Die Rolle der Caspase-4 und -5 in renalen Schädigungsmodellen - OPUS 4
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Die Rolle der Caspase-4 und -5 in renalen Schädigungsmodellen
Medizinische Klinik IV, Universitätsklinikum Erlangen
Der Medizinischen Fakultät
der Friedrich-Alexander-Universität
Erlangen-Nürnberg
zur
Erlangung des Doktorgrades Dr. med.
vorgelegt von
Daniel Göth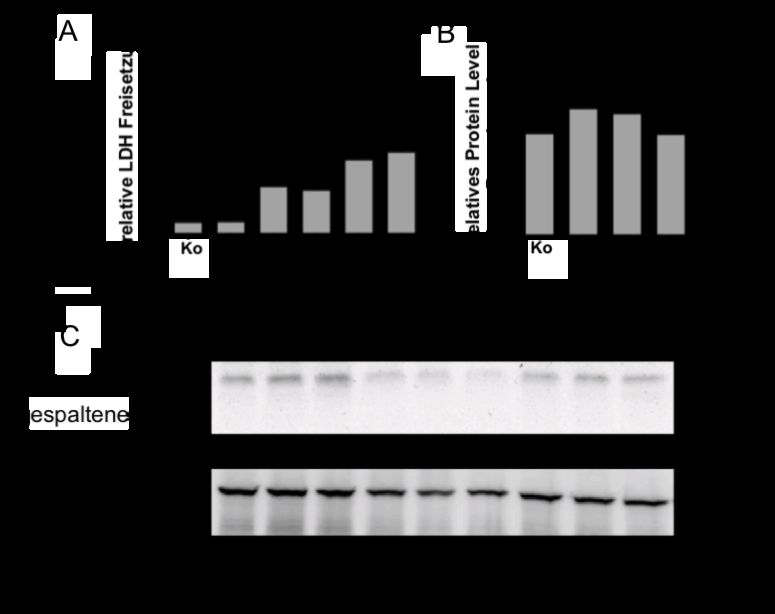
Als Dissertation genehmigt von der
Medizinischen Fakultät der Friedrich-Alexander-Universität
Erlangen-Nürnberg
Vorsitzender des Promotionsorgans: Prof. Dr. Markus F. Neurath
Gutachter: Prof. Dr. Margarete Goppelt-Strübe
Gutachter: Prof. Dr. Felix Knauf
Tag der mündlichen Prüfung: 21. September 2021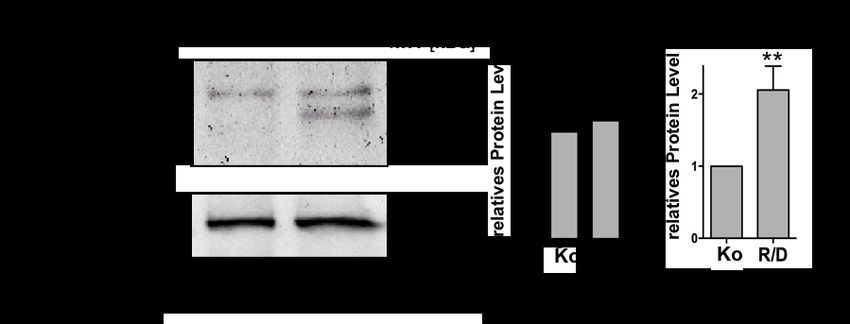
Inhaltsverzeichnis ZUSAMMENFASSUNG 1 ABSTRACT 2 EINLEITUNG 3 Caspasen 3 Inflammatorische Caspasen 3 Inflammasomen 3 Kanonische Inflammasomaktivierung 4 Caspase-11 / Nicht-kanonische Inflammasomaktivierung 4 Caspase-4/-5 6 Pyroptose 6 Interleukine 7 IL-1 Familie 7 Interleukin-1a 8 Schädigungsmodelle 9 Kalziumoxalat-Nephropathie 9 Ischämie-Reperfusionsschaden 11 EGTA 12 LPS 12 Hypertonizität/Hypernatriämie 13 Hyperalbuminurie 13 MATERIAL UND METHODEN 14 ERGEBNISSE 32 Oxalatnephropathie 32 Ischämie-Reperfusionsschaden 41 Weitere Schädigungsmodelle 44 EGTA 44 LPS 46 Hypernatriämie / Hypertonizität 49 Bovines Serum Albumin 50 H2O2 – oxidative Schädigung 50 Inserts 51 Caspase-5 52 IL-1a 52 Caspase-4-abhängige Hochregulation von pro-IL-1a 53 Nukleäre Translokation von IL-1a 55 Zusammenfassung der Hauptergebnisse 58
DISKUSSION 59 Murine Caspase-11 59 Humane Caspase-4 und Caspase-5 im Vergleich zur murinen Caspase-11 60 Aktivierung der Caspase-4 62 Schädigungsmodelle 62 Einfluss von Kalziumoxalat-Kristallen auf hpTEC 62 Die Rolle von Caspase-4 und -5 im Modell der Oxalatnephropathie 64 Ischämie-Reperfusions-Modell 64 Hyperalbuminurie 68 EGTA 68 Hypernatriämie / Hypertonizität 70 Polarisiertes Tubulusepithel 71 Zusammenfassung und Ausblick 72 LITERATURVERZEICHNIS 73 ABKÜRZUNGSVERZEICHNIS 83 ABBILDUNGSVERZEICHNIS 85 TABELLENVERZEICHNIS 86 DANKSAGUNG FEHLER! TEXTMARKE NICHT DEFINIERT. LEBENSLAUF FEHLER! TEXTMARKE NICHT DEFINIERT.
Zusammenfassung
Hintergrund und Ziele: Die murine Caspase-11 und ihre humanen Gegenstücke, Caspase-4 und
-5, sind bei Entzündungsreaktionen an der Regulation von Zelluntergang und damit verbunden
an der Freisetzung von Interleukin-1a beteiligt. Während die Aktivierung der Caspasen durch
zytosolisches Lipopolysaccharid sehr gut untersucht ist, existieren bislang nur wenige Daten zur
Rolle dieser Enzyme in anderen zellulären Schädigungsmodellen. In der Promotionsarbeit sollte
deshalb die Bedeutung der Caspasen 11, 4 und 5 in renalen Schädigungsmodellen in
Zusammenhang mit der Regulation von IL-1a untersucht werden.
Methoden: Die Expression von Caspase-11 wurde in Nierenhomogenaten aus Mausmodellen
gemessen. Die humanen Caspasen wurden in primären Tubulusepithelzellen untersucht, die aus
gesunden Anteilen von Tumornephrektomien isoliert wurden. Es wurden verschiedene in-vitro-
Modelle renaler Schädigung etabliert und analysiert: Oxalatnephropathie (Natrium-
/Kalziumoxalat), Ischämie-Reperfusionsschaden (Ringer/DMOG), LPS, EGTA, Albuminurie (BSA),
Hypertonizität (NaCl) sowie oxidative Schädigung (H2O2). In Western Blot Analysen wurden
Caspase-4, -5 sowie IL-1a bestimmt. IL-1a-Translokation in den Kern wurde mittels IL-1a-
Immunfluoreszenzfärbung sowie Western Blot von Kern-Zytosol-Fraktionierungen gezeigt.
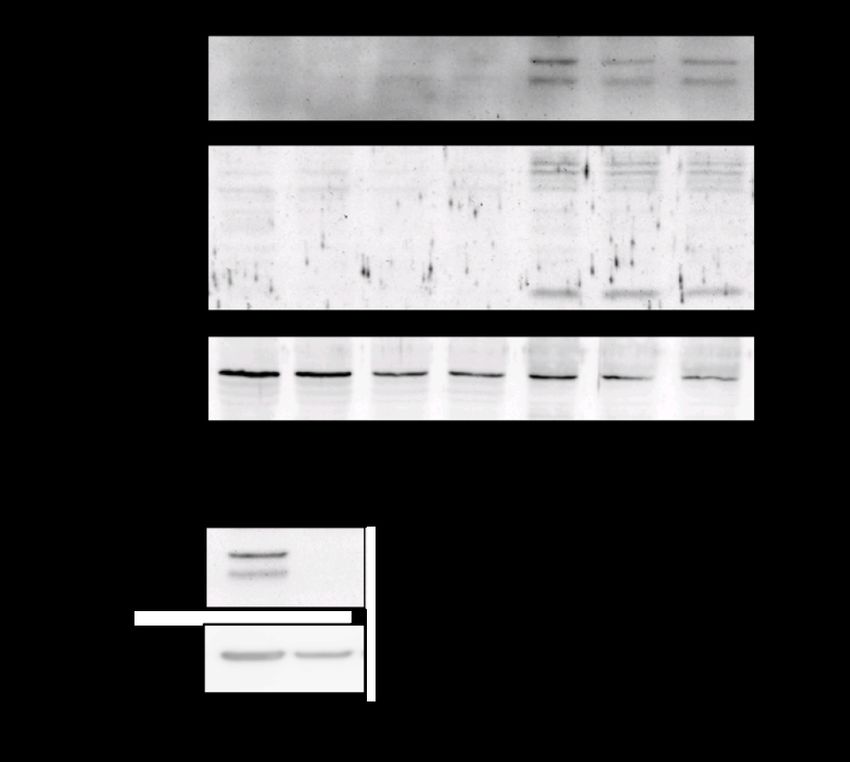
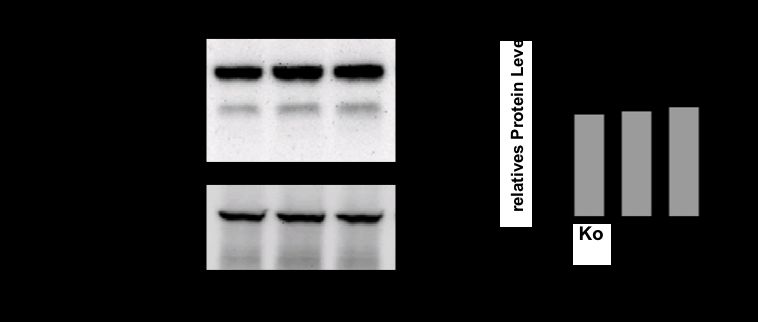
Ergebnisse und Beobachtungen: Im murinen Modell eines IRI war pro-Caspase-11 im Western
Blot hochreguliert, während das Ringer/DMOG-Modell in Lysaten aus hpTEC eine
Hochregulation der gespaltenen Caspase-4 bei 37 kDa sowie eine Hochregulation von pro-IL-1a
zeigte. Im Überstand der Zellen fand sich kein freigesetztes IL-1a. In siCaspase-4-transfizierten
hpTEC war die Hochregulation von IL-1a unter Ringer/DMOG deutlich geringer ausgeprägt.
Kern-Zytosol-Fraktionierungen sowie IL-1a-Immunfluoreszenzfärbung zeigten eine
Translokation von IL-1a in den Nukleus unter Ringer/DMOG. Im murinen Modell einer
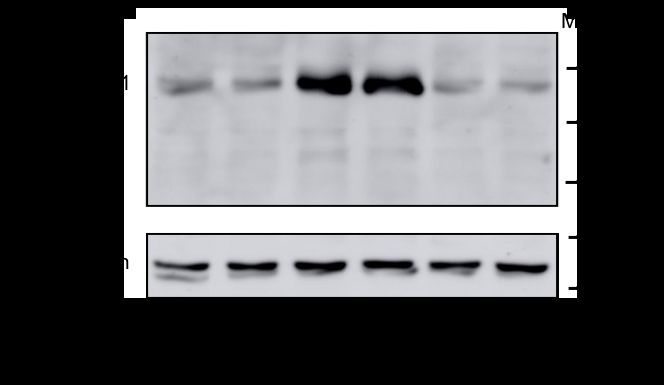
Oxalatnephropathie waren sowohl pro-Caspase-11 als auch die gespaltene Form bei 37 kDa
sowie IL-1a in den Gesamtnierenhomogenaten hochreguliert. Im Gegensatz dazu wurde
Caspase-4 in Natrium- sowie Kalziumoxalat-stimulierten hpTEC nicht reguliert. Auch in den
weiteren überprüften in-vitro Schädigungsmodellen (EGTA, LPS, NaCl, BSA und H2O2) ließ sich
keine Regulation der Caspase-4 nachweisen. Caspase-5 war in hpTEC im Western Blot nicht
nachweisbar.
Schlussfolgerungen: Nur im Modell eines renalen IRI konnte in humanen renalen Epithelzellen
eine Spaltung und vermutlich Aktivierung der Caspase-4 nachgewiesen werden. Die
Hochregulation von IL-1a war in diesem Modell Caspase-4-abhängig. Interessanterweise wurde
IL-1a nicht freigesetzt, sondern in den Kern transloziert. In Zusammenhang mit Literaturdaten
1aus andern Zellarten könnte diese Translokation bei apoptotischem Zelluntergang im IRI eine
Rolle spielen.
Abstract
Background: Murine caspase-11 and its human orthologue, caspase-4 and -5, are part of
inflammation and regulation of cell death. They are connected to release of interleukin-1a.
While there is much evidence for cytosolic LPS-activation of these caspases, to date little is
known about the role of these caspases in other damage models. This dissertation examined the
role of human caspase-4 and -5 in multiple human renal damage models and the link to
regulation of IL-1a.
Methods: We detected the expression of caspase-11 in in renal homogenates in mouse models.
Further experiments were conducted with human primary tubular epithelial cells isolated from
healthy tissue of tumornephrectomized kidneys. In-vitro damage models were oxalate
nephropathy (sodium oxalate, calcium oxalate), ischemia reperfusion injury (IRI, Ringer/DMOG),
LPS, EGTA, albuminuria (BSA), hypertonicity (NaCl) and oxidative damage (H2O2). We identified
caspase-4, -5 and IL-1a by western blot analysis. The IL-1a-translocation into the nucleus was
detected via IL-1a-immunofluorescence and western blot of fractionated nuclear and cytosolic
compartments.
Results: In the murine model of IRI western blot analysis showed upregulation of pro-caspase-
11, while we detected upregulation of cleaved caspase-4 at 37 kDa and pro-IL-1a in lysates of
the human in-vitro Ringer/DMOG-model. There was no evidence of IL-1a-release into the
supernatant. Upregulation of pro-IL-1a was reduced in sicaspase-4 transfected hpTEC. The IL-
1a-immunofluorescence and western blot of fractionated nuclear and cytosolic compartments
showed translocation of pro-IL-1a into the nucleus upon stimulation with Ringer/DMOG. In the
murine model of oxalate nephropathy both pro-caspase-11 and the cleaved form at 37kDa was
upregulated in kidney homogenates. In the sodium- and calcium oxalate stimulated hpTEC was
no regulation of caspase-4, as well as in the other damage models (EGTA, LPS, NaCl, BSA und
H2O2). There was no detection of Caspase-5 in hpTEC in western blot analysis.
Conclusions: In the in-vitro model of renal IRI we detected cleavage and presumed activation of
caspase-4. In this model, upregulation of IL-1a was caspase-4-dependend. Interestingly, IL-1a
was not released into the supernatant as predicted but translocated into the nucleus. According
to literature this could possibly play a role in apoptotic cell death in IRI.
2Einleitung
Caspasen
Caspasen sind eine Gruppe von Cysteinproteasen, deren proteolytischer Angriffspunkt C-
Terminal von Aspartat gelegen ist. Davon lässt sich aus dem Englischen der Name cysteinyl-
aspartate specific protease (Caspase) ableiten. Funktionell lassen sie sich in Apoptose-
Initiatoren, Apoptose-Exekutoren sowie inflammatorische Caspasen unterteilen (Tabelle 1)
(McIlwain et al., 2013).
Tabelle 1: Funktionelle Gliederung der humanen Caspasen
Inflammatorische Caspasen
Zu den inflammatorischen Caspasen zählen im Menschen Caspase-1, -4, -5 und -12 sowie in der
Maus Caspase-1, -11 und -12. Während jedoch die humanen Caspasen auf Chromosom 11
kodiert liegen, findet man diese murin auf Chromosom 9. Dies legt nahe, dass sie
entwicklungsgeschichtlich im Rahmen einer Genduplikation entstanden sind (McIlwain et al.,
2013). Sie alle werden als inaktive Proform synthetisiert und als Reaktion auf einen Stimulus
mittels proteolytischer Spaltung aktiviert (Davis et al., 2011).
Inflammasomen
Inflammasomen sind Multiproteinkomplexe, die sich nach einer Stimulation zytosolisch
zusammensetzen. Am besten untersucht sind die NALP bzw. NLRP-Inflammasomen. Die
Abkürzungen setzen sich zusammen aus nucleotide-binding domain, leucin-rich repeat-
containing oder NOD-like receptor (NLR) oder NACHT, LRR, PYD domains-containing protein. Im
Folgenden wird zur Vereinfachung nur vom NLRP-Inflammasom die Rede sein, auch wenn je
nach Autor NALP oder NLRP genutzt wird.
3Kanonische Inflammasomaktivierung
Die dieser Promotionsarbeit zugrunde liegenden Arbeiten der AG Knauf beschäftigten sich mit
dem NLRP3 Inflammasom in der Kalziumoxalat-Nephropathie (Knauf et al., 2013). Die
Aktivierung des Inflammasoms geschieht über zwei Schritte. Das auf einem niedrigen Level
exprimierte NLRP3 benötigt zur Hochregulation ein Priming-Signal wie den TLR-Agonisten LPS
(Lipopolysaccharid), ATP (Adenosintriphosphat) via P2X7 oder HMGB1 (High-Mobility-Group-
Protein B1). Diese können als danger associated molecular patterns (DAMPs) oder pathogen
associated molecular patterns (PAMPs) subsumiert werden (Darisipudi and Knauf, 2016). Zur
vollständigen Aktivierung ist ein zweites Signal notwendig. Bereits identifiziert sind hierfür
Pathogene bakteriellen (Duncan et al., 2009, Harder et al., 2009) oder viralen Ursprungs (Rajan
et al., 2011) und Kristalle (Darisipudi and Knauf, 2016). Diskutiert werden weiterhin reaktive
Sauerstoffspezies im Sinne eines oxidativen Schadens (Li et al., 2018).
Das aktivierte NLRP3 Inflammasom mit der apoptosis-associated speck-like protein containing
the caspase-recruitment domain (ASC) aktiviert pro-Caspase-1 zu Caspase-1. Diese spaltet
wiederum pro-IL-1b und pro-IL-18 zu den aktiven Formen IL-1b und IL-18. Durch diesen Vorgang
wird in der Zelle eine Pyroptose (siehe Seite 6) initiiert. Dabei erhöht sich die
Membranpermeabilität und die beiden proinflammatorischen Interleukine IL-1b und IL-18
werden freigesetzt.
Caspase-11 / Nicht-kanonische Inflammasomaktivierung
Der Hauptteil dieser Arbeit beschäftigt sich mit der Rolle eines Nebenpfades der beschriebenen
Inflammasomaktivierung. Den bisher am besten untersuchten Teil dieser Kaskade bildet die
inflammatorische Caspase-11. Die ungespaltene pro-Caspase-11 besteht aus einer p20- und
einer p10-Untereinheit sowie einer Single Caspase Recruitment Domain (CARD). Jeweils
zwischen den Untereinheiten und zwischen CARD und der p20-Untereinheit besteht die
Möglichkeit der Spaltung (Abbildung 1). Insgesamt weist die pro-Caspase-11 eine Größe von 43
kDa auf (Yazdi et al., 2010).
Abbildung 1: Aufbau Pro-Caspase-11
4Die murine Caspase-11 ist ausführlich beschrieben als ein Teil der nichtkanonischen NLRP3-
Inflammasomaktivierung. Sie ist in der Lage, zytosolisch LPS zu binden und toll-like Rezeptor
(TLR) unabhängig pro-Caspase-1 zu aktiver Caspase-1 zu spalten. Dies geschieht über die
Spaltung von Gasdermin D (GSDMD) (Kayagaki et al., 2015, Feng et al., 2018) und eine
Interaktion mit dem NLRP3-Inflammasom. Caspase-1 entfaltet seine proinflammatorische
Wirkung über eine Aktivierung von IL-1b und IL-18 (Abbildung 2). GSDMD kann jedoch auch
ohne NLRP3 zelluläre Pyroptose induzieren. Somit bewirkt Caspase-11 eine Caspase-1-
unabhängige Freisetzung von IL-1a und HMGB1 sowie einen pyroptotischen Zelluntergang.
Abbildung 2: Murine nicht-kanonische NLRP3 Inflammasomaktivierung über Caspase-11-Aktivierung
Eine weitere Funktion von Caspase-11 ist die Caspase-1-unabhängige Spaltung und Aktivierung
von Caspase-3 und Caspase-7. Bei beiden handelt es sich um Exekutoren-Caspasen der
Apoptose. Caspase-11 -/- Mäuse zeigen nach einer Ischämie der Arteria cerebri media und
Arteria splenica weniger apoptotische Zellen in der TUNEL-Färbung und reduzierte Caspase-3-
Aktivität zerebral sowie splenal. Aufgereinigte rekombinante Caspase-11 ist alleine in der Lage,
Caspase-3 zu spalten und zu aktivieren (Kang et al., 2000, Lee et al., 2008). Die LPS-induzierte
Apoptose in Lymphozyten scheint ebenfalls Caspase-11-abhängig zu sein (Kang et al., 2002).
Außerdem scheinen Caspase-11 -/- Mäuse vor einem LPS-induzierten tödlichen septischen
Schock geschützt zu sein (Kayagaki et al., 2011).
5Zusammenfassend ist die murine Caspase-11 Initiator von sowohl der nicht-kanonischen
Inflammasomaktivierung als auch von der Inflammasom-unabhängigen Pyroptose und
Apoptose.
Caspase-4/-5
Die humanen Äquivalente zur murinen Caspase-11 sind Caspase-4 und Caspase-5. Beide
entstanden durch eine Genduplikation von Caspase-11 (Martinon et al, 2007). Sie sind
zelltypspezifisch fähig, intrazellulär LPS zu binden und nicht-kanonisch das NLRP3-Inflammasom
zu aktivieren (Kajiwara et al., 2014, Baker et al., 2015).
Die Regulation beider Caspasen wurde bisher jedoch vor allem in monozytären Zelllinien
untersucht. In Monozyten scheint Caspase-5 im Gegensatz zu Caspase-4 auf einem konstanten
Level exprimiert zu werden und LPS eine Spaltung der Caspase-5 zu induzieren. Beide sind
jedoch nach einer LPS-Stimulation zur Freisetzung von IL-1a notwendig (Vigano et al., 2015).
Caspase-4 aktiviert, wie Caspase-11, nicht-kanonisch das NLRP3-Inflammasom nach Stimulation
von Makrophagen durch gram-negative Bakterien wie Salmonella oder Legionella (Casson et al.,
2015). In Leukozyten generell zeigen Caspase-4 und -5 unterschiedliche Expressions- und
Aktivierungsmuster (Casson et al., 2015, Knodler et al., 2014, Roberts and Yilmaz, 2015). In
Tumorzellen des kolorektalen Karzinoms hingegen sind sowohl Caspase-4 als auch -5
gleichermaßen erhöht (Flood et al., 2015).
Zu den bereits untersuchten epithelialen Zelllinien gehören in erster Linie Enterozyten (Sellin et
al., 2015), Keratinozyten (Shi et al., 2014) und das retinale Pigmentepithel (Bian et al., 2011).
Bisher unklar ist, inwieweit Caspase-4 und -5 im renalen Tubulusepithel eine Rolle spielen.
Pyroptose
Der pyroptotische Zelltod vereinigt Aspekte von Apoptose und Nekrose. Es handelt sich dabei
um einen programmierten über inflammatorische Caspasen vermittelten Zelltod, der im
Gegensatz zur Apoptose proinflammatorisch wirkt (Jorgensen and Miao, 2015). Nach
Ausbildung einer Porenformation in der Plasmamembran betroffener Zellen mit
Permeabilisierung der Membran und Konzentrationsausgleich von Ionen und Wasser kommt es
zur Schwellung mit letztendlich osmotischer Lyse der Zelle (Fink and Cookson, 2006). Die
freigesetzten proinflammatorischen Zytokine induzieren eine Leukozyteninvasion und
Inflammation (Man and Kanneganti, 2016).
6Interleukine
Ein in dieser Arbeit untersuchter Effekt der Aktivierung inflammatorischer Caspasen, hier
Caspase-4 und -5, ist die Regulation von Interleukinen. Diese sind eine heterogene Gruppe von
Zytokinen. Aus dem lateinischen inter = zwischen und dem griechischen leukos = weiß ergibt
sich die wesentliche funktionelle Gemeinsamkeit der Mitglieder dieser Gruppe. So fungieren sie
als Botenstoffe zwischen Leukozyten wie B- und T-Lymphozyten, Monozyten und Makrophagen.
Es handelt sich hierbei jedoch um eine eher historische Beschreibung. Heute ist bekannt, dass
sie darüber hinaus der Kommunikation zwischen Epithel, Endothel und bindegewebigen Zellen,
unter anderem Fibroblasten und Chondrozyten, untereinander sowie mit Leukozyten dienen.
(Murphy et al., 2016)
Relativ wenig ist bisher über die Bildung und funktionelle Bedeutung von Interleukinen im
renalen Tubulusepithel bekannt. Beispiele hierfür sind unter zellulärem Stress gebildetes IL-6
mit Induktion einer Inflammation und Fibrose (Su et al., 2017) sowie die Hochregulation von IL-
34 nach Ischämie-Reperfusionsschaden und nachfolgender Infiltration von Neutrophilen und
Makrophagen (Sanchez-Nino et al., 2016).
IL-1 Familie
Die Interleukin-1 Familie ist eine Gruppe aus 11 Zytokinen. Abhängig von der Größe und Länge
ihres Precursors werden diese in Untergruppen eingeteilt (Tabelle 2) (Palomo et al., 2015, Sims
and Smith, 2010). Allen Mitgliedern dieser Gruppe ist gemein, dass sie proinflammatorische
Prozesse initiieren und einen eigenen Rezeptor auf den entsprechenden Zielstrukturen besitzen
(Netea et al., 2015).
Tabelle 2: Funktionelle Gliederung der IL-1-Familie
7Interleukin-1a
Interleukin-1a (IL-1a) ist Teil der IL-1 Familie sowie der IL-1 Subfamilie. Es existiert als 31 kDa
große Proform, welche mittels der Protease Calpain in die kleinere C-terminal gelegene 17 kDa
Form und die N-terminale Proform (NTP) gespalten werden kann (Abbildung 3) (Kobayashi et
al., 1990, Carruth et al., 1991). Beide Formen sind funktionell aktiv. N-terminal besitzt pro-IL-1a
das sogenannte nucleus localization signal (NLS) (Wessendorf et al., 1993). Dieses ermöglicht
über einen bisher noch unbekannten Mechanismus eine Translokation von pro-IL-1a oder NTP
in den Nukleus. IL-1a-NTP kann anschließend proteasomal abgebaut werden (Ainscough et al.,
2014).
Abbildung 3: Aufbau, Spaltung und Aktivierung von IL-1⍺
IL-1a wird konstitutiv von vielen Zellreihen exprimiert. Hierzu gehören Epithelien wie
Keratinozyten (Ansel et al., 1988) oder Enterozyten, Pneumozyten, Hepatozyten, Endothel
(Garlanda et al., 2013) und immunkompetente Zellen wie T-Zellen, Monozyten und
Makrophagen (Afonina et al., 2015). Neben einer konstanten Expression im steady state
existieren Stimuli wie Wachstumsfaktoren, proinflammatorische Zytokine und Stressoren,
beispielsweise oxidativer Stress oder Aktivierung von Toll-like-Rezeptoren (TLR).
Hinsichtlich Expressionsmuster, -menge und regulierender Faktoren von IL-1a unterscheiden
sich die diversen Zelltypen voneinander. Während die im vorherigen Absatz erwähnten
Entitäten gut beschrieben sind, ist wenig für renales Tubulusepithel bekannt.
Funktionell gehört IL-1a zur Gruppe der „dual function Zytokine“ (Luheshi et al., 2009, Rider et
al., 2013). Einerseits beeinflusst es nach der Translokation in den Nukleus die Transkription
diverser Gene. Andererseits fungiert es als proinflammatorisches Zytokin. Als unkonventionell
sezerniertes Protein wird es unabhängig vom Golgi-Apparat direkt in das Zytoplasma
synthetisiert (Monteleone et al., 2015). Somit ist es unter physiologischen Bedingungen stets
zytoplasmatisch vorhanden und wird erst bei unkontrolliertem Untergang der Zelle mit Verlust
8der membranösen Barrierefunktion in das umgebende Milieu freigesetzt, wo es eine sterile
Inflammation induziert (Gabay et al., 2010). Damit gehört es zur Gruppe der Alarmine. Die
wichtigsten Formen des hierbei relevanten Zelltodes sind die Nekrose sowie die Pyroptose.
Seine Wirkung entfaltet IL-1a über eine Bindung des membranständigen IL-1R1-Rezeptors auf
Effektorzellen (Stylianou and Saklatvala, 1998). Interessanterweise sind beide Formen, pro-IL-
1a und die gespaltene 17 kDa große Form, funktionell aktiv und binden mit unterschiedlicher
Affinität an den IL-1R1-Rezeptor. Im Falle eines kontrollierten Zelluntergangs wie der Apoptose
transloziert IL-1a in den Nukleus. Auf diese Weise wird eine Freisetzung von IL-1a und somit
eine Entzündungsinduktion verhindert. Weiterhin interagiert es nukleär, gebunden an
Chromatin (Lamacchia et al., 2013, Cohen et al., 2010), mit Transkriptionsfaktoren wie p300
oder Gcn5 (Werman et al., 2004) und induziert proinflammatorische Kaskaden, Proliferation (Hu
et al., 2003), Migration und Zellseneszenz (Werman et al., 2004). Außerdem existiert neben der
sezernierten eine membrangebundene Form, die ebenfalls direkt mit IL-1R interagieren kann
(Orjalo et al., 2009). Auch IL-1a-NTP kann mithilfe der NLS nach nukleär translozieren. Die
funktionelle Bedeutung ist noch nicht genau verstanden.
Schädigungsmodelle
Untersucht wurde die Rolle der inflammatorischen Caspasen 4, 5 und 11 sowie der Einfluss auf
IL-1a in renalen Schädigungsmodellen. Folgende Modelle mit Hinweis auf eine Beteiligung
dieser Caspasen wurden überprüft.
Kalziumoxalat-Nephropathie
Kalziumoxalat ist Bestandteil von 70% aller Nierensteine (Daudon et al., 2015). Im Kindesalter
ist die Nephrolithiasis in etwa 30 % der Fälle Ursache für ein akutes Nierenversagen (Jamal and
Ramzan, 2004). Im Erwachsenenalter ist dies mit 1-2 % deutlich geringer, aber dennoch relevant
(Organ and Norman, 2011). Ein Übergang in eine chronische Nierenerkrankung (CKD) ist
möglich.
Abhängig vom Alter der Patienten kann man die Oxalatnephropathie unterschiedlichen
Ätiologien zuordnen. So unterscheidet man die primäre Hyperoxalurie mit angeborenen
Enzymdefekten im hepatischen Glyoxalat-Stoffwechsel von der sekundären Hyperoxalurie.
Diese kann diätetisch durch eine chronisch erhöhte Aufnahme bedingt sein. Aber auch
Erkrankungen des Gastrointestinaltraktes wie Morbus Crohn, Morbus Hirschsprung oder
Zystische Fibrose bewirken eine gesteigerte Aufnahme von Oxalat ebenso wie stattgehabte
9bariatrische Operationen oder Ileozökalresektionen (Nazzal et al., 2016). Weiterhin führt eine
Intoxikation mit Ethylenglykol zur Bildung von Oxalat (Pomara et al., 2008).
Übersteigt die Konzentration von Kalziumoxalat (Abbildung 4) einen kritischen Wert, fällt dieses
aus und bildet ausgehend von einem Kristallisationskeim Kalziumoxalatkristalle (Kolbach-
Mandel et al., 2015).
Abbildung 4: Strukturformel Kalziumoxalat
Klinisch tritt neben der Bildung von Nierensteinen, Nephrokalzinose und progredientem
Nierenfunktionsverlust eine Ablagerung von Kalziumoxalatkristallen in Knochen, Herz und
peripheren Nerven auf. Dies beschränkt sich jedoch hauptsächlich auf Fälle von primärer
Hyperoxalurie (Hoppe, 2012).
Oxalatnephropathie und NLRP3
Die Ablagerung von Oxalatkristallen sowie deren Interaktion mit renalem Tubulusepithel
bewirken zum einen eine massive Schädigung des betroffenen Epithels. Zum anderen induzieren
Kristallopathien generell und Kalziumoxalat im Speziellen eine Entzündungsreaktion (Rubio et
al., 2009). Es kommt zur Immigration von Lymphozyten, Monozyten beziehungsweise
Makrophagen und Granulozyten (Wilson et al., 2018). Bei längerfristig bestehendem Steinleiden
fibrosiert die Niere zunehmend und es kommt zur progredienten Funktionseinschränkung und
einer CKD (Ermer et al., 2016). Es konnte bereits ein Zusammenhang zwischen dem renalen
Schaden, der Entzündung und dem NLRP3-Inflammasom gezeigt werden. NLRP3 -/- Mäuse sind,
gemessen an der glomerulären Filtrationsrate (GFR) und Blut-Harnstoff-Stickstoff-Werten
(BUN), besser vor Nierenversagen und Funktionsverlust geschützt und haben eine geringere
Mortalität (Knauf et al., 2013). In-vitro Versuche mit murinen Monozyten, Makrophagen und
dendritischen Zellen zeigen hier eine reduzierte Freisetzung von IL-1b und IL-18 sowohl unter
Caspase-1- als auch Caspase-11-Knockdown.
10Unklar bleibt zum einen die Frage, inwieweit hierbei der nicht-kanonische Signalweg via
Caspase-4/-5/-11 im Menschen eine Rolle spielt und zum anderen ob neben den genannten
immunkompetenten Zellen auch das Tubulusepithel Caspase-4/-5/-11-abhängig auf
Oxalatkristalle reagiert. Daten aus anderen vergleichbaren Erkrankungen wie der renalen
Cystinose zeigen bereits eine entsprechende Beteiligung (Sansanwal et al., 2010).
Ischämie-Reperfusionsschaden
Der renale Ischämie-Reperfusionsschaden (ischemia reperfusion injury, IRI) ist einer der
führenden Gründe für das akute Nierenversagen (Xue et al., 2006). Er tritt auf nach einer Phase
reduzierter Blutversorgung gefolgt von einer Reperfusion. So handelt es sich beispielsweise bei
einer Nierentransplantation, einem Herzkreislaufstillstand mit folgender Reanimation, einer
Sepsis, einer länger andauernden arteriellen Hypotonie oder bei kardiochirurgischen Eingriffen
um Situationen, die einen renalen IRI bedingen können (Kramer et al., 2015). Die Folgen eines
renalen IRI reichen von akutem Nierenversagen, der Abstoßungsreaktion eines Transplantats
über chronischen Funktionsverlust bis hin zur terminalen Niereninsuffizienz mit
Dialysepflichtigkeit des Patienten.
Zum jetzigen Zeitpunkt sind bereits einige Bausteine der Pathophysiologie des IRI verstanden.
Hierzu gehören die Akkumulation toxischer Metabolite, die Aktivierung des Renin-Angiotensin-
Aldosteron-Systems (RAAS), eine mitochondriale Dysfunktion sowie Stress des
endoplasmatischen Retikulums (ER) (Bush et al., 2000) mit der Produktion reaktiver
Sauerstoffspezies (ROS, reactive oxygen species). Betroffenes Tubulusepithel verliert die
Zellpolarität, zytoskelettale Integrität (Molitoris et al., 1992), aber auch Zell-Zell-
Adhärenzkontake wie Tight junctions mit Desquamation der Zellen und erhöhter Permeabilität
der tubulären Epithelformation (Thadhani et al., 1996). Letztendlich mündet diese Schädigung
in Apoptose oder Nekrose. Beschrieben ist sogar eine Caspase-11-abhängige Pyroptose im
renalen Tubulusepithel (Yang et al., 2014). Gleichzeitig werden proinflammatorische Zytokine
wie IL-1b, IL-6, IL-8, TNF-a und IFN-g freigesetzt. Im Anschluss infiltrieren Leukozyten das
umliegende Gewebe und werden TLR-abhängig aktiviert (Bonventre and Zuk, 2004, Kezic et al.,
2017). Die postischämische Hochregulation des intracellular adhesion molecules ICM-1
unterstützt die Immigration von Leukozyten (Kelly et al., 1994, Kelly et al., 1996). Eine Übersicht
über die Pathophysiologie der Ischämie-Reperfusion bietet Abbildung 5.
11Abbildung 5: Pathophysiologie der Ischämie-Reperfusion
Die Bildung toxischer Sauerstoffspezies (Roberts and Yilmaz, 2015) aktiviert, zumindest im
Rahmen bakterieller gram-negativer Infektionen, Caspase-4 und -5. Unklar bleibt, ob die Bildung
von ROS im Zusammenhang mit Caspase-4 und -5 ebenfalls eine Rolle im Ischämie-
Reperfusionsschaden spielt.
EGTA
Ethylenglykoltetraessigsäure (EGTA) ist eine Polyamino-Carboxylsäure und als solche ein
Kalziumchelator. Nach der Stimulation von Tubulusepithelzellen kommt es in vitro zur
Internalisierung kalziumabhängiger Zell-Zell-Kontakte (Rothen-Rutishauser et al., 2002, Mattey
and Garrod, 1986). Innerhalb der Arbeitsgruppe Goppelt-Strübe sind hier als Zell-Zell-Kontakte
tubuläre E- und N-Cadherine gut untersucht (Goppelt-Struebe and Stroebel, 1998).
LPS
Die murine Caspase-11 wird als zytosolischer LPS-Rezeptor beschrieben. So bewirken gram-
negative intrazelluläre Erreger wie beispielsweise Legionella Caspase-11-abhängig eine nicht-
kanonische NLRP3-Aktivierung (Rathinam et al., 2012, Case et al., 2013, Broz et al., 2012). Durch
die Stimulation von Makrophagen oder dendritischen Zellen mit LPS wird eine solche Infektion
simuliert und das NLRP3-Inflammasom geprimed. In humanen Modellen werden Caspase-4 und
-5 als Äquivalent zur murinen Caspase-11 ebenfalls als LPS-Sensoren definiert (Vigano et al.,
2015). Daher handelt es sich bei der Stimulation von hpTEC mit LPS um ein weiteres renales
Schädigungsmodell zur Rolle der Caspase-4 und -5.
12Hypertonizität/Hypernatriämie
Ein hoher osmotischer Druck beziehungsweise hohe Natriumkonzentrationen steigern den
tubulären Energieverbrauch aufgrund intensivierter energieverbrauchender
Transportvorgänge. Der hierdurch erhöhte Sauerstoffbedarf bewirkt ein hypoxisches Milieu, in
dem die Tubulusepithelzellen unter Stress des endoplasmatischen Retikulums und der
Produktion von ROS geschädigt werden. Es kommt in der Folge zu einer Entzündungsreaktion
und Fibrosierung (Roson et al., 2006). Der durch eine Hypernatriämie ausgelöste oxidative Stress
aggraviert die Nierenschädigung einer Kalziumoxalat-Nephropathie im Rattenmodell (Huang
and Ma, 2015).
Hyperalbuminurie
Die chronische Albuminurie ist eine häufige Folge zahlreicher nephrologischer Erkrankungen mit
Schädigung der glomerulären Barrierefunktion wie bei der diabetischen Nephropathie mit
Glomerulosklerose (Gorriz and Martinez-Castelao, 2012). Unter physiologischen Bedingungen
werden glomerulär filtrierte Proteine wieder tubulär rückresorbiert (Hackbarth et al., 1982, Birn
and Christensen, 2006). Nach Überschreiten eines Transportmaximums fällt zunehmend
Albumin im Tubulussystem an und wirkt toxisch auf die Tubulusepithelzellen. Nach der
Akkumulation fehlgefalteter Proteine kommt es zur Sekretion proinflammatorischer
Mediatoren und oxidativem Stress des ER (Lee, 1992, Kaufman, 1999) sowie zum apoptotischen
Zelluntergang (Ohse et al., 2006). Zusätzlich scheinen intratubulär erhöhte, an Albumin
gebundene Fettsäuren den Schaden durch eine Hyperalbuminämie zu intensivieren (Kamijo et
al., 2002, van der Vusse, 2009).
13Material und Methoden
Gefördert durch das Interdisziplinäre Zentrum für Klinische Forschung (IZKF) im Klinikum der
Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU).
Material
Medium, Puffer, Gele und andere Reagenzien
Nachfolgend wird die Zusammensetzung der verwendeten Reagenzien angegeben, die nicht
bereits gebrauchsfertig erworben wurden.
Tabelle 3: Blotpuffer
TRIS 7,25 g
Glycin 3,65 g
SDS 0,47 g
VE-Wasser 1l
Tabelle 4: 10x Laufpuffer
TRIS 3,026 g
Glycin 14,32 g
SDS 1g
VE-Wasser 1l
Tabelle 5: Mowiol
Glycerin 6g
Mowiol 2,5 g
0,2M TRIS pH 8,5 12 ml
VE-II-Wasser 6 ml
Tabelle 6: PBS
KCl 0,2 g
KH2PO4 0,2 g
NaCl 8,0 g
Na2HPO4 (wasserfrei) 1,15 g
VE-Wasser 1l
14Tabelle 7: 10x TBS
TRIS 302,75 g/5 l VE-Wasser
NaCl 426 g/5 l VE-Wasser
Die Lösung wurde auf pH 7,6 eingestellt.
Tabelle 8: TBS/T
10x TBS 500 ml
Tween 20 20% 25 ml
VE-Wasser 4500 ml
Tabelle 9: SDS-Lysepuffer
4% SDS in PBS, pH6,5 100 μl/Probe
Complete 40 μl/1ml 4% SDS
Natriumorthovanadat 10 μl/1ml 4% SDS
Tabelle 10: Kern-Zytosol-Isolation Puffer A
Hepes 10 mM pH 7,9
EDTA 0,1 mM
KCL 10 mM
DTT 1 mM
Complete 40 μl/mL
Tabelle 11: Kern-Zytosol-Isolation Puffer C
Hepes 20 mM pH 7,9
NaCl 400 mM
EDTA 0,1 mM
DTT 1 mM
Complete 40 μl/ml
15Medium und Bestandteile
§ DMEM/Ham’s F-12 Medium; Biochrom
§ Fetal Bovine Serum Clone, PAA Laboratories
§ Hydrocortisone ≥98%; Sigma-Aldrich Co. LLC.
§ Insulin-Transferrin-Selenium (ITS-G); gibco® by life technologies™
§ L-Glutamin (200mM); Biochrom AG
§ Penicillin / Streptomycin, 10.000 U/ml/10.000μg/ml; Biochrom AG
§ Recombinant Human EGF, 1 mg/ml; PeproTech
§ 3,3’,5-Triiodo-L-thyronine sodium salt; Sigma-Aldrich Co. LLC.
§ Trypsin/EDTA solution (10x), 0,5%/0,2%; Biochrom AG
Antikörper
Primärantikörper
§ Anti-E Cadherin antibody [HECD-1], ab1416, 0,050 mg/ml; abcam
§ Caspase-4 (4B9), sc-56056, 100 μg/ml; Santa Cruz Biotechnology, Inc.
§ Caspase-11 (17D9), NB120-10454, 1:2.000, Novus Biologicals
§ CTGF (L-20), sc-14939, 200 μg/ml; Santa Cruz Biotechnology, Inc.
§ IL-1 alpha Polyclonal Antibody, 25 μg/150μl; proteintechTM
§ IL1A mouse monoclonal antibody, TA506853, 1:2.000, OriGene
Technologies, Inc.
§ N-Cadherin (H-63), sc-7939, 200 μg/ml; Santa Cruz Biotechnology, Inc.
§ Caspase-5 (D3G4W) Rabbit mAb, 46680; 1:1.000, Cell Signalling Technology
§ Caspase-5 p20 (H-2), sc-393346, 1:500, Santa Cruz Biotechnology, Inc.
Sekundärantikörper
§ Alexa Fluor® 488 donkey anti-goat IgG (H+L) 2 ml/ml, life technologies
§ Alexa Fluor® 488 donkey anti-mouse IgG (H+L) 2 ml/ml, life technologies
§ Alexa Fluor® 488 donkey anti-rabbit IgG (H+L) 2 ml/ml, life technologies
§ Alexa Fluor® 555 donkey anti-goat IgG (H+L) 2 ml/ml, life technologies
§ Alexa Fluor® 555 donkey anti-mouse IgG (H+L) 2 ml/ml, life technologies
§ Alexa Fluor® 555 donkey anti-rabbit IgG (H+L) 2 ml/ml, life technologies
§ Donkey anti-goat IgG-HRP, sc-2020 200μg/0,5ml, Santa Cruz Biotechnology,
Inc.
§ Western Sure HRP Goat, Anti-Rabbit IgG (H+L), 200μg/0,5ml, 926-80011, Li-
Cor GmbH Germany
§ Goat Anti-Rat IgG H&L (HRP) (ab97057), 1:10.000, Abcam
16siRNA
§ siCaspase-4, EHU156131-20UG, Sigma-Aldrich Co. LLC.
§ siGFP, SR-CL020-005, Eurogentec
Blockierlösungen
§ Bovine Serum Albumin; PAA Laboratories
§ Milchpulver; Carl Roth GmbH + Co. KG
§ Roti®-Block ready-to-use, 10x Konzentrat; Carl Roth GmbH + Co. KG
Sonstige Reagenzien
§ Ammoniumpersulfat; AppliChem GmbH
§ Bromphenolblau Standard; Fluka AG
§ Collagen from human placenta, 1 mg/ml; Sigma-Aldrich Co. LLC.
§ Collagenase, Type I, Clostridium histolyticum, Calbiochem
§ Complete, EDTA-free, 1873580, Sigma-Aldrich Co. LLC.
§ Cyclosporin A, ≥98.5% (TLC); Sigma-Aldrich Co. LLC.
§ Cytotoxicity Detection Kit (LDH), 88953; Roche Diagnostics
§ DAPI; Sigma-Aldrich Co. LLC.
§ Di-Natriumhydrogenphosphat; Carl Roth GmbH + Co. KG
§ DMOG, 89464-63-1, Biozol Diagnostica Vertrieb GmbH
§ Ethanol absolut reinst (min 99,5%); CHEMSOLUTE® Th. Geyer GmbH + Co.
KG
§ Glycerin; Carl Roth GmbH + Co. KG
§ Glycine; Merck Millipore
§ Human IL-1a ELISA MAX Deluxe, 434904, Biolegend
§ Kaliumdihydrogenphosphat; Merck Millipore
§ Lysophosphatic Acid, 5mg/ml; StressMarq Biosciences Inc.
§ LysoTracker® Red DND-99, 1mM; molecular probes® by life technologiesTM
§ Methanol reins, ACS, ISO, PH. Eur. (min. 99,7%); CHEMSOLUTE® Th. Geyer
GmbH + Co. KG
§ MitoTracker® Mitochondrion-Selective Probes; 50 μg (1 mM); Molecular
Probes® Invitrogen detection technologies
§ Mowiol® 4-88; Carl Roth GmbH + Co. KG
§ Natriumchlorid; ChemSolute®
§ Oil Red O; Sigma-Aldrich Co. LLC.
§ Oregon Green® 488 Phalloidin; Thermo Fisher Scientific
17§ PageRulerTM Prestained Protein Ladder, 10 to 180 kDa; Thermo Fisher
Scientific
§ Paraformaldehyde; Merck Millipore
§ PierceTM ECL Western Blotting Substrate; Thermo Fisher Scientific
§ 2-Propanol reins, PH. Eur. (min. 99,5 %); CHEMSOLUTE® Th. Geyer GmbH +
Co. KG
§ Ringer-Infusionslösung Ecobag, B.Braun
§ Roti®-Load 1, Proteinauftragspuffer, reduzierend; Carl Roth GmbH Co. KG
§ Rotiphorese® Gel 30 (37,5:1); Carl Roth GmbH Co. KG
§ Saint-Red, SR-1003, Synvolux Therapeutics B.V.
§ Salzsäure reinst (min. 25 %); CHEMSOLUTE® Th. Geyer GmbH + Co. KG
§ SDS Pellets; Carl Roth GmbH Co. KG
§ Sodium Oxalate ACS reagent, ≥ 99,5 %; Sigma-Aldrich Co. LLC.
§ TEMED; Carl Roth GmbH Co. KG
§ Triethyl phosphate; Sigma-Aldrich Co. LLC.
§ TRIS Pufferan®; Carl Roth GmbH Co. KG
§ Triton X-100 detergent; Bio-Rad Laboratories, Inc.
§ TWEEN® 20; Carl Roth GmbH Co. KG
Technische Geräte
§ BZ-9000 (BIOREVO), Keyence
§ ECLIPSE 80i; Nicon
§ Electrophoresis Power Supply EPS 600; Pharmacia Biotech
§ IPC High Precision Multichannel dispenser, 8 Kanäle; Ismatec/IDEX
§ Luminiscent Image Analyser LAS-1000; Fujifilm
Software
§ AIDA-BIO, Analysis Program For Biological Applications
§ ImageJ, NIH
§ Graphpad Prism 5.0
Zellkultur
Allgemeines
Sämtliche Versuche wurden zur Vermeidung von Kontaminationen mit Pilzen oder Bakterien
unter sterilen Bedingungen auf einer Sicherheitswerkbank durchgeführt. Die verwendeten
Einmalhandschuhe wurden vor jeder Benutzung mit 70%igem Ethanol desinfiziert und eine
18Einwirkzeit von 30 Sekunden eingehalten. Unter diesen keimfreien Bedingungen wurden die
Zellkulturen in einer Zellkulturschale kultiviert und zur Weiterzucht zwei- bis dreimal pro Woche
passagiert.
Zunächst wurde hierbei das Medium abgenommen und die Zellen wurden zweimal mit 10 ml
PBS gewaschen. Im Anschluss erfolgte eine Behandlung mit 5mL Trypsin-EDTA, um die Zellen für
die weitere Verarbeitung von der Schale abzulösen. Per Lichtmikroskop ließ sich die Ablösung
der Zellen vom Schalenboden qualitativ bewerten.
Da sich proximales Tubulusepithel nach 3-4 Minuten und distales nach etwa 8-10 Minuten löst,
konnte anhand des Zeitpunkts der Aufnahme der gelösten Zellen in Trypsin in ein Falcon-Tube
und Stoppen der Trypsin-Reaktion mittels Zugabe von 2,5 % FCS-Medium (Tabelle 2.X) bestimmt
werden, ob die aufgenommenen Zellen aus überwiegend distalem, proximalem oder
gemischtem Tubulusepithel bestand. Mittels Immunfluoreszenzfärbung auf N- und E-Cadherin
konnte der Anteil an distalem und proximalem Epithel quantifiziert werden. Distales Epithel
exprimiert E-Cadherin, proximales N-Cadherin.
Nach Überführung der Suspension in ein steriles Falcon-Tube wurde dieses 5 Minuten bei
Raumtemperatur und 1.000 rpm zentrifugiert. Der Überstand wurde heraus pipettiert und das
gewonnene Zell-Pellet in 8 ml 2,5 % FCS-Medium resuspendiert. Mittels Neubauer-Zählkammer
konnten 10 μl der Suspension in vier Quadranten ausgezählt, das Ergebnis gemittelt und auf die
Zellanzahl pro ml Suspension geschlossen werden (Ergebnis*10^4 Zellen/ml). Nun konnten die
Zellen sowohl für Versuche als auch für die Weiterzucht ausgesät werden.
Einfrieren von Zellen
Zum Einfrieren der Zellen wurden diese zunächst wie zuvor beschrieben gesplittet. Nach der
Zentrifugation wurde das Zellpellet in 1:1 FCS und 20 % DMSO/FCS resuspendiert. Die
Suspension wurde dann in einem Kryoröhrchen zunächst bei -80°C tiefgefroren und nach einigen
Tagen Akklimatisierung in flüssigen Stickstoff bei -196°C umgelagert. In diesem Zustand war eine
Aufbewahrung für einen längeren Zeitraum möglich.
Auftauen von Zellen
Zum Auftauen eines unter -196°C in flüssigem Stickstoff gelagerten Kryoröhrchens wurde dieses
für rund 10 Sekunden in 37°C warmes Wasser gehalten, um die Zellsuspension zunächst
19anzutauen und im Anschluss möglichst rasch mithilfe von vorgewärmtem 2,5 % FCS-Medium in
ein Falcon-Tube zu überführen. Nach 5 Minuten Zentrifugation bei Raumtemperatur und
1000rpm wurde der Überstand vorsichtig abgenommen und das Zellpellet in 2,5 % FCS-Medium
resuspendiert. Die Zellsuspension konnte so für Versuche oder die Weiterzucht verwendet
werden.
Humane primäre Tubulusepithelzellen (hpTEC)
Herkunft
Die in dieser Dissertation verwendeten hpTEC wurden in freundlicher Zusammenarbeit mit der
Abteilung für Urologie des Universitätsklinikums Erlangen unter der Leitung von Herrn Prof. Dr.
med. Bernd Wullich gewonnen. Die Zellen wurden aus Nieren im Rahmen einer
Tumornephrektomie isoliert. Im Anschluss an die operative Nierenentnahme wurde die Niere
an das Pathologische Institut des Universitätsklinikums Erlangen unter der Leitung von Herrn
Prof. Dr. med. Arndt Hartmann gesendet und dort von dem jeweils diensthabenden Pathologen
im Schnellschnittlabor entgegengenommen. Dieser entfernte maximal tumorfern etwa 10-15 ml
Nierenparenchym, welches direkt in 15ml Hank’s Balanced Salt Solution (HBSS) aufgenommen
und bei 4°C auf Eis gelagert wurde. Die Weiterverarbeitung erfolgte möglichst zeitnah nach der
Gewebeentnahme.
Präparation und Kultivierung
Die Präparation des Nierengewebes erfolgte stets unter sterilen Bedingungen auf einer
Sicherheitswerkbank. Zusätzlich wurden alle Falcon-Tubes und Pinzetten zunächst mit 70%igem
Ethanol desinfiziert. Das Nierenpräparat wurde in eine, auf einem 4°C kalten Kühlelement
stehende, sterile Glaspetrischale gegeben und einmal mit 10ml 4°C kaltem HBSS gewaschen.
Anschließend wurde das Gewebe mittels zweier Skalpelle in möglichst kleine Stücke
geschnitten, um eine maximal große Oberfläche zu generieren. Nach Aufnahme der
Nierenfragmente in 15ml 37°C vorgewärmtes HBSS wurde 1ml Kollagenase sowie 100μl DNAse
zugegeben und alles für etwa eine Stunde im 37°C warmen Wasserbad inkubiert. Während
dieser Zeit wurde die Suspension alle 10 Minuten leicht geschüttelt, um eine möglichst gute
Durchmischung aller Bestandteile zu gewährleisten.
Mithilfe eines sterilen Spritzenstempels einer 20ml Spritze sowie HBSS als Spülflüssigkeit
wurden die nun überwiegend aus dem Zellverband gelösten Zellen durch Cellstrainer gedrückt.
Das zunächst verwendete Sieb mit einer Porengröße von 100 μm diente der mechanischen
Zerkleinerung und dem Zurückhalten von Glomeruli und anderer größerer Bestandteile der
20Suspension. Das zweite Sieb mit einer Porengröße von 70 μm wurde verwendet, um
verbliebene, größere Tubulusfragmente zu entfernen.
Das passierte Nierengewebe wurde in ein steriles Falcon-Tube gegeben und 5 Minuten bei
1000 rpm und 4°C zentrifugiert. Der abpipettierte Überstand wurde verworfen und das Pellet in
10 ml Erythrolyse-Puffer (Tabelle 1.2) aufgenommen und bei Raumtemperatur 8 Minuten
inkubiert. Anschließend wurde die Erythrolyse durch Zugabe von 40 ml 4°C kaltem HBSS
gestoppt.
Es erfolgte eine erneute 5-minütige Zentrifugation bei 1000 rpm und 4°C. Nach abermaligem
Entfernen des Überstandes konnte das gewonnene Pellet in 20-30 ml 0,5 % FCS-Medium
resuspendiert und, je nach Größe des Ausgangsmaterials, in zwei bis drei 100mm Petrischalen
ausgesät und bei 37°C und 5 % CO2 im Brutschrank kultiviert werden.
Nach 1-2 Tagen wurden die nun angewachsenen Zellen dreimal mit 37°C warmem PBS
gewaschen und anschließend ein Mediumwechsel auf 0 % FCS-Medium durchgeführt. Während
der nächsten 3-4 Tage bildeten die Zellen ein Monolayer aus, konnten gesplittet und für
Versuche oder die Weiterzucht verwendet werden. Für die Versuche wurden die Zellen bis
maximal zur Passage 4 verwendet, da innerhalb der Arbeitsgruppe frühere Versuche gezeigt
hatten, dass die Zellen mit zunehmender Passagierung dedifferenzieren und es zu einem
Abstoppen des Wachstums kommt.
Für die Versuchsaussaat wurden die Zellen zunächst in 2,5 % FCS-Medium ausgesät. Am
folgenden Tag erfolgte ein Mediumwechsel auf 0 % FCS-Medium. In der Regel lagen nach einem
weiteren Tag konfluente Zellen vor, die nun je nach Versuch stimuliert werden konnten.
Tabelle 12: PTZ-Medium für hpTEC
DMEM-HAMS-F12 500 ml
Insulin-Transferrin-Selenium 1%
Penicillin / Streptomycin 1%
Glutamin 1%
T3 10 ng/ml
EGF 200 μg/ml
Hydrocortison 500 μg/ml
Tabelle 13: Erythrozyten-Lysepuffer
NH4Cl 150 mM
KHCO3 10 mM
EDTA 0,1 mM
Mit VE1-Wasser auf pH 7,2 titriert und steril filtriert
21Murines Tubuluszell-Lysat
Murine Oxalatnephropathie
Die Lysate und Gewebeschnitte des murinen Modells einer Oxalatnephropathie wurden
freundlicherweise von Prof. Dr. med. Felix Knauf (Nephrologie und Internistische
Intensivmedizin, Charité-Universitätsmedizin Berlin) zur Verfügung gestellt. Die Tiere wurden
jeweils 2 Tage oder 3 Wochen mit Oxalat-angereicherter Nahrung behandelt. Als Kontrolle
dienten Mäuse mit normaler Nahrung ohne Zusatz löslichen Oxalats. Die Gewinnung der Proben
lief gemäß Protokoll nach Knauf et al. (Knauf et al., 2013).
Muriner Ischämie-Reperfusionsschaden
Die Lysate des murinen Modells eines renalen IRI wurden freundlicherweise von Dr. med.
Gunnar Schley (Medizinische Klinik 4, Universitätsklinikum Erlangen) zur Verfügung gestellt. Die
Arteria renalis der untersuchten Tiere wurde für jeweils 10 Minuten, 20 Minuten oder SHAM-
Prozedur als Kontrolle abgeklemmt und anschließend wieder geöffnet. Nach 3 Tagen
Reperfusion wurden die Mäuse geopfert. Die gesamte Probengewinnung erfolgte nach dem
Protokoll nach Schley et al. ab (Schley et al., 2011).
Stimulatoren
Als Stimulatoren kamen die folgenden Substanzen in den angegebenen Konzentrationen zum
Einsatz:
Tabelle 14: Stimulatoren
Substanz Stock-Konzentration Finale Konzentration
Kalziumoxalat 95 mM 10 mM
Calpain-Inhibitor 20 mM 100 nM
DMOG 1 M in VE-Wasser 1 mM
EGTA 95 mM 1 mM
H2O2 3,09 μg/nl 300 μM
LPS 1 mg/ml 5 μg/ml
Natriumoxalat 200 mM 1 mM oder 2 mM
TSA 1 mg/ml 100 ng/ml
22Das Herstellen der Arbeitslösung aus der Stocklösung erfolgte durch die Verdünnung in sterilem
0 % FCS-Medium.
Fluoreszenzfärbung der Zellen
Vor Aussaat der Zellen musste pro Schale oder Well ein Deckglas mit Kollagen beschichtet
werden. Hierzu wurden zunächst 140 μl Kollagen IV + HCl (1:20 in PBS) auf ein Deckglas pipettiert
und dies mindestens 2 Stunden bei 37°C im Brutschrank inkubiert. Nach anschließendem
zweimaligem Waschen mit PBS konnte das Deckglas in eine Schale oder ein Well gelegt und die
benötigten Zellen je nach Versuchsaufbau ausgesät werden.
Nach Abschluss der Zellstimulation wurden die Zellen 10 Minuten lang bei Raumtemperatur in
PFA fixiert und dreimal jeweils 5 Minuten mit PBS gewaschen. Zum Aufschluss der
Zellmembranen inkubierten die Proben daraufhin 10 Minuten bei Raumtemperatur in 0,5 %
Triton-X-Detergenz und wurden zweimal kurz in PBS gewaschen. Nun konnte der benötigte
Primär-Antikörper nach Protokoll angesetzt und pro Probe ein Tropfen von 70μl auf den Deckel
einer 24 Well-Platte pipettiert werden. Die Deckgläser wurden mit Hilfe einer Pinzette
umgedreht und auf dem Primär-Antikörper abgelegt. Das Ganze verblieb über Nacht bei 4°C in
einer feuchten Kammer, um das Austrocknen zu verhindern.
Am nächsten Morgen wurden die Deckgläser wieder zurückgedreht und dreimal 5 Minuten in
PBS gewaschen. Entsprechend dem Primär-Antikörper wurden 70 μl Sekundär-Antikörper pro
Probe aufgebracht und eine Stunde bei Raumtemperatur inkubiert. Falls nötig konnte zur
Kernfärbung DAPI 1:2000 in PBS mit unter den Sekundär-Antikörper gegeben werden. Nach
viermaligem jeweils 5 Minuten andauerndem Waschen mit PBS wurden die Deckgläser in 10 μl
Mowiol auf einem Objektträger eingedeckelt und über Nacht bei 4°C zum Aushärten belassen.
Fluoreszenzfärbung der Gewebeschnitte
Die verwendeten Gewebeschnitte waren allesamt in Paraffin aufgenommen und bedurften
daher zunächst der Inkubation von jeweils 5 Minuten in dreimal Xylol 100 % und Ethanol 100 %
mit abnehmender Konzentration. So wurde sichergestellt, dass die Schnitte vorsichtig wieder
bewässert wurden.
Der nächste Schritt lief je nach Versuch unterschiedlich ab:
1) Die Gewebeschnitte wurden in 250 ml Citratpuffer aufgenommen, 2-4 Minuten in der
Mikrowelle bei 80 W aufgekocht und anschließend für weitere 10 Minuten bei 60 W
belassen.
232) Die Gewebeschnitte wurden in 250 ml TRS-Lösung aufgenommen und für den Zeitraum
von 10 Minuten im Schnellkochtopf bei etwa 100°C gekocht.
Anschließend wurden die gekochten Schnitte für etwa zwei Stunden auf der Werkbank zum
Abkühlen stehen gelassen. Nach 5 Minuten in VE-H2O konnten die Objektträger mit einem
fusselfreien Tuch von Flüssigkeit befreit und mittels Liquid-Blocker-Stift Kreise um die
Gewebeschnitte gezogen werden. In jeden dieser Ringe wurden etwa 70-80 μl Blockierlösung
pipettiert.
Nun blockierten die Schnitte für eine Stunde bei Raumtemperatur in einer feuchten Kammer
und wurden daraufhin dreimal 5 Minuten mit PBS bei Raumtemperatur gewaschen. Der
Primärantikörper wurde ebenfalls zu jeweils 70-80 μl in die gezeichneten Kreise pipettiert und
inkubierte über Nacht in einer feuchten Kammer bei 4°C. Nach dreimaligem Waschen für jeweils
5 Minuten mit PBS verblieb der Sekundärantikörper für eine Stunde in der feuchten Kammer bei
Raumtemperatur. Nun wurde alles abermals dreimal 5 Minuten mit PBS gewaschen, mit einem
fusselfreien Tuch getrocknet und mit Mowiol unter einem Deckglas eingedeckelt.
War es notwendig, einen Objektträger auszudeckeln, um einen weiteren Antikörper auf die
Gewebeschnitte zu geben, wurde auf den Objektträger bei Raumtemperatur PBS gegeben und
für mindestens 2-3 Stunden belassen. Anschließend wurde das Deckglas vorsichtig
abgenommen. Zur Lösung des Mowiols wurde bei Raumtemperatur PBS auf die Gewebeschnitte
pipettiert und für eine weitere Stunde inkubiert. Daraufhin wurde der neue gewünschte Primär-
Antikörper aufgetragen und es wurde nach dem jeweiligen Protokoll weiter verfahren.
Western Blot
Proteinernte
Zur Proteinernte wurde zunächst der Überstand jeder Probe abgenommen und in ein
beschriftetes Eppendorf Tube überführt. Nachdem dieses 5 Minuten bei 2.400 rpm bei
Raumtemperatur zentrifugiert wurde, konnte der Überstand in ein weiteres Tube pipettiert und
das Zellpellet verworfen werden.
Die verbliebenen adhärenten Zellen am Boden der Schalen oder Wells wurden zweimal kurz mit
PBS gewaschen und daraufhin wurde die Schale für 1 Minute schräg auf dem jeweiligen Deckel
abgestellt. Daraufhin wurde der letzte verbliebene Rest PBS abpipettiert. Anschließend
inkubierte jede Schale beziehungsweise jedes Well für 10 Minuten in 100 μl SDS-Lysepuffer
(Tabelle 9). Mittels eines Gummischabers wurden die Zellen nun mechanisch gelöst und jede
Probe in jeweils ein Eppendorf Tube gegeben.
24Alle Proben wurden nun zusätzlich bei 50% Intensität dreimal 5 Sekunden per Ultraschall lysiert
und dabei nach jedem Durchgang kurz in einer auf -20°C temperierten Kühleinheit abgekühlt.
Die Proben zentrifugierten daraufhin 5 Minuten lang bei 13.000 rpm und Raumtemperatur. Die
Überstände wurden in ein weiteres Tube überführt und das Pellet verworfen.
Proteinbestimmung nach Pierce
5 μl jeder zu bestimmenden Probe wurde in jeweils ein Tube pipettiert. Die parallel dazu
angefertigte Eichreihe bestand aus fünf Eppendorf Tubes mit 0, 5, 10, 20 und 30 μl Albumin-
Standard (Stockkonzentration: 1 mg/ml). Alle Proben inklusive der Eichreihe wurden nun auf 50
μl mit VE-Wasser aufgefüllt und es wurden jeweils 750 μl BCA-Reagenz hinzugegeben (Tabelle
15).
Alles inkubierte anschließend 30 Minuten im Wasserbad bei 37°C oder alternativ 1 Stunde bei
Raumtemperatur. Anschließend wurde die Proteinkonzentration photometrisch bei 562 nm
gemessen.
Tabelle 15: BCA-Reagenz
Reagenz A (Bicinchoninic Acid Solution) 500 μl
VE-H2O 250 μl
Reagenz B (CuSO4) 10 μl
Vorbereiten der Proben
Die gewünschte Masse an Lysat wurde anhand der jeweiligen Proteinkonzentration errechnet
und in ein Tube pipettiert. Nach Zugabe von 8 μl 4x Roti-Load wurden alle so vorbereiteten
Proben 10 Minuten lang bei 95°C gekocht und anschließend auf Eis für 3-4 Minuten herunter
gekühlt.
Überstände wurden entweder mit 100 % Ethanol oder Acetat gefällt. Zur Ethanol-Fällung
wurden 200 μl Überstand zu 800μl reinem Ethanol gegeben, die Proben gevortext und über
Nacht bei -20°C belassen. Am folgenden Morgen wurde alles 1 Stunde bei 13.000 rpm und 4°C
zentrifugiert und anschließend der Überstand abgenommen. Das Pellet trocknete nun etwa 20
Minuten unter dem Abzug, bevor 15 μl 2x Rotiload hinzupipettiert wurde und die Proben bei
95°C für 10 Minuten gekocht wurden. Auch hier wurde alles nach dem Kochen für 3-4 Minuten
auf Eis herunter gekühlt. Die Acetat-Fällung gestaltete sich leicht abweichend. Zu 200 μl
Überstand wurden 800 μl -20°C kaltes Acetat gegeben und alles über Nacht bei -20°C inkubiert.
25Nun wurden die gefällten Überstände für 15 Minuten bei 15.000 rpm bei 4°C zentrifugiert, der
Überstand abgenommen und das Pellet 20 Minuten unter dem Abzug getrocknet. Nach Zugabe
von 15 μl 2x Rotiload wurden die Proben bei 95°C 10 Minuten lang gekocht und anschließend
für 3-4 Minuten auf Eis gekühlt.
Gele gießen
Vor Beginn wurden alle zu verwendenden Glasplatten und Abstandhalter mit Spülmittel, einem
Schwamm und einem fusselfreien Tuch gründlich gereinigt, damit keine Fasern oder Fettreste
mehr auf den Utensilien zu finden waren. Anschließend wurden pro Gel eine Glasplatte, eine
Plastikplatte sowie zwei Abstandhalter zusammengesetzt und vier solcher Gießvorlagen in einer
passenden Gussbox eingespannt. Die benötigten Reagenzien wurden anschließend
zusammenpipettiert (Tabelle 16) und eine halbe Minute mit Hilfe eines Magnetrührers
durchmischt. Nach langsamem Einfüllen des Trenngels in die Gelformen wurde nach einer
Minute 1 ml 70% Isopropanol vorsichtig auf die nun leicht erhärteten Gele pipettiert. Bei
Raumtemperatur polymerisierte das Gemisch, mittels feuchtem Papiertuch unter Luftabschluss
gehalten, aus. Danach wurde das auf dem Trenngel befindliche Isopropanol abdekantiert und
das Sammelgel nach Protokoll gemischt (
26Tabelle 17). Nach der Zugabe der Lösung auf das Trenngel mussten sofort 10- oder 15-Taschen-
Kämme in das Gemisch gedrückt werden, damit das Sammelgel um die Taschen herum
auspolymerisieren konnte. Alles wurde mit einem feuchten Papiertuch abgedeckt und in einer
Plastiktüte luftdicht bei 4°C belassen.
Tabelle 16: 12% Trenngel, für 4 Gele
Acrylamid, bis 30 % 18 ml
TRIS 3M, pH 8,9 5,6 ml
SDS 10 % 0,45 ml
VE Wasser 21 ml
TEMED 67,5 µl
APS 10 % 225 µl
27Tabelle 17: Sammelgel, für 4 Gele
Acrylamid, bis 30 % 2 ml
TRIS 1M, pH 8,9 2 ml
SDS 10 % 160 µl
VE Wasser 12 ml
TEMED 24 µl
APS 10 % 200 µl
Bromphenolblau 40 µl
Gelelektrophorese
Die zuvor gegossenen Gele wurden mitsamt den Glasplatten gesäubert und in die
Elektrophoreseapparatur eingespannt. Diese wurde auf ein -20°C vorgekühltes Coolpack
gestellt. Nun wurde der 10x Laufpuffer (Tabelle 4) 1:10 mit VE-Wasser verdünnt und über die
eingespannten Gele gegeben, sodass alle Kammern komplett bedeckt waren und kein
Kurzschluss zwischen Anode und Kathode bestand. Jede Kammer wurde einmal mit 1ml
Laufpuffer gespült und alle gekochten Proben einmal kurz zentrifugiert. Je nach
Versuchsprotokoll konnten jetzt 2,5 μl Page Ruler Marker und die Proben in die Kammern
pipettiert werden. Der Start der Gelelektrophorese erfolgte bei 120 V sowie 25 mA pro Gel. Nach
etwa 1,5 Stunden waren gerade so noch alle Markerbanden auf dem Gel sichtbar und die
Phorese wurde gestoppt.
Blot-Verfahren
Zunächst wurde die PVDF-Membran zurechtgeschnitten und je nach Versuch beschriftet. Auf
diese wurde 10 ml Methanol und anschließend 40 ml Blotpuffer (Tabelle 3) gegeben und für 5
Minuten inkubiert. In den Blotpuffer wurden weiterhin pro Membran zwei Blotpapiere
getaucht. Im Blotter wurde nun von unten nach oben ein Blotpapier, die PVDF-Membran, das
Gel und wieder ein Blotpapier gestapelt. Alles war dabei von Blotpuffer benetzt und trocknete
nicht aus. Zusätzlich wurde jede dieser Schichten mithilfe eines Reagenzglases vorsichtig
ausgerollt, damit keine Luftblasen mehr vorhanden waren. Der Start des Blotters erfolgte bei
25V sowie 60mA pro Gel. Die Zeit wurde auf 1,5 Stunden gesetzt. Nach Ablauf dieser wurde das
Gel in Coomassie-Blau überführt und die Membran kurz in TBS/T gewaschen. Anschließend
wurde die Membran mindestens 1 Stunde in 20 ml Blockierlösung blockiert.
28Sie können auch lesen

















































