärer Risikofaktor Interessanterweise sind Diabetes mellitus und arterielle Hypertonie nicht nur direkte Auslöser von Gefäßerkrankungen [5,6], sondern stellen auch die beiden häufigsten Ursachen der Entwicklung einer chronischen Nierenerkrankung dar [7,8]. Eine chronische Nierenerkrankung (chronic kidney disease, CKD) liegt nach Definition der KDIGO dann vor, wenn eine Störung der Nierenstruktur oder -funktion über einen Zeitraum von länger als drei Monaten negative Auswirkungen auf den Gesundheitszustand ausübt [9]. Ihre Klassifikation beruht neben der Ursache der Nierenschädigung (z.B. Glomerulonephritiden, diabetische Nephropathie, hyperten- 10
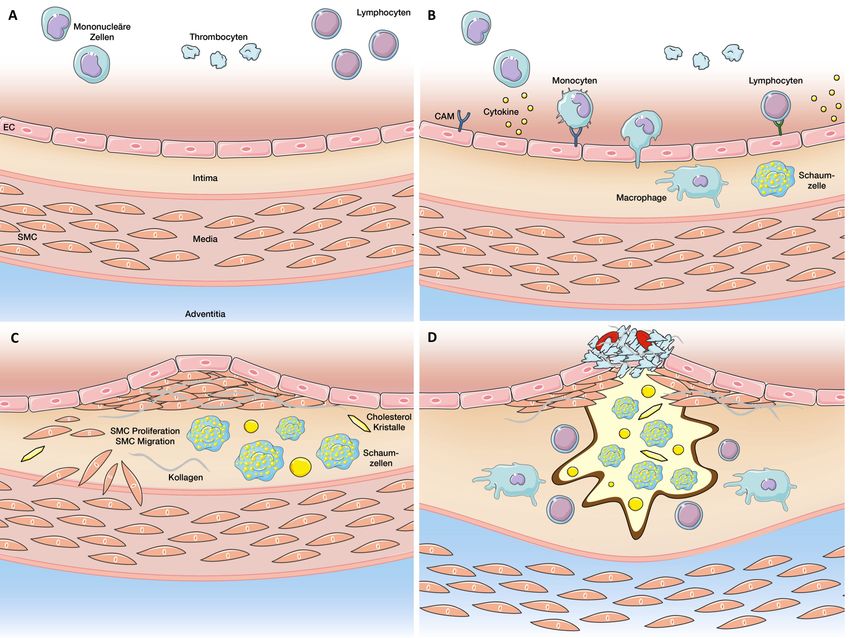
sive Nephrosklerose) auf der verbleibenden glomerulären Filtrationsrate (GFR) und der Albuminurie. Gemeinsamer Endpunkt jeder über mehrere Jahre hinweg bestehender chronischen Nierenschädigung ist dabei das Stadium der terminalen Niereninsuffizienz (GFR < 15 ml/h) mit progressiver Glomerulosklerose und der Entwicklung von Schrumpfnieren [10]. Neben einer Beeinträchtigung der ex- und inkretorischen Nierenfunktion sind eine Salz- und Wasserretention durch Störungen im Wasser-/Elektrolyt- und Säuren-Basen-Haushalt sowie Endorganschäden durch retinierte harnpflichtige Substanzen die Folge [10]. So wird ersichtlich, weshalb die CKD mittlerweile als eine der stärksten unabhängigen kardiovaskulären Risikofaktoren angesehen wird und ihrerseits selbst im Sinne eines circulus vitiosus beispielsweise die Entstehung einer arteriellen Hypertonie begünstigt [11]. CKD kann daher per se und nicht nur im Zusammenspiel mit den Komorbiditäten Diabetes mellitus und arterieller Hypertonie als hauptverantwortlich für die Pathogenese atherosklerotischer Gefäßerkrankungen betrachtet werden. Dies gilt insbesondere für eine alternde Gesellschaft, da die Prävalenz einer chronischen Nierenerkrankung mit zunehmendem Alter steigt [12]. Dennoch scheint die mit einer chronischen Nierenerkrankung assoziierte hohe kardiovaskuläre Mortalität und Morbidität nicht alleine durch klassische Risikofaktoren erklärt werden zu können [13]. 2.2 Grundprinzipien der Atherogenese 2.2.1 Pathophysiologie der Atherosklerose Die Entstehung atherosklerotischer vaskulärer Läsionen stellt ein Grundprinzip kardiovaskulärer Erkrankungen wie koronarer Herzkrankheit, ischämischem Schlaganfall oder peripher-arterieller Verschlusskrankheit dar. Der in Abbildung 1 illustrierte Prozess der Atherogenese zeigt ein komplexes Geschehen in einem durch multiple Risikofaktoren bestimmten Milieu, welches durch die Ausbildung atheromatöser Plaques in Gefäßwänden geprägt ist. Infolge von Plaqueruptur und Thrombusbildung kommt es zu flusslimitierenden Stenosen und Gewebsischämie, welche sich klinisch als Myokardinfarkt oder Schlaganfall präsentieren können [14]. 11
Abbildung 1: Pathophysiologie der Atherogenese. EC= endothelial cells (Endothelzellen), SMC= smooth muscle cells (glatte Muskelzellen), CAM= cell adhesion molecule (Zelladhäsionsmoleküle). Abbildung von Dr. Dr. T. Speer zur Verfügung gestellt. Im gesunden Zustand (Abb. 1A) verhindert eine intakte Endothelzellschicht (EC) als Barriere zwischen Blutstrom und Gefäßwand die Adhäsion und Aktivierung korpuskulärer Blutbestandteile. Insbesondere modifizierte Lipoproteine wie Low- density lipoprotein (LDL) können die endotheliale Barriere überwinden und gelangen über verschiedene Prozesse in den subendothelialen Raum [15]. Diese subendotheliale Retention von LDL und Lipidpartikeln wird heute als Kernprozess zur Initiierung der Atherogenese diskutiert [16]. Im Subendothelraum aggregieren die LDL-Partikel, werden zusätzlich durch Oxidation zu einem pro-atherogenen Partikel und können so eine inflammatorische Reaktionskaskade anstoßen. Modifizierte LDL- Partikel aktivieren Endothelzellen (analog der Wirkung von Zytokinen wie TNF-α und IL-1β) und induzieren damit die endotheliale Expression verschiedener Zelladhäsionsmoleküle (CAM) (Abb. 1B) [17]. Dies ermöglicht zirkulierenden Monozyten die Adhäsion an das Endothel. Die gleichzeitige Freisetzung von chemotaktischen Botenstoffen, Wachstumsfaktoren und proinflammatorischen Zytokinen aktiviert die Monozyten und fördert damit deren Adhäsion und 12
Transmigration in den subendothelialen Raum, wodurch ein circulus vitiosus entsteht [18]. Im subendothelialen Raum differenzieren Monozyten zu Makrophagen und werden durch Einlagerung von Cholesterinestern und freien Fettsäuren in Schaumzellen umgewandelt. Wachstumsfaktoren stimulieren zudem die Proliferation glatter Gefäßmuskelzellen (SMC), die aus der Media in die Intima einsprossen (Abb. 1C). Gleichzeitig setzen sie extrazelluläre Matrixbestandteile wie Proteoglykane, Elastin und Kollagen frei. Die dadurch gebildete fibröse Kappe dient der schützenden Abkapselung des Konglomerats aus Schaumzellen, extrazellulären Lipidablage- rungen, Cholesterinkristallen und apoptotischen Zelltrümmern, sodass diese Plaques zunächst asymptomatisch bleiben. Vermutlich durch eine Störung der apoptotischen Beseitigung toter Makrophagen („Efferozytose“) kann es innerhalb der Plaque zur Fortdauer und Intensivierung der Inflammation kommen [19]. Als erste Anhalte für das Fortschreiten einer Läsion zur so genannten vulnerablen Plaque konnten eine pathologische Intimaverdickung (PIT) [20] und Akkumulation unveresterter Cholesterine beobachtet werden. Durch Umwandlung des Lipidkerns in ein nekrotisches Zentrum [21] und Ausdünnung der fibrösen Kappe zeigen sich solche vulnerablen Plaques (Abb. 1D) anfällig gegenüber Scherkräften, was zur Plaque- fissur oder -ruptur, mit der Konsequenz tödlicher thrombotischer Gefäßverschlüsse, führen kann [22]. 2.2.2 Endotheliale Dysfunktion initiiert Atherogenese Neben der rein physikalischen Barrierefunktion stellen Endothelzellen ein komplexes Kontrollorgan der vaskulären Homöostase dar. Durch parakrine Faktoren regulieren sie u.a. den Gefäßtonus, inhibieren die Thrombozytenaggregation, verhindern die Adhäsion von Leukozyten und limitieren die Proliferation glatter Gefäßmuskelzellen [23]. Das Schlüsselmolekül dieser Aufgaben stellt dabei das endothelstämmige freie Radikal Stickstoffmonoxid (NO) dar, welches die Relaxation glatter Gefäßmuskel- zellen anregt, die Expression vaskulärer Zelladhäsionsmoleküle inhibiert, anti- inflammatorisch und anti-koagulatorisch wirkt [24,25] sowie die Oxidation von LDL unterdrückt [26]. Endotheliale Dysfunktion, welche als erster Schritt der Atherogenese angesehen wird [27], entsteht vorwiegend durch ein Ungleichgewicht zwischen der Produktion von NO und konkurrierender reaktiver Sauerstoffspezies (ROS) [28]. Der resultierende oxidative Stress stimuliert zudem die oben beschriebene pro-inflammatorische Zellaktivierung und wirkt so pro-atherogen. 13
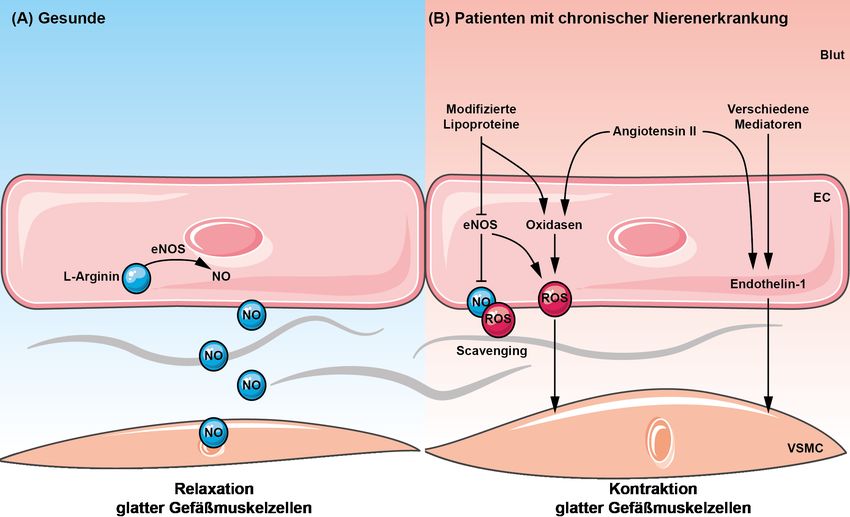
Tatsächlich sind alle kardiovaskulären Risikofaktoren, wie arterielle Hypertonie, Hypercholesterinämie, Diabetes mellitus, Raucherstatus sowie manifeste kardio- vaskuläre Erkrankungen selbst mit einer erhöhten ROS-Produktion in der Gefäßwand assoziiert, wodurch die Bioverfügbarkeit von NO am Endothel zumindest teilweise reduziert wird [23,29]. Es überrascht daher nicht, dass insbesondere das urämische Milieu einer chronischen Nierenerkrankung oxidativen Stress generiert. Dieser entsteht erstens durch ROS-Produktion infolge Aktivierung pro-oxidativer endothelialer Enzyme (NADPH-Oxidase, Xanthin-Oxidase, Enzyme der mitochondrialen Atmungskette), zweitens durch Inhibition anti-oxidativer Enzyme wie der endothelialen NO-Synthase durch sog. Entkopplung [23]. Letztlich führt diese Dysfunktion des Endothels zu Vasospasmen, Thromboseneigung, vaskulärer Inflammation sowie Intimaproliferation und induziert somit die Atherogenese. Abbildung 2: Auswirkungen des Milieus chronischer Nierenerkrankungen auf die Endothelfunktion. VSMC = vascular smooth muscle cells (glatte Gefäßmuskelzellen). Abbildung modifiziert nach Speer et al. (Oxford Textbook of Clinical Nephrology 2015). 2.3 Lipoproteine 2.3.1 Lipoproteine als Regulatoren der Endothelfunktion Die Vertreter der heterogenen Lipoprotein-Familie nehmen eine bedeutsame Rolle als Modulatoren der Endothelfunktion ein. 14
So werden dem High Density Lipoprotein (HDL) als Vehikel des reversen Cholesterintransports aus der Körperperipherie zur Leber hauptsächlich anti- atherogene Eigenschaften zugeschrieben. Darüber hinaus übt HDL vasoprotektive Effekte durch gesteigerte NO-Produktion via Hochregulation und Stimulation der endothelialen NO-Synthase (eNOS) aus, schützt Endothelzellen vor Apoptose und fördert deren Wachstum sowie Migration. Zuletzt weist HDL durch Stimulation der endothelialen Prostazyklin-Synthese und Herabregulation von Selectinen anti- thrombotische Eigenschaften auf [30,31]. Demgegenüber gilt eine Erhöhung des Low Density Lipoproteins (LDL) seit vielen Jahren als kardiovaskulärer Risikofaktor. Da jedoch annäherungsweise die Hälfte aller Patienten mit klinischer Manifestation der Atherosklerose normale bis lediglich geringfügig erhöhte LDL-Cholesterinspiegel aufweisen [27], müssen abseits der LDL- C-Konzentration auch qualitative oder strukturelle Merkmale der Partikel oder Umweltfaktoren von Bedeutung sein. Ebenso erscheint es auf mechanistischer Ebene im Rahmen der Schaumzellen-Hypothese zunächst paradox, dass erhöhte LDL-Spiegel zur Bildung atherogener Schaumzellen führen, da Makrophagen durch einen negativen Feedbackmechanismus des LDL-Rezeptors vor einer Cholesterinüberladung geschützt werden. Um diese Downregulation zu umgehen und pro-atherogen zu wirken, müssen native LDL-Partikel somit zusätzlich strukturell modifiziert werden [17]. Gegenwärtig sind mehrere solcher posttranslationalen Proteinmodifikationen bekannt, welche LDL in ein pro-atherogenes Lipoprotein umwandeln und für dessen Effekte auf das Gefäßsystem verantwortlich sind. Bislang am besten charakterisiert ist die ROS-vermittelte Oxidation von LDL, die zur Bildung von oxidiertem LDL (oxLDL) führt. Dieses wird von Makrophagen über Scavenger-Rezeptoren konzentrations- unabhängig aufgenommen [32] und vermittelt somit frühe Auswirkungen in der Entwicklung endothelialer Dysfunktion. Auch oxLDL induziert einen Teufelskreis, indem es u.a. durch Aktivierung der NADPH-Oxidase und eNOS-Uncoupling selbst als Stimulus zur Bildung weiterer freier Sauerstoffradikale fungiert. Durch Sensibilisierung des kontraktilen Gefäßapparates werden der Gefäßtonus gesteigert, Vasospamen begünstigt und Remodelling-Vorgänge angestoßen [33]. 15
2.3.2 Carbamylierung von Lipoproteinen
Neben der Oxidation wurde kürzlich die Carbamylierung von Lipoproteinen als
weitere, deren Funktion beeinflussende Modifikation identifiziert [34]. Unter
Carbamylierung versteht man eine nicht-enzymatische Proteinmodifikation durch
langanhaltende Exposition zu im Rahmen nachlassender Nierenfunktion zunehmend
aus Harnstoff spontan freigesetzten Cyanaten. Deren aktive Form, Isocyansäure
(HNCO), reagiert irreversibel mit freien Aminosäuren (RNH2) bzw.
Aminosäureseitenketten von Proteinen und anderen Molekülen zu Carbamyl-
Aminosäuren(seitenketten) (RNHC(O)NH2).
HNCO + RNH2 → RNHC(O)NH2
Die resultierende Carbamid-Verbindung verändert nicht nur die Struktur
carbamylierter Substanzen, sondern zugleich auch deren Ladung und Funktion. In
vivo kann so die molekulare Aktivität von Enzymen, Co-Faktoren, Hormonen,
Antikörpern, Rezeptoren und Transportproteinen beeinflusst werden [34].
Bereits vor vielen Jahren wurde der Mechanismus der Carbamylierung am
Hämoglobin von Patienten mit terminaler Nierenerkrankung beschrieben [35] und mit
der Toxizität der Urämie in Verbindung gebracht [34]. Jedoch scheint der Vorgang
der Carbamylierung nicht unbedingt Harnstoff-spezifisch zu sein, da Cyanate auch
bei Nierengesunden durch die Myeloperoxidase und Peroxid-katalysierte Oxidation
von Thiocyanaten (beispielsweise aus Tabakrauch) in Entzündungsherden anfallen
können [36,37]. In der jüngeren Vergangenheit ist nun insbesondere die Harnstoff-
bzw. Myeloperoxid-abhängige Carbamylierung von Lysin-Resten an Lipoproteinen
chronisch nierenkranker Patienten in den Fokus der Forschung gerückt, da sowohl
bei dieser Population, als auch Patienten mit koronarer Herzkrankheit, erhöhte
Serumspiegel carbamylierter Proteine (insbesondere Lipoproteine) detektiert wurden
[38-40]. Durch Nachweis der Beeinträchtigung funktionaler Integrität von HDL- und
LDL-Partikeln konnte hierbei ein erster Zusammenhang zur Atherosklerose-
Entwicklung bei chronischer Nierenerkrankung nachgewiesen werden [39-41].
Carbamyliertes cLDL-Cholesterin scheint damit eine dem oxLDL ähnliche Rolle
wahrzunehmen, wobei Studien zeigten, dass einzelne LDL-Partikel auch simultan
carbamyliert und oxidiert werden können und beide Modifikationen im Rahmen der
Atherogenese in vielen Partikeln voraussichtlich koexistieren [42]. Die Vermutung,
cLDL führe im Rahmen chronischer Nierenerkrankung in Analogie zu oxLDL zu
16endothelialer Dysfunktion, galt daher bislang als plausibler Erklärungsversuch für das hohe kardiovaskuläre Risikoprofil dieser Patientengruppe. Der zugrundeliegende molekulare Mechanismus konnte aber nie schlüssig bewiesen werden [36]. 2.4 Voruntersuchungen der AG Dr. Dr. Speer: cLDL induziert endotheliale Dysfunktion in Aortenringen ex vivo Welche exakten Effekte cLDL auf die Endothelzellfunktion ausübt, blieb unklar, bis es unserer Arbeitsgruppe in Organkammerexperimenten kürzlich gelang, zu zeigen, dass cLDL die endotheliale Funktion in isolierten Aortenringen beeinträchtigt [43]. Die folgenden Abbildungen sind der entsprechenden Publikation der Arbeitsgruppe von Dr. Dr. T. Speer [43] entnommen. In Voruntersuchungen an Aortenringen von Wildtyp C57Bl/6J-Mäusen konnte durch Messung der isometrischen Wandspannung beobachtet werden, dass in vitro carbamyliertes LDL (siehe Kapitel 4.3) im Gegensatz zu nativem LDL gesunder Pro- banden spezifisch die durch Acetylcholin und dem Kalzium-Ionophor A23187 vermit- telte vaskuläre Relaxationsfähigkeit beeinträchtigen kann (Abb. 3). Die Wirkung auf Rezeptor-abhängige und –unabhängige Signalwege gleichermaßen zeigt, dass der Mechanismus nicht vom muskarinergen Acetylcholin-Rezeptor abhängig sein kann. 17
Abbildung 3: Behandlung mit cLDL hemmt gegenüber nLDL sowohl rezeptorvermittelte
(Acetylcholin) als auch rezeptorunabhängige Vasodilatation (A23187) in Aortenringen ex vivo.
(A) Messung der isometrischen Wandspannung an Aortenringen von C57Bl/6J-Mäusen mit
-9 -5
Acetylcholin in aufsteigenden Konzentrationen von 10 bis 10 mol/l während submaximaler
-6
Kontraktion durch Noradrenalin (10 mol/l) ± nLDL/cLDL (je 100 µg/ml) (B) Analoger Versuchsaufbau
-9 -5
unter dem Kalzium-Ionophor A23187 (10 bis 10 mol/l). Relaxation je prozentual zur Vorkontraktion
angegeben. AUC als –log(ED50). Abbildung entnommen aus Speer et al. Eur Heart J 2014.
Eine Versuchsserie mit dem Vasokonstriktor KCl zeigte keine Änderung der
Gefäßkontraktilität in Anwesenheit von nLDL und cLDL gegenüber der Kontrolle (Abb.
4), sodass die ausbleibende Relaxation der ersten Versuchsreihe nicht im Rahmen
einer Zunahme der Vasokonstriktion durch cLDL erklärt werden kann.
Abbildung 4: Kein signifikanter Unterschied in der Gefäßkontraktion durch KCl in Gegenwart
von nLDL und cLDL. Messung der Zunahme isometrischer Wandspannung von Aortenringen unter
-6
dem Vasokonstriktor KCl (80 mmol/l) während submaximaler Kontraktion durch Noradrenalin (10
mol/l) ± nLDL/cLDL (je 100 µg/ml). AUC als –log(ED50). Abbildung entnommen aus Speer et al. Eur
Heart J 2014.
Der Frage, ob die Einschränkung der vaskulären Relaxationsfähigkeit durch cLDL via
Endothel oder glatte Gefäßmuskelzellen vermittelt wird, ging unsere Arbeitsgruppe
durch isometrische Spannungsmessungen unter dem NO-Donor Nitroprussid-
Natrium (Sodium nitroprusside, SNP) nach (Abb. 5). Da sich nach Zugabe von cLDL
gegenüber Nitroprussid alleine kein signifikanter Unterschied in der gemessenen
Wandspannung zeigte, kann der von cLDL ausgeübte Effekt nicht in einer Störung
der vasodilatativen Eigenschaften glatter Gefäßmuskelzellen als Rezeptororgane von
NO liegen, sondern muss spezifisch durch Beeinträchtigung der Endothelfunktion
durch eine verminderte Bioverfügbarkeit von NO begründet sein.
18Abbildung 5: Kein signifikanter Unterschied in der Gefäßrelaxation durch Nitroprussid nach
Zugabe von cLDL. Messung der isometrischen Wandspannung von Aortenringen unter Nitroprussid
10 -5 -6
(10- bis 10 mol/l), während submaximaler Kontraktion durch Noradrenalin (10 mol/l) ± cLDL (100
µg/ml). Relaxation prozentual zur Vorkontraktion angegeben. AUC als –log(ED50). Abbildung
entnommen aus Speer et al. Eur Heart J 2014.
Diese Ergebnisse legen nahe, dass cLDL spezifisch endothelabhängige, nicht jedoch
endothelunabhängige Gefäßrelaxationen durch sowohl rezeptorvermittelte, als auch
rezeptorunabhängige Agonisten inhibiert (Abb. 3-5).
Durch Zugabe des ROS-Scavengers Polyethylenglykol-Superoxid-Dismutase (PEG-
SOD) konnte die endothelvermittelte Relaxation gegenüber Acetylcholin in mit cLDL-
behandelten Aortenringen wiederhergestellt werden (Abb. 6), was auf eine potentielle
Schlüsselrolle von ROS in der Induktion endothelialer Dysfunktion durch cLDL
hinweist.
Abbildung 6: Wiederherstellung der endothelvermittelten Relaxation gegenüber Acetylcholin ±
nLDL/cLDL nach Zugabe von PEG-SOD. Messung der isometrischen Wandspannung an
-9 -5
Aortenringen mit Acetylcholin in aufsteigenden Konzentrationen von 10 bis 10 mol/l während
-6
submaximaler Kontraktion durch Noradrenalin (10 mol/l) ± nLDL/cLDL (je 100 µg/ml) ± PEG-SOD
(150 U/ml). Relaxation prozentual zur Vorkontraktion angegeben. AUC als –log(ED50). Abbildung
entnommen aus Speer et al. Eur Heart J 2014.
19Der Effekt von cLDL auf die Gefäßdilatation erschien unserer Arbeitsgruppe somit
vergleichbar mit derer von oxLDL, was auf die Möglichkeit verschiedenartiger
Modifikationen von LDL hinweist, welche über ähnliche Signalwege gleichartige
Auswirkungen auf die vaskuläre Homöostase ausüben [32]. Die Tatsache, dass
natives LDL gesunder Probanden in diesen Pionierversuchen jedoch keine
vergleichbaren Auswirkungen zeigte, legt nahe, dass nicht der LDL-Partikel selbst,
sondern dessen Modifikationen für die Endothelfunktion maßgeblich verantwortlich
sein müssen [44,45].
2.5 Fragestellung und Zielsetzung der Arbeit
Die gezeigten deskriptiven Ergebnisse unserer Arbeitsgruppe zeigen deutliche
Effekte von carbamyliertem LDL auf die Endothelfunktion in isolierten Aortenringen
ex vivo. Die zugrundeliegenden molekularen Mechanismen wurden bislang allerdings
nicht charakterisiert.
Das Ziel dieser Arbeit soll daher die Bearbeitung der folgenden Fragestellungen sein:
1. Wie beeinflusst cLDL die endotheliale Homöostase in vitro und in vivo?
2. Welche molekularen Mechanismen liegen den Effekten von cLDL auf das
Endothel zugrunde?
3. Welche klinische Relevanz haben diese Ergebnisse? Stellt carbamyliertes
LDL einen Prädiktor für das Auftreten kardiovaskulärer Ereignisse bei
Nierenkranken dar?
203. Material
3.1 Antikörper
3.1.1 Primärantikörper
Name Spezies Hersteller Verdünnung Diluent Inkubation Einsatz
Anti-Carbamyl Lysine goat Academy Bio- 1:200 Diluent 1h RT FLISA
(CBL) Medical (FLISA)
(CBL 30S-G1a)
anti-eNOS (pS1177), mouse BD Transduction 1:2.000 5% BSA ü.N. WB
Phospho-Specific Lab (#612393)
anti-eNOS (pT495), mouse BD Transduction 1:2.000 5% BSA ü.N. WB
Phospho-Specific Lab (#612706)
anti-eNOS III mouse BD Transduction 1:1.000 5% BSA 1h RT IF
Lab (#610297)
anti-GPX rabbit Millipore (AB 1:1.000 5% BSA 1h RT IF
5010)
GAPDH (6C5) mouse Millipore 1:20.000 5% milk 1h RT WB
(MAB374)
Phospho-p38 MAPK rabbit Cell Signaling 1:1.000 5% BSA ü.N. WB
(Thr180/Tyr182) (#9211)
total-p38 MAPK rabbit Cell Signaling 1:1.000 5% BSA ü.N. WB
(#9212)
3.1.2 Sekundärantikörper
Name Spezies Hersteller Verdünnung Diluent Inkubation Einsatz
Alexa Fluor 488 Anti- donkey Invitrogen 1:1.000 1% BSA 1h RT IF
mouse IgG1 (A21202)
Alexa Fluor 594 Anti- donkey Invitrogen 1:1.000 1% BSA 1h RT IF
rabbit IgG (H+L) (A21207)
Anti-Goat IgG IRDye donkey LI-COR 1:500 Diluent 1h RT FLISA
800CW Biosciences (FLISA)
Anti-Goat IgG, HRP donkey Santa cruz 1:4.000 5% milk 1h RT WB
(sc-2020)
ECL Anti-Rabbit IgG, donkey GE Healthcare 1:10.000 5% milk 1h RT WB
HRP (NA934)
ECL Anti-Mouse IgG, sheep GE Healthcare 1:20.000 5% milk 1h RT WB
HRP (NA931)
213.2 Kits und siRNA
Produkt Hersteller
N-TER Nanoparticle siRNA Sigma Aldrich, St. Louis, MO, USA
Transfection System
scrambled small interfering GAPDH Sigma Aldrich, St. Louis, MO, USA
(5'-GGUUUACAUGUUCCAAUAU[dT][dT]-3')
siRNA against LOX-1 Sigma Aldrich, St. Louis, MO, USA
(5'-GAAUUUGAAGGCUCUGGAA[dT][dT]-3')
3.3 Medien, Lösungen, Puffer
Medien, Lösungen, Puffer Zusammensetzung
Block-Puffer (FLISA) PBS
1% (w/v) BSA
Block-Puffer (WB) Waschpuffer (WB)
5% (w/v) BSA oder Magermilchpulver
Diluent (FLISA) TBS
0,05% (v/v) Tween-20
0,1% BSA
pH 7,4
EBM-Starvationmedium EBM (Lonza, # CC-3121)
0,5% (v/v) FBS
EGM2-Gefriermedium EGM2-Vollmedium
10% (v/v) DMSO
EGM2-Transfektionsmedium EBM-2 (Lonza, # CC-3156)
EGM-2 Single Quots (Lonza, # CC-4176)
10% (v/v) FBS
kein Gentamicin
EGM2-Vollmedium EBM-2 (Lonza, # CC-3156)
EGM-2 Single Quots (Lonza, # CC-4176)
10% (v/v) FBS
Elektrophoresepuffer Aqua dest.
25 mM Tris
192 mM Glycin
0,1% (w/v) SDS
pH 8,3
Fibronectin Gebrauchslösung DMEM + Glucose 1 g/l; + L-Glutamin; - L-Pyruvat
1% (w/v) Fibronectin
22KBr-Dichtelösung 1,063 g/cm3 Aqua dest.
94,26 g/l KBr
KBr-Dichtelösung 1,210 g/cm3 Aqua dest.
337,36 g/l KBr
KHB-Chelatorenlösung Krebs HEPES Puffer
5 µmol/l DETC
25 µmol/l DFO
Lämmli-Puffer (10x) Aqua dest.
50% (v/v) 1 M Tris (pH 6,8)
20% (v/v) Glycerol
2 g/l Bromphenolblau
200 g/l SDS
10% (v/v) β-Mercapto-Ethanol
LDL-Puffer Aqua dest.
0,9% (w/v) NaCl
0,05 mM Natrium-EDTA
Lysepuffer (WB) Aqua. dest
50 mmol/l Tris (pH 7,5)
150 mmol/l NaCl
1 mmol/l EDTA
0,5% (v/v) IGEPAL CA-630
0,1% (w/v) 1 M DTT
10 µg/ml Aprotinin
10 µg/ml Leupeptin
0,1 mmol/l Na3VO4
1 mmol/l NaF
1 mmol/l PMSF
Lysepuffer (eNOS-Entkopplung) Aqua dest.
50 mmol/l Tris (pH 7,5)
150 mmol/l NaCl
1 mmol/l EDTA
0,5% (v/v) IGEPAL CA-630
10 µg/ml Aprotinin
10 µg/ml Leupeptin
0,1 mmol/l Na3VO4
1 mmol/l NaF
1 mmol/l PMSF
TBS (10x) Aqua dest.
24 g/l Tris
80 g/l NaCl
Transferpuffer (10x) Aqua dest.
390 mM Glycin
480 mM Tris
0,037% (w/v) SDS
23Transferpuffer (1x) Aqua dest.
10% (v/v) Transferpuffer (10x)
20% (v/v) Methanol
Waschpuffer (FLISA) PBS
0,05% (v/v) Tween-20
Waschpuffer (WB) Aqua dest.
10% (v/v) TBS (10x)
0,1% (v/v) Tween-20
3.4 Geräte
Produkt Hersteller
Anästhesie-System Fluovac Harvard Apparatus, Holliston, MA, USA
- mit IMS Fluosorber Harvard Apparatus, Holliston, MA, USA
CO2-Inkubator - Serie CB Binder, Tuttlingen
CCD-Kamera Fusion Fx7 peQLab Biotechnologie GmbH, Erlangen
Dichtemessgerät Density Meter DMA 500 Anton Paar GmbH, Graz, Österreich
ESR-Spektrometer e-scan Bruker BioSpin GmbH, Rheinstetten
Fluoreszenzmikroskop Biozero BZ-8100 Keyence, Ōsaka, Japan
mit Objektiv PlanApo 40x Nikon, Tokyo, Japan
mit Objektiv PlanFluor ELWD DM 20xC Nikon, Tokyo, Japan
Metallblockthermostat BIO-1V VLM GmbH, Bielefeld
Microplate Reader sunrise-basic Tecan, Männedorf, Schweiz
Mikrozentrifuge PicoFuge II Stratagene, CA, USA
Mini-PROTEAN Tetra Cell Elektrophorese- Bio-Rad Laboratories, Hercules, CA, USA
System mit PowerPac Basic Power Supply
Odyssey Sa Infrared Imaging System LI-COR Biosciences, Bad Homburg
Pipettierhilfe Pipetman Gilson International, Limburg an der Lahn
Pipettierhilfe Pipetus-akku Hirschmann Laborgeräte, Eberstadt
Pipettierhilfe Swift PET Abimed Analysen-Technik, Langenfeld
Quick-Seal Cordless Tube Topper 7700 Beckman Coulter, Brea, CA, USA
Semi-Dry Blotter TE 77 XP Hoefer, San Francisco, USA
Multipipette Eppendorf, Hamburg
Thermomixer comfort Eppendorf, Hamburg
Thermomixer ThermoStat plus Eppendorf, Hamburg
Tube Slicer Beckman Coulter, Brea, CA, USA
Vakuumpumpe EcoVac schuett-biotech, Göttingen
Vortex REAX 2000 Heidolph Instruments, Schwabach
24Waagen:
Präzisionswaage PCB 100-3 (0,001g) Kern & Sohn GmbH, Balingen-Frommern
Semi-Mikrowaage R 160P-D1 (0,00001g) Sartorius AG, Göttingen
Wipptisch Rocker PMR-30 Grant Instruments, Cambridgeshire, UK
Wipptisch ST5 Cat Fröbel Labortechnik GmbH, Lindau
Zentrifugen:
Heraeus Fresco 17 Centrifuge Thermo Scientific, Waltham, MA, USA
Heraeus Megafuge 1.0R Heraeus instruments, Osterode
Optima XPN 90 Ultracentrifuge Beckman Coulter, Brea, CA, USA
mit Ultrazentrifugenrotor Type 70.1 Ti
3.5 Verbrauchsmaterialen
Produkt Hersteller
Aspirationspipette (2 ml) Sarstedt, Newton, NC, USA
BD Falcon Culture Slides (8 Kammer) BD Biosciences, Heidelberg
Deckgläser (50x24 mm) Carl Roth, Karlsruhe
Eppendorf-Mikroreaktionsgefäß (1,5 ml) Sarstedt, Newton, NC, USA
Falcon-Zentrifugationsröhrchen (50 ml) Greiner-bio.one, Frickenhausen
Gel saver Tip II (200 µl) Starlab, Hamburg
Hämatokrit-Versiegelungskitt VWR International GmbH, Darmstadt
Immobilon-P Transfer-Membran Merck Millipore, Billerica, MA, USA
(Porengröße 0,45 µm)
Microplatte (96-well) Microlon 200 Greiner-bio.one, Frickenhausen
(Med. Bindung, U-Boden)
Microvette CB 300 Sarstedt, Newton, NC, USA
Mikro-Kapillaren Hirschmann Laborgeräte, Eberstadt
Mikrotiterplatte Nunc F Maxisorb Thermo Fisher Scientific, Waltham, MA,
(96 well, flat bottom) USA
Neubauer "improved" Zählkammer Biochrom AG, Berlin
Quick Seal Centrifuge Tubes (16x76 mm) Beckman Coulter, Brea, CA, USA
serologische Pipette Cellstar (10/ 20 ml) Greiner-bio.one, Frickenhausen
Spitzen für Multipipette (50/ 100 µl) VWR International GmbH, Darmstadt
TipOne Filterspitzen Starlab, Hamburg
(10/ 20/ 100/ 200/ 1000 µl)
Ultrafree-Mc Centrifugal Filter Units Merck Millipore, Billerica, MA, USA
(0,22 µm) GV durapore
Whatman-Papier Schleicher&Schuell Bioscience, Dassel
Zellkulturflasche (175 cm2) Sarstedt, Newton, NC, USA
25Zellkulturflasche Cellstar (25,75 cm2) Greiner-bio.one, Frickenhausen
Zellkulturplatte (12-well) Greiner-bio.one, Frickenhausen
Zellkulturplatte (6-well) TPP Switzerland, Trasadingen, Schweiz
Zellschaber SPL Life sciences, Korea
3.6 Chemikalien und sonstige Reagenzien
Chemikalien, sonstige Reagenzien Hersteller
2-Propanol VWR International GmbH, Darmstadt
Acrylamid (30%, Rotiphorese Gel A 30) Carl Roth, Karlsruhe
APS Sigma Aldrich, St Louis, MO, USA
Aprotinin Sigma Aldrich, St Louis, MO, USA
Aqua dest. Apotheke des Universitätsklinikums des
Saarlandes
BCNU Sigma Aldrich, St Louis, MO, USA
Bio-Rad Protein Assay Farbstoff-Konz. Bio-Rad, Hercules, CA, USA
Bromphenolblau AppliChem, Darmstadt
BSA VWR International GmbH, Darmstadt
Captopril Sigma Aldrich, St Louis, MO, USA
CMH Noxygen, Elzach
DETC Noxygen, Elzach
DFO Noxygen, Elzach
DMEM Low Glucose Gibco/Life Technologies, Carslbad, CA, USA
(+ L-Glutamin; - L-Pyruvat)
DMSO Sigma Aldrich, St Louis, MO, USA
DPI Sigma Aldrich, St Louis, MO, USA
Eisensulfat (FeSO4) Sigma Aldrich, St Louis, MO, USA
Essigsäure Sigma Aldrich, St Louis, MO, USA
Ethanol Sigma Aldrich, St. Louis, MO, USA
FBS Gibco/Life Technologies, Carslbad, CA, USA
Fibronectin Sigma Aldrich, St. Louis, MO, USA
Glycin Carl Roth, Karlsruhe
HBSS (- CaCl2; - MgCl2) Gibco/Life Technologies, Carslbad, CA, USA
IGEPAL CA-630 Sigma Aldrich, St Louis, MO, USA
Isofluran Baxter International, Deerfield, IL, USA
Isopropanol Sigma Aldrich, St. Louis, MO, USA
KBr AppliChem, Darmstadt
KHB Noxygen, Elzach
26Kaliumcyanat (KOCN) Sigma Aldrich, St Louis, MO, USA
L-Arginin Sigma Aldrich, St Louis, MO, USA
L-Cystein Sigma Aldrich, St Louis, MO, USA
L-Histidin Sigma Aldrich, St Louis, MO, USA
L-Lysin Sigma Aldrich, St Louis, MO, USA
L-NAME Sigma Aldrich, St Louis, MO, USA
L-Serin Sigma Aldrich, St Louis, MO, USA
L-Threonin Sigma Aldrich, St Louis, MO, USA
Leupeptin Sigma Aldrich, St Louis, MO, USA
Luminata Forte Western HRP Substrat Merck Millipore, Billerica, MA, USA
Magermilchpulver AppliChem, Darmstadt
Methanol VWR International GmbH, Darmstadt
NaF Sigma Aldrich, St Louis, MO, USA
Natrium-EDTA Sigma Aldrich, St Louis, MO, USA
Natriumorthovanadat (Na3VO4) Sigma Aldrich, St Louis, MO, USA
Natriumphosphat (Na3PO4) Sigma Aldrich, St Louis, MO, USA
Pageruler Plus Prestained Protein ladder Thermo Fisher Scientific, Waltham, MA, USA
PBS (- CaCl2, - MgCl2) Gibco/Life Technologies, Carslbad, CA, USA
PFA Sigma Aldrich, St Louis, MO, USA
PMSF Carl Roth, Karlsruhe
RNAse freies H2O Sigma Aldrich, St Louis, MO, USA
SB202190 Sigma Aldrich, St Louis, MO, USA
SDS Carl Roth, Karlsruhe
Taurin Sigma Aldrich, St Louis, MO, USA
TEMED AppliChem, Darmstadt
Trichloressigsäure Sigma Aldrich, St Louis, MO, USA
Tris Base Thermo Fisher Scientific, Waltham, MA, USA
Triton X-100 Sigma Aldrich, St Louis, MO, USA
Trypanblau (0,4%) Invitrogen/Life Technologies, Carlsbad, CA,
USA
Trypsin-EDTA (0,05%) Sigma Aldrich, St Louis, MO, USA
Tween-20 AppliChem, Darmstadt
Vectashield Mounting-Medium mit DAPI Vector Labs, Burlingame, CA, USA
β-Mercapto-Ethanol Sigma Aldrich, St Louis, MO, USA
273.7 Analyse- und Statistikprogramme Alle Auswertungs- und Statistikprogramme wurden unter den Betriebssystemen Microsoft Windows XP respektive Mac OSX 10.6.8 ausgeführt. Dabei dienten MS Office 2007, MS Office mac2011, SPSS 20.0 und Graph Pad Prism 6.0 der statistischen Erhebung und Diagrammerstellung Zusätzlich wurde zur Bedienung einiger Geräte spezielle Software verwendet, wie beispielsweise das Programm Fusion zur Belichtung und Aufnahme von Western Blot-Membranen mit der CCD- Kamera Fusion Fx7, während die integrierte Analysesoftware der anschließenden Densitometrie diente. Die Auswertung des Carbamyl-Lysin-FLISA mit dem Odyssey Sa Infrared Imaging System (Fa. LI-COR Biosciences) erfolgte mit der zugehörigen Software Image Studio, während ESR-Spektren mittels Software Bruker e-scan Research aufgezeichnet wurden. Weiterhin wurde die BioZero Observation Application zur Anfertigung von Bildern mittels Fluoreszenzmikroskop Biozero BZ- 8100 (Fa. Keyence) benutzt und die Daten-Analyse-Software Magellan 6.0 zur Konzentrationsbestimmung mit dem Microplate Reader sunrise-basic (Fa. Tecan). 28
4. Methoden 4.1 LDL-Isolation, -Analyse und -Modifikation 4.1.1 LDL-Isolation mittels Ultrazentrifugation Die Isolation von LDL aus dem Serum von Patienten und gesunden Probanden erfolgte mittels Dichtegradienten-Ultrazentrifugation, wie zuvor durch Havel RJ et al. beschrieben [46]. Bei dieser Technik macht man sich zu Nutze, dass die LDL- Fraktion der Lipoproteine eine Dichte zwischen 1,019 g/cm3 und 1,062 g/cm3 aufweist, während die Dichte der HDL-Fraktion zwischen 1,063 g/cm3 und 1,210 g/cm3 liegt. 4.1.1.1 Langzeitprotokoll Jegliches, zur weiteren ex vivo-Carbamylierung benötigte LDL gesunder Probanden wurde mittels eines viertägigen Langzeitprotokolls isoliert, welches ebenso die Möglichkeit bietet, in einem zusätzlichen Arbeitstag HDL zu isolieren. Folgende Abbildung stellt den Prozess der LDL-/ HDL-Isolation aus Serum mittels Ultrazentrifugation dar: Abbildung 7: Schematische Darstellung der LDL- / HDL-Isolation mittels Ultrazentrifugation. 29
Tag 1
Zunächst wurde zu 5 ml des verwendeten Patienten-Serums 8,5 ml einer KBr
Lösung mit einer Dichte von 1,006 g/cm3 gegeben. Das Serum mit der Dichtelösung
wurde dann mittels Spritze und Kanüle in die Zentrifugationsröhrchen (Quick Seal
Centrifuge Tube) verfüllt. Anschließend wurden die Zentrifugationsröhrchen
abgewogen und zum Austarieren der Ultrazentrifuge gewichtsgleiche Paare mit
maximaler Differenz von 0,02 g gebildet. Zuletzt wurden die Röhrchen mit dem
Quick-Seal Cordless Tube Topper 7700 (Fa. Beckman Coulter) verschweißt und
nach Dichtigkeitsprüfung mind. 20 h bei 59.000 RPM und 4 °C ultrazentrifugiert.
Tag 2
Nach Zentrifugation befanden sich nun im Unterstand die Lipoproteine HDL und LDL,
während sich die Chylomikronen und VLDL (Dichte < 1,006 g/cm3) im Überstand
befanden. Nach Schneiden der Röhrchen mit dem Tube Slicer (Fa. Beckman
Coulter) wurde der Überstand verworfen und der Unterstand (HDL + LDL)
weiterverwendet. Dazu erfolgte zunächst die Messung der Dichte im Unterstand
mittels eines Dichtemessgerätes (Fa. Anton Paar). Nach Messung der Dichte wurde
mit folgender Formel berechnet, wie viel KBr zugegeben werden musste, um eine
Zieldichte von 1,063 g/cm3 zu erzielen.
Berechnung der benötigten KBr-Menge [g]:
(
"#$%&'() *+'$,+-./' "0'11,+-./' )
!! (
2 " 34526)*+'$,+-./' )
Nach Zugabe der berechneten Menge KBr zu dem Unterstand erfolgte dessen
Suspension mittels Magnetrührer. Nach Lösung des KBr wurde die entstandene
!
Suspension zusammen mit Dichtelösung (Dichte 1,063 g/cm3) in die
Zentrifugationsröhrchen überführt, diese gewogen und verschlossen. Es schloss sich
danach ein weiterer Zentrifugationsschritt für mind. 20 h bei 59.000 RPM und 4 °C an.
Tag 3
Nach Zentrifugation befanden sich nun im Überstand die LDL-Fraktion (Dichte: 1,019
bis 1,062 g/cm3) und im Unterstand die HDL-Fraktion. Während die HDL-Fraktion
verworfen wurde, wurde im Überstand – wie bereits beschrieben – mittels KBr und
einer entsprechenden Dichtelösung erneut eine Zieldichte von 1,063 g/cm3
eingestellt, um Kontaminationen mit HDL zu entfernen. Die Berechnung der
benötigten KBr-Menge erfolgte nach Dichtemessung unter Verwendung der obigen
30Formel. Es schloss sich ein erneuter Zentrifugationsschritt für 24 h bei 59.000 RPM und 4 °C an. Tag 4 Nach erfolgter Zentrifugation befand sich die LDL-Fraktion erneut im Überstand. Dieser wurde separiert und nach Verwerfen des restlichen Röhrcheninhalts konzentriert und sterilfiltriert (siehe 4.1.2). 4.1.1.2 Kurzzeitprotokoll Die Isolation von LDL zur Datenerhebung für die klinische Studie an Prä-Dialyse- Patienten konnte aufgrund der größeren Probandenzahl nur mittels eines Kurzzeitprotokolls zügig verwirklicht werden, da hier im Gegensatz zum Langzeitprotokoll LDL innerhalb eines einzigen Tages isoliert werden konnte. Neben Analogien im allgemeinen Ablauf, waren die weiteren Konzentrations- und Sterilfiltrationsverfahren in beiden Protokollen identisch (siehe 4.1.2). Abbildung 8: Schematische Darstellung des Kurzzeitprotokolls zur LDL-Isolation mittels Ultrazentrifu- gation. Zur Probenvorbereitung und -auftrennung wurden zunächst zu 500 µl des zu verwendenden Serums nüchterner Probanden 3,3 ml einer KBr-Lösung mit einer 31
Dichte von 1,21 g/cm3 gegeben. Serum und Dichtelösung wurden resuspendiert und
die Dichte der Suspension gemessen. Anschließend wurde mit folgender Formel
berechnet, wie viel KBr zugegeben werden musste, um eine Zieldichte von 1,21
g/cm3 zu erzielen.
Berechnung der benötigten KBr-Menge [g]:
(
"#$%&'() *+'$,+-./' "0'11,+-./' )
!! (
2 " 34526)*+'$,+-./' )
Die Serum-KBr Suspension wurde dann mittels Spritze und Kanüle in die
Zentrifugationsröhrchen (Quick Seal Centrifuge Tube) verfüllt und vorsichtig mit 1
!
mM EDTA in PBS (Dichte 1,007 g/cm3) luftblasenfrei überschichtet, sodass eine
Trennschicht sichtbar wurde (Abbildung 9a). Anschließend wurden die
Zentrifugationsröhrchen abgewogen und zum Austarieren der Ultrazentrifuge
gewichtsgleiche Paare mit maximaler Differenz von 0,02 g gebildet. Zuletzt wurden
die Röhrchen mit dem Quick-Seal Cordless Tube Topper 7700 verschweißt und nach
Dichtigkeitsprüfung mind. 3 h bei 65.000 RPM und 15 °C ultrazentrifugiert.
Nach Zentrifugation befanden sich die Chylomikronen und VLDL (Dichte < 1,006
g/cm3) als weiße Schicht am oberen Ende des Röhrchen, während sich HDL
zusammen mit KBr am Boden des Röhrchens abgesetzt hatten und die LDL-Fraktion
als gelbe Schicht einem Nimbus gleich in der oberen Hälfte sichtbar war (Abbildung
9b). Nach Öffnen der Röhrchen mittels Rasierklinge wurde die gelbe LDL-Schicht mit
Hilfe einer Kanüle sorgfältig aufgenommen der restliche Röhrchen-Inhalt verworfen.
Abbildung 9: a Quick-Seal Zentrifugationsröhrchen mit Blutplasma-KBr-Suspension (untere Hälfte)
3
und scharfer Trennschicht nach Überschichtung mit 1 mM EDTA in PBS (Dichte 1,007 g/cm ) b nach
Zentrifugation klare Trennung der Lipoprotein-Fraktionen, LDL mittig als gelblicher Ring sichtbar
324.1.2 LDL-Konzentration und -Sterilfiltration Unabhängig von der Art des verwendeten Protokolls zur Isolation wurde die entnommene LDL-Fraktion abschließend in Filter-Zentrifugationsröhrchen (Amicon Ultra 4 ml Centrifuge Filter Units, 30K, Fa. Millipore) zum Entfernen des verbliebenen KBr überführt. Die Filtergefäße wurden mit LDL-Puffer (Rezept siehe 3.3) aufgefüllt und 10 min bei 4.000 RPM und 4 °C zentrifugiert. Dieser Schritt wurde zweimal wiederholt. Im nächsten Schritt wurde das im Filter verbleibende LDL resuspendiert und in Mikroreaktionsgefäße mit einem integrierten 0,22 µm großen Filter (Ultrafree- Mc Centrifugal Filter Units (0,22 µm), Fa. Millipore) pipettiert und zur Sterilisation 10 min bei 3.000 RPM und 4 °C zentrifugiert. Der Filter wurde verworfen und das Zentrifugat als Endprodukt behalten. 4.1.3 LDL-Konzentrationsbestimmung Zuletzt wurden die Proteinkonzentrationen der fertigen Proben nach dem Bradford- Assay bestimmt (siehe 4.6.2), wobei eine Standardreihe von 0-20 µg/µl BSA eingesetzt wurde. Danach wurden die Proben bis zur weiteren Verwendung bei 4 °C gelagert. Für die im Weiteren beschriebenen Assays wurde die LDL-Präparation binnen 14 Tagen nach Isolation verwendet, um eine Degradierung zur vermeiden. 4.1.4 LDL-Carbamylierung ex vivo Das zuvor aus dem Serum gesunder Spender isolierte LDL (siehe 4.1.1) wurde im nächsten Schritt ex vivo carbamyliert, gemäß eines Protokolls von Wang et al. [40]. Hierzu wurde das LDL in einem Puffer aus 50 µmol/l Natriumphosphat bei pH 7,0 auf eine Konzentration von 3 mg/ml LDL-Protein verdünnt und anschließend mit 20 mg Kaliumcyanat pro mg LDL für 4 h bei 37 °C inkubiert. In mind. fünf Waschschritten unter Verwendung der Filter-Zentrifugationsröhrchen (Amicon Ultra 4 ml Centrifuge Filter Units, 30K, Fa. Millipore) wurde das Kaliumcyanat danach wieder aus den LDL- Proben entfernt und die Proteinkonzentration erneut gemäß Bradford-Methode bestimmt (siehe 4.6.2). Das als Kontrolle zu verwendende nLDL wurde ebenso behandelt, nur wurde hierbei auf die Zugabe von Kaliumcyanat verzichtet. Für ein weiteres Experiment wurden LDL-Proben in Gegenwart von je 10 mM des L- Enantiomers der Aminosäuren Arginin, Cystein, Histidin, Lysin, Serin, Threonin oder Taurin (Fa. Sigma Aldrich) carbamyliert [47]. 33
Sie können auch lesen

















































