Aus der Klinik für Hämatologie, Hämostaseologie, Onkologie und Stammzelltransplantation der Medizinischen Hochschule Hannover Klinische Bedeutung ...
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Aus der Klinik für Hämatologie, Hämostaseologie, Onkologie
und Stammzelltransplantation
der Medizinischen Hochschule Hannover
Klinische Bedeutung von persistierender klonaler Hämatopoese
nach Chemotherapie bei Patienten mit akuter myeloischer
Leukämie
Dissertation
zur Erlangung des Doktorgrades der Medizin in der
Medizinischen Hochschule Hannover
vorgelegt von Piroska Klement
aus Gießen
Hannover 2021
Angenommen vom Senat am: 23.03.2021 Gedruckt mit der Genehmigung der Medizinischen Hochschule Hannover Präsident: Prof. Dr. med. Michael P. Manns Betreuer/in der Arbeit: Prof. ‘in Dr. med. Felicitas Thol 1. Referent/in: Prof. ‘in Dr. med. Britta Maecker-Kolhoff 2. Referent/in: Prof. ‘in Dr. rer. nat. Britta Eiz-Vesper Tag der mündlichen Prüfung: 23.03.2021 Prüfungsausschuss Vorsitz: Prof. Dr. med. Thomas Werfel 1. Prüfer/in: Prof. ‘in Dr. med. Heike Bantel 2. Prüfer/in: Prof. Dr. med. Torsten Witte
Inhaltsverzeichnis
1. Einleitung .......................................................................................................................................... 1
1.1 Akute myeloische Leukämie .................................................................................................. 1
1.1.1 Definition und klinische Symptome ............................................................................... 1
1.1.2 Epidemiologie ................................................................................................................... 2
1.1.3 Diagnose und Klassifikation ........................................................................................... 2
1.1.4 Pathogenese ..................................................................................................................... 4
1.1.5 Risikostrazifizierung ......................................................................................................... 8
1.1.6 Therapie und Prognose................................................................................................. 10
1.2 Klonale Hämatopoese unbestimmten Potentials .............................................................. 11
1.2.1 Definition .......................................................................................................................... 11
1.2.2 Epidemiologie ................................................................................................................. 13
1.2.3 Bedeutung ....................................................................................................................... 13
1.3 Minimale Resterkrankung ..................................................................................................... 14
1.3.1 Definition .......................................................................................................................... 14
1.3.2 MRD-Messmethoden..................................................................................................... 16
1.4 Zielsetzung der Arbeit ........................................................................................................... 18
2. Patienten, Material und Methoden............................................................................................... 20
2.1 Patientenkohorte ...................................................................................................................... 20
2.2 Material ...................................................................................................................................... 21
2.3 Methoden .................................................................................................................................. 23
2.3.1 Grundschema .................................................................................................................... 23
2.3.2 DNA Isolation aus Vollblut und Knochenmark ............................................................. 23
2.3.3 Library Präparation mit TruSight Myeloid Sequencing Panel .................................... 24
2.3.4 Molekulare Analyse mittels Sanger Sequenzierung.................................................... 27
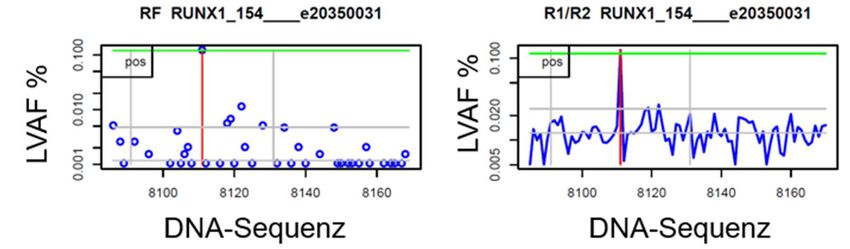
2.3.5. Molekulare MRD Analyse mittels Next Generation Sequenzierung ........................ 29
2.4 Bioinformatische Auswertung .............................................................................................. 36
2.4.1 Bioinformatische Auswertung der Myeloid Panel Sequenzierung und Auswahl der
personalisierten MRD Marker ................................................................................................... 36
2.4.2 Bioinformatische Auswertung des error- corrected sequencing für sensitive MRD
Detektion ...................................................................................................................................... 37
2.5 Statistische Auswertung .......................................................................................................... 39
3. Ergebnisse ...................................................................................................................................... 40
3.1 Deskriptive Patientendarstellung ........................................................................................... 40
3.2 Klinische Parameter abhängig von der VAF zum Zeitpunkt der kompletten Remission
vor allogener Stammzelltransplantation ...................................................................................... 41
3.3 Molekulare Charakteristika abhängig von der VAF zum Zeitpunkt der kompletten
Remission vor allogener Stammzelltransplantation .................................................................. 46
3.4 Assoziationen zwischen minimaler Resterkrankung in kompletter Remission und
Rezidivfreiem Überleben ............................................................................................................... 51
3.5 Prognostische Effekte von minimaler Resterkrankung bei kompletter Remission vor
allogener Stammzelltransplantation............................................................................................. 60
3.6 Subgruppenanalyse zur Evaluierung von DNMT3A als MRD-Marker ............................. 64
4. Diskussion ...................................................................................................................................... 69
4.1 Diskussion der Methoden ..................................................................................................... 69
4.1.1 NGS als zuverlässige MRD Messmethode ................................................................ 69
4.1.2 Vergleich der MRD Methode mit Durchflusszytometrie ........................................... 72
4.1.3 Vergleichbarkeit mit rt-PCR .......................................................................................... 73
4.2 Diskussion der Ergebnisse ................................................................................................... 73
4.2.1 Charakteristika MRD-positiver Patienten und Patienten mit hoher VAF in CR ....... 73
4.2.2 Bedeutung von persistierender Resterkrankung in CR für die Prognose ................ 75
4.2.3 Bedeutung von persistierender klonaler Hämatopoese in CR ................................ 76
5. Zusammenfassung......................................................................................................................... 79
6. Literaturverzeichnis ........................................................................................................................ 80
7. Abkürzungsverzeichnis ................................................................................................................. 89
8. Lebenslauf ....................................................................................................................................... 92
9. Erklärung nach § 2 Absatz 2 Nummer 7 und 8 der Promotionsordnung ............................... 94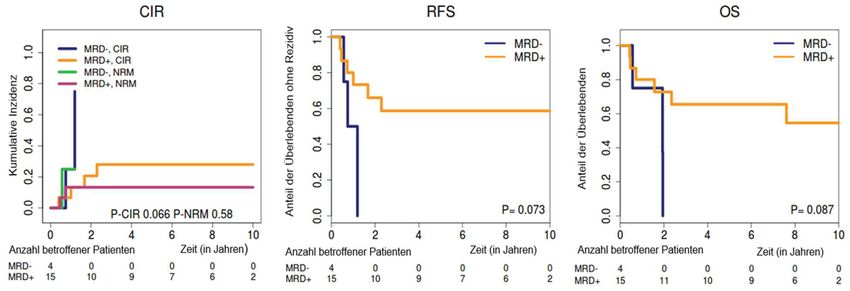
10. Danksagung .................................................................................................................................. 95
1. Einleitung
1.1 Akute myeloische Leukämie
1.1.1 Definition und klinische Symptome
Die akute myeloische Leukämie (AML) ist eine Form von Blutkrebs, die die myeloische
Zellreihe betrifft. Durch das Zusammenwirken von erworbenen somatischen
Mutationen auf genomischer Ebene verlieren die hämatopoetischen Stammzellen ihre
Fähigkeit zur Differenzierung und proliferieren unkontrolliert. Dadurch verdrängen die
unreifen Vorläuferzellen (Blasten) die physiologische Hämatopoese, was zu einer
schwerwiegenden Störung in der Blutbildung führt. (1)
Das klinische Bild der AML beruht auf einer Infiltration des Knochenmarks durch
unreife Blasten. Die Symptome können sich sehr unterschiedlich darstellen, je nach
betroffener Zellreihe und der Schwere der hämatopoetischen Insuffizienz. Zu Beginn
der Erkrankungen stehen unspezifische Symptome wie Abgeschlagenheit, vermehrte
Müdigkeit und Gewichtsverlust im Vordergrund. Im Verlauf kommt es durch
ausgeprägte Zytopenien in allen Zellreihen zu den typischen Symptomen. Störungen
in der Erythropoese äußern sich u.a. durch Anämien, Blässe, Belastungsdyspnoe und
geringere Belastbarkeit. Die verminderte Zahl an Thrombozyten führt zu einer
gesteigerten Blutungsneigung, was mit Hämatomen, Petechien und
Schleimhautblutungen einher gehen kann. Zudem äußert sich die massive
Verringerung der Leukozyten durch eine erhöhte Infektanfälligkeit und Fieber, wobei
die Infekte sehr viel schwerwiegendere Verläufe haben können. In seltenen Fällen
kommt es zu Hyperleukozytosen, bei denen die Leukozytenzahl auf mehr als 100.000
pro µl ansteigt. In Zuge dessen treten Leukostasen auf, bei denen es zu einer Adhäsion
von Leukozyten an die Gefäßwände kommt. Dies kann zu lebensbedrohlichen
cerebralen Mikrozirkulationsstörungen führen. Auch extramedulläre Manifestationen
sind möglich, sodass unterschiedliche Gewebe mit leukämischen Blasten infiltriert
werden. Diese können z. B. in Leber und Milz sein und sich als Hepato- und
Splenomegalie manifestieren. Häufig sind auch die Lymphknoten befallen, die dann
zunehmend, schmerzlos anschwellen (Lymphadenopathie). Ebenso können die
Blasten Haut, Gingiva und das zentrale Nervensystem infiltrieren. (2)
1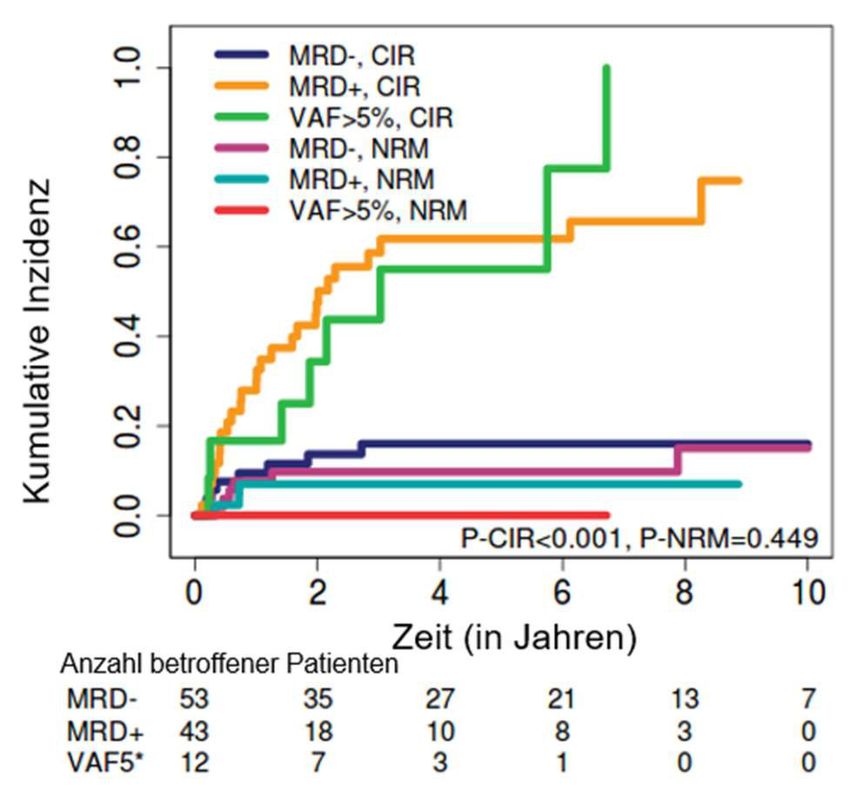
1.1.2 Epidemiologie
Die Inzidenz der AML beträgt 3,7 Erkrankungen pro 100.000 Einwohner im Jahr und
ist damit die häufigste Form von akutem Blutkrebs im Erwachsenenalter. Akute
Leukämien zeigen zwei Häufigkeitsgipfel, wobei die Erkrankung zum einen vermehrt
in der frühen Kindheit auftritt und zum anderen bei den 80- bis 84-Jährigen. (3) Die
Inzidenz der akuten myeloischen Leukämie steigt mit zunehmendem Alter fortlaufend
an. So beträgt sie bei den 35- bis 40- Jährigen 1/100.000 pro Jahr. Bei der Gruppe der
80- bis 85- Jährigen sind es hingegen 17/100.000 Erkrankte pro Jahr, womit die AML
ein Leiden des höheren Alters ist. (4) Im Kindesalter ist Leukämie die häufigste
Krebsdiagnose bei den unter 15-Jährigen. Jedoch ist die akute lymphatische Leukämie
bis zu fünfmal häufiger als die AML und macht damit 76% aller kindlichen Leukämien
aus. (3)
1.1.3 Diagnose und Klassifikation
Zur Sicherung der Diagnose spielt neben dem Blutbild und dem Differentialblutbild die
Untersuchung des Knochenmarks eine zentrale Rolle. Hierfür wird eine
Knochenmarkpunktion mit Aspiration durchgeführt, um Material für die Zytologie zu
gewinnen. Wenn dies nicht möglich sein sollte („punctio sicca“), wird eine
Knochenmarksstanze im Beckenkamm entnommen. Neben der zytologischen
Beurteilung des Blutausstrichs unter dem Mikroskop erfolgen zytochemische und
immunphänotypische Untersuchungen zur Einordnung der Blasten. Die
molekulargenetische Analyse gewinnt zunehmend an Bedeutung. (5)
Für die Diagnose einer akuten myeloischen Leukämie ist ein Blastenanteil von >/=
20% im Knochenmark oder peripheren Blut nötig. Liegen die Translokationen t(15;17),
t(8;21), t(16;16) oder die Inversion inv(16) vor, kann die Diagnose der AML auch mit
einem geringeren Blastenanteil gesichert werden. (6)
Da sich die AML vor allem durch ihre Heterogenität auszeichnet und vielfältig
präsentieren kann, hat ihre Klassifikation bereits einige Änderungen durchlaufen. Die
aus dem Jahre 1976 stammende French-America-British-Klassifikation (FAB)
unterteilte die Leukämien anhand von morphologischen und zytochemischen Kriterien
in die Subgruppen M0-M7. Da in dieser Klassifikation zytogenetische und
molekulargenetische Merkmale keine Berücksichtigung fanden, wurde 2008 eine neue
WHO Klassifikation eingeführt. Hier finden sich sowohl zytogenetische Veränderungen
wie die oben erwähnten Translokationen und Inversionen wieder, als auch Mutationen
2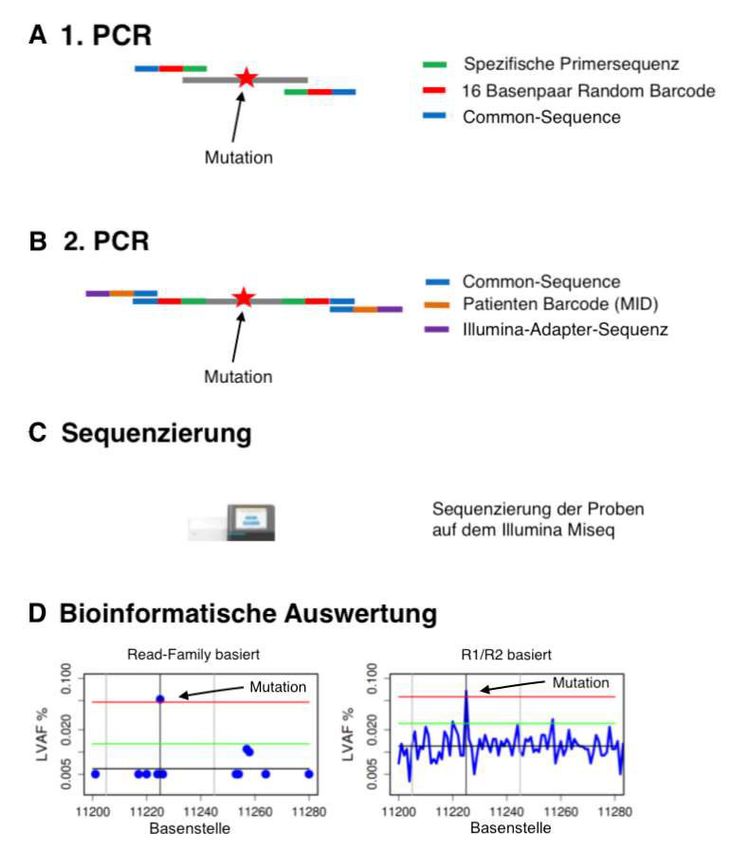
in den Genen CEBPA und NPM1. (7) Die aktuelle WHO Klassifikation von 2016 wurde
mehrfach überarbeitet und beinhaltet zudem zahlreiche molekulargenetische
Veränderungen. Anhand dieser Fassung kann die Mehrheit der AML-Fälle klassifiziert
werden. (8)
Tabelle 1. Auszug aus der WHO-Klassifikation der myeloischen Neoplasien und akuten
Leukämien von 2016.
AML mit wiederkehrenden zytogenetischen Aberrationen
AML mit t(8;21)(q22;q22), RUNX1-RUNX1T1 unabhängig
AML mit inv(16)(p13.1;q22) oder t(16;16)(p13.1;q22), von
CBFb/MYH11 Blastenanzahl
akute Promyelozyten-Leukämie mit t(15;17)(q22;q12), in PB oder KM
PML/RARA
AML mit t(9;11)(p22;q23), MLLT3-KMT2A alle weiteren
AML mit t(6;9)(p23;q34), DEK-NUP214 AML-
AML mit inv(3)(q21;q26.2) oder t(3;3)(q21;q26.2), GATA2, Subtypen:
MECOM >/= 20%
AML (megakaryoblastisch) mit t(1;22)(p13;q13), RBM15-MKL1 Blasten in PB
Provisorische Entität: AML mit BCR-ABL1 oder KM
AML mit mutiertem NPM1
AML mit biallelischer Mutation von CEBPA
Provisorische Entität: AML mit mutiertem RUNX1
AML mit myelodysplasieverwandten Veränderungen
Therapieassoziierte AML
AML, nicht weiter spezifiziert (NOS)
mit minimaler Differenzierung (ehemals M0)
ohne Ausreifung (ehemals M1)
mit Ausreifung (ehemals M2)
akute myelomonozytäre Leukämie (ehemals M4)
akute monoblastische und monozytäre Leukämie (ehemals M5)
rein erythroide Leukämie (ehemals M6)
akute Megakaryoblasten-Leukämie (ehemals M7)
akute Basophilen-Leukämie
akute Panmyelosis mit Myelofibrose
3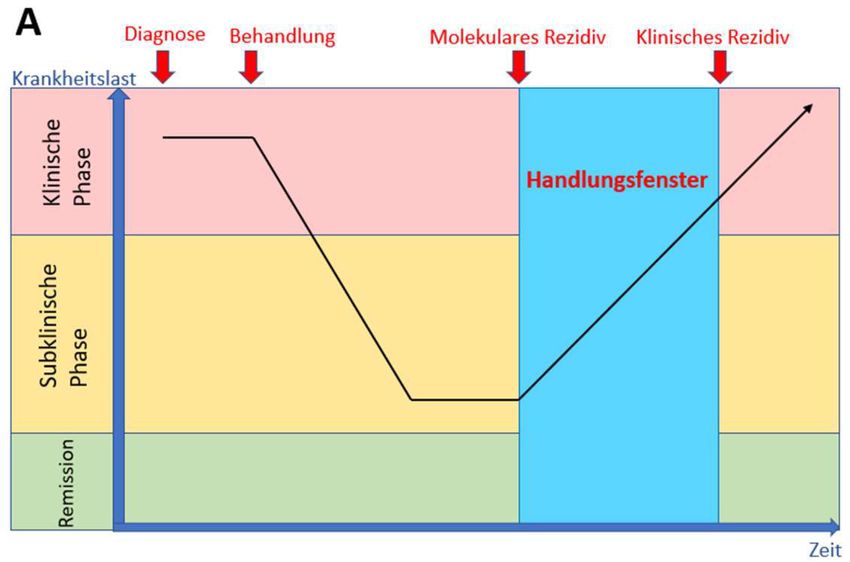
Myeloisches Sarkom
Myeloische Proliferation, assoziiert mit Down-Syndrom
transiente, abnormale Myelopoiese
AML, assoziiert mit Down-Syndrom
Blastische plasmazytoide dendritische Zell-Neoplasien
t: Translokation; inv: Inversion; APL: Akute Promyelozytenleukämie; PB: peripheres Blut; KM:
Knochenmark. Tabelle nach der WHO Klassifikation für myeloische Neoplasien und akute Leukämien
von 2016 (8)
1.1.4 Pathogenese
Die Leukämogenese ist ein komplexer Prozess auf zytogenetischer und molekularer
Ebene. Grundlage der Erkrankung sind Veränderungen in den hämatopoetischen
Stammzellen durch Onkogene bzw. den Verlust von Tumorsuppressorgenen, welche
diese in leukämische Stammzellen transformieren. (9) Krebszellen besitzen ähnlich
wie Stammzellen die Eigenschaft sich unbegrenzt zu teilen und sich selbst zu
erneuern. Bezüglich der Krankheitsentstehung gibt es mehrere Ansatzpunkte, die dies
zu erklären versuchen. Ein Punkt ist, dass die zyto- und molekulargenetischen
Veränderungen primär in der leukämischen Stammzelle selbst stattfinden. Ein anderer
besagt, dass die Mutationen vorwiegend während der Differenzierung der Stammzelle
auftreten und sich somit erst in späteren Reifestadien manifestieren. (10) Vorarbeiten
der Arbeitsgruppe Heuser et al. belegen, dass Vorläuferzellen stammzellähnliche
Eigenschaften entwickeln, und so zu leukämischen Stammzellen werden können. (9)
Derzeit wird von einem mehrstufigen Prozess ausgegangen, bei dem im Rahmen der
klonalen Hämatopoese Genmutationen und chromosomale Aberrationen erworben
werden und zu Veränderungen im Genom und dessen Funktion führen. (11) Daher
kann bereits vor Beginn der Krankheitssymptomen ein präleukämischer Klon, der nur
anfängliche Mutationen enthält, nachgewiesen werden. Diese präleukämischen
Stammzellen können auch bei AML Patienten in kompletter Remission noch
nachgewiesen werden. (12)
1.1.4.1 Zytogenetische Aberrationen
Die Bestimmung von zytogenetischen Aberrationen ist in Bezug auf die diagnostische
und prognostische Aussagekraft unverzichtbar geworden. Die genaue Bedeutung
hinsichtlich der Risikostratifizierung ist in Abschnitt 1.1.5 erläutert. Anders als bei der
chronischen myeloischen Leukämie (CML), bei der fast immer die Translokation
4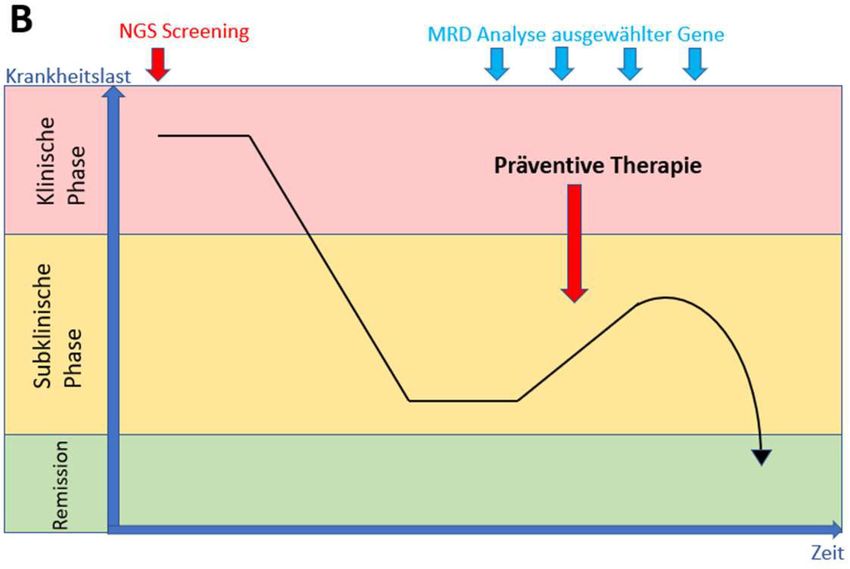
t(9;22) nachgewiesen werden kann, sind die zytogenetischen Veränderungen der AML
sehr viel komplexer und heterogener. Mehr als 200 verschiedene strukturelle und
numerische Aberrationen sind bei der AML bekannt und umfassen u.a. reziproke
Translokationen, Inversionen, Insertionen, Deletionen, Monosomien und Trisomien.
Chromosomale Aberrationen finden sich bei etwa 55% aller erwachsenen AML-
Patienten. (13) Bei diesen unterscheidet man zwischen balancierten und
unbalancierten Translokationen. Balancierte Translokationen können bei ungefähr
jeder fünften akuten myeloischen Leukämie gefunden werden. Zu ihnen zählen u.a.
reziproke Translokationen und Inversionen wie t(8;21) RUNX1RUNX1T1, t(15;17)
PML-RARα, inv(16;16) oder t(16;16) CBFβ-MYH11, 11q23 MLL-Anomalien, t(9;11)
MLLT3-KMT2A, t(6;9) DEK-NUP214 und t(1;22) RBM15-MKL1. (14) Ebenso können
unbalancierte Translokationen auftreten, die zu einer quantitativen Veränderung des
Erbgutes führen. Üblicherweise treten der Verlust von Erbgut in den Regionen 8q, 11q
und 21q auf. Ab drei oder mehr chromosomalen Aberrationen handelt es sich um einen
komplex aberranten Karyotyp. (15)
Diese chromosomalen Veränderungen können sogenannte Fusionsproteine, die zu
Veränderung von Transkriptionsfaktoren führen, generieren und somit Regulations-
und Reparaturprozesse der Zellen verändern. (16)
1.1.4.2 Molekulargenetische Veränderungen
Bei ungefähr der Hälfte aller AML-Patienten können keine chromosomalen
Aberrationen nachgewiesen werden. Für dieses Patientenkollektiv mit normalem
Karyotyp ist die Analyse von molekularen Markern in mehreren Hinsichten relevant.
Zum einen kann anhand der molekulargenetischen Veränderungen eine
Charakterisierung der Leukämie erfolgen. Außerdem können diese zur
Risikostratifizierung herangezogen werden und beeinflussen somit wichtige klinische
Entscheidungen wie eine Stammzelltransplantation oder den Einsatz von
genspezifischen „Target“-Therapien. (17) Jedoch spielen molekulare Marker auch für
Patienten mit chromosomalen Veränderungen eine Rolle.
Mittlerweile sind zahlreiche Mutationen bekannt, die zunehmend mit AML assoziiert
sind. Die Veränderungen im Genom wirken über verschiedene Mechanismen. So
kommt es zu einer übermäßigen Proliferation der hämatopoetischen Progenitorzellen.
Dies ist häufig auf eine Aktivierung von Signalwegen, die die Signaltransduktion
(Rezeptortyrosinkinasen) steuern zurückzuführen, sowie auf eine Hemmung der
5Apoptose. Andere Mutationen beeinflussen transkriptionsfaktorcodierende Gene und
verhindern so die Differenzierung der Stammzellen. Ein weiterer Effekt der mutierten
Gene zeigt sich auf epigenetischer Ebene. Durch Veränderungen des Chromatins und
der Histone können ganze Genabschnitte veränderte Transkriptionsaktivitäten
aufweisen. Methylierungen in der Promotorregion verändern die Expression des
darauffolgenden Genabschnittes. (18)
In fast allen AML Fällen kann eine „Driver“-Mutation detektiert werden, die dann in
einem komplexen Zusammenspiel mit anderen Mutationen zur Pathogenese der
Erkrankung führt. The Cancer Genome ATLAS (TCGA) teilte die Mutationen in
verschiede Kategorien ein. Dazu zählten die Transkriptionsfaktorfusionen in den
Genen PML-RARα, MYH11-CEfβ, RUNX1RUNX1T1 und PICALM-MLLT10, die in
ungefähr jedem fünften Patienten auftraten. Myeloische
Transkriptionsfaktormutationen fanden sich in CEBPα und RUNX1 mit einer ähnlichen
Frequenz. NPM1 war bei 27% der Patienten mutiert. Auch Tumorsuppressorgene, wie
TP53, WT1 und PHF6 waren mutiert (16%), sowie Gene der DNA Methylierung (TET1,
TET2, IDH1, IDH2, DNMT3A, DNMT3A, DNMT1) (44%). Mit 59% aller Erkrankten wies
ein Großteil der Patienten Mutationen in den Genen der Signaltransduktion auf (FLT3,
KIT, KRAS/NRAS, PTPS und andere Tyrosinkinasen). Ein Drittel war in den
chromatinmodifizierenden Genen (ASXL1, EZH2, KDM6A, MLL-PTD) mutiert. (19)
Einige Gene, werden im Folgenden näher erläutert, da sie eine wichtige Rolle in der
Entstehung der Leukämie einnehmen und häufig bei Patienten identifiziert wurden.
NPM1
Das Nucleophosmin1- Gen ist bei ungefähr 27% aller AML-Patienten mutiert und stellt
somit das am häufigsten mutierte Gen dar. (19) Die Mutationen treten besonders bei
Patienten mit normalem Karyotyp auf. Es handelt sich vornehmlich um drei
verschiedene Mutationen, wobei die Typ A Mutation, eine Insertion der Basen TCTG
zwischen den Basenstellen 960 und 961 mehrheitlich vorliegt. (20) Deutlich seltenere
Varianten sind Mutationen B und D. (21) Sie bewirken eine Frameshift-Mutation im
Bereich des C-Terminus. Die Mutationen führen zu fehlerhaften Codierungen im
Nukleophosminprotein, welches überwiegend im Zellkern vorkommt und an
verschiedenen Transportprozessen beteiligt ist. Das Protein reguliert unter anderem
den ARF-p53-Tumorsuppressor-Signalweg. (22) NPM1-Mutationen treten häufig
zusammen mit FLT3-Mutationen auf und haben eine wichtige prognostische
6Bedeutung. Patienten mit mutiertem NPM1 und einem FLT3 Wildtyp zeigen eine
wesentlich bessere Prognose als andere Konstellationen. Sie machen knapp ein
Viertel der AML-Patienten mit normalem Karyotyp aus und wirken sich deshalb auf die
therapeutische Planung aus. (23)
FLT3
Mutationen im fms-related Tyrosinkinase 3 Gen gehören ebenfalls zu den häufigsten
Mutationen bei AML-Patienten. Sie führen zu einer Aktivierung der Tyrosinkinase und
somit zu einer unkontrollierten Proliferation von hämatopoetischen Vorläuferzellen.
(18) Es werden hauptsächlich zwei Mutationen beschrieben. Bei der internen
Tandemduplikation (ITD) werden 3-400 Basen in die codierende Region der
Juxtamembrandomäne eingefügt. Außerdem treten Punktmutationen in der
Tyrosinkinasedomäne (TKD) auf. Diese sind mit einer Prävalenz von 5-10% bei den
Erkrankten seltener als die FLT3 ITD Mutation, die bei 15-35% auftritt. (24) Mehrere
Analysen zeigen, dass Patienten mit normalem Karyotyp und mutiertem FLT3 ITD eine
signifikant schlechtere Prognose haben als Patienten mit FLT3 Wildtyp. (25)
DNMT3A
Mutationen im Demethyltransferase3A Gen treten bei fast jedem fünften AML-
Patienten auf. (19) Das Gen codiert eine Transferase, welche sogenannte CpG-Inseln
methyliert, sodass das darauffolgende Gen weniger exprimiert wird. (26) In den
meisten Fällen liegt eine Punktmutation in der Aminosäure Arginin R882 vor, die dann
zu Histidin codiert wird. (27) Wie genau die Mutation wirkt, ist noch nicht geklärt. Ältere
Patienten weisen signifikant häufiger Mutationen im DNMT3A-Gen auf, als jüngere.
Die Mutationsrate nimmt mit zunehmendem Alter bis zum 40. Lebensjahr zu, und
stagniert in der Altersgruppe der 40-bis 60-Jährigen. (28) Mehrere Studien zeigen,
dass DNMT3A-Mutationen mit einer schlechten Prognose assoziiert sind. (17,27) Dies
gilt besonders für ältere Patienten mit Mutationen im Hotspot R882. (29) DNMT3A-
Mutationen werden im Zusammenhang mit klonaler Hämatopoese diskutiert, was
genauer in Abschnitt 1.2 erläutert wird.
Zusammenspiel der molekulargenetischen Veränderungen im Rahmen der
Pathogenese
Die Pathogenese der AML beruht auf einem komplexen Zusammenspiel mehrerer
individueller Mutationen. Insgesamt kommen bei der AML weniger Mutationen als bei
anderen adulten Krebsformen vor. (19) Bei nahezu jedem Patienten kann mindestens
7eine Drivermutation, die der Zelle einen Wachstumsvorteil verleiht, gefunden werden.
(30) Es wird angenommen, dass die Erkrankung sich über einen längeren Zeitraum
hinweg entwickelt und die meisten Mutationen lange bevor die Krankheit
symptomatisch wird auftreten. So können verschiedene Klone mit unterschiedlichen
Mutationen nebeneinander existieren. (31)
Zum besseren Verständnis der klonalen Evolution wurden diese Subklone näher
untersucht. Dabei stellte sich heraus, dass Mutationen in den Genen DNMT3A,
ASXL1, ASXL2, IDH1, IDH2 und TET2, die hauptsächlich an der epigenetischen
Regulierung beteiligt sind, am frühesten erworben werden. Da sie fast immer in den
präleukämischen Stammzellen gefunden werden, können sie als frühester Schritt in
der Leukämogenese betrachtet werden. Sie treten selten allein auf, sondern immer im
Zusammenhang mit anderen Mutationen. Deshalb besteht die Annahme, dass sie
allein nicht zur Leukämogenese fähig sind, sondern weitere Mutationen nötig sind. (30)
Veränderungen in Tyrosinkinaserezeptoren wie FLT3, NRAS und KRAS entwickelten
sich erst im späteren Krankheitsverlauf und zeigten häufig mehrere Mutationen pro
Patient. Mutationen in NPM1 entwickelten sich meist erst nachdem Mutationen in
DNMT3A, IDH1 oder NRAS auftraten. Insgesamt lassen sich bei der Leukämogenese
gewisse Muster in der Molekulargenetik erkennen. (30)
1.1.5 Risikostratifizierung
Die zwei wichtigsten Parameter zur Risikostratifizierung stellen das Alter und die
zytogenetischen, sowie die molekulargenetischen Veränderungen dar. Ältere
Patienten präsentieren sich klinisch in der Regel schlechter. Damit einher geht ein
kürzeres Gesamtüberleben, da die Patienten weniger auf die Therapie ansprechen
und diese schlechter vertragen. (32) Mit zunehmendem Alter erreichen weniger
Patienten eine komplette Remission, wobei gleichzeitig das Rezidivrisiko steigt. (33)
Zudem bildet der Karyotyp eine fundamentale Säule hinsichtlich der prognostischen
Aussagekraft für das Überleben und Therapieansprechen. (34)
Die Empfehlungen des European LeukemiaNet (ELN) für die Diagnostik und das
Management der AML wurden mehrfach überarbeitet, zuletzt 2017. Jüngste
Erkenntnisse bezüglich der Genetik der Erkrankung sowie neue Technologien der
Sequenzierung und Therapien haben dazu beigetragen. Auch die molekular-
8zytogenetischen Risikogruppen wurden überarbeitet. Diese beinhalten nun auch
Ansprechkriterien, die auf dem MRD-Status basieren. (6)
Tabelle 2. Auszug aus der European LeukemiaNet (ELN) Klassifikation zur Risikostratifizierung
anhand von zyto- und molekulargenetischen Veränderungen.
Risikogruppe Genetische Veränderungen
günstig t(8;21)(q22;q22.1); RUNX1-RUNX1T1
Inv(16)(p1.1q22) oder t(16;16)(p13.1;q22); CBFB-MYH11
NPM1 Mutation ohne FLT3-ITD (normaler Karyotyp) oder FLT3-
ITDniedrig
Biallelisch mutiertes CEPBA
intermediär Mutiertes NPM1 mit FLT3-ITDhoch
Wildtyp NPM1 ohne FLT3-ITD oder mit FLT3-ITDniedrig
t(9;11)(p22;q23); MLLT3-KMT2A
Zytogenetische Abnormitäten, die nicht als günstig oder ungünstig
eingeteilt werden
ungünstig t(6;9)(p23;q34); DEK-NUP214
t(v;11)(v;q23); KMT2A-Genumlagerung
t(9;22)(q34.1;q11.2); BCR-ABL1
inv(3)(q21q26.2) oder t(3;3)(q21;q26.2); GATA2, MECOM (EVI1)
-5 oder del(5q); -7; -17/abn(17p)
komplexer Karyotyp (3 Aberrationen)
monosomaler Karyotyp (eine Monosomie, assoziiert mit mindestens
einer weiteren Monosomie oder einer anderen strukturellen,
chromosomalen Aberration (außer CBF-AML))
Wildtyp-NPM1 mit FLT3-ITDhoch
Mutiertes RUNX1
Mutiertes ASXL1
9Mutiertes TP53
FLT3-ITDniedrig: Mutant-Wildtyp-Allel-Quotient /=0,5;
t: Translokation; inv: Inversion; del: Deletion; abn: abnormal. Nach der genetischen Risikostratifizierung
des European Leukemia Net von 2017 (6)
1.1.6 Therapie und Prognose
Da die Erkrankung schwerwiegende Störungen des Knochenmarks hervorruft, verläuft
sie unbehandelt innerhalb kürzester Zeit zum Tode. Die Auswahl der Therapie erfolgt
individuell anhand des zyto- und molekulargenetischen Befundes. (35) Ziel der
Therapie ist das Erreichen einer kompletten Remission (CR). Davon spricht man, wenn
der Blastenanteil im Knochenmark auf unter 5% fällt, weder Auerstäbchen noch
extramedulläre Manifestationen nachweisbar sind, die Neutrophilen auf >/=1000/µl
angestiegen sind und die Thrombozyten >/=100.000/µl erreicht haben. (6) Seit 2017
findet sich auch der negative Status der minimalen Resterkrankung (MRD) als Teil der
CR wieder. Ein Rezidiv liegt vor, wenn die Blasten einen Anteil von >/=5% im
Knochenmark ausmachen, sie im peripheren Blut auftreten, sich extramedulläre
Manifestationen entwickeln oder der MRD-Marker wieder auftritt. (6)
Zu Beginn muss evaluiert werden, ob der Patient für eine intensive Chemotherapie
geeignet ist. Dabei ist nicht nur das Alter des Patienten entscheidend, sondern
vielmehr die klinische Präsentation, die Komorbiditäten und ein ungünstiges
zytogenetisches Risikoprofil nach ELN. (6)
Entscheidet man sich für eine intensive Induktion, wird die Chemotherapie meistens
nach dem 3+7-Schema verabreicht. Hierbei erhält der Patient drei Tage lang ein
Anthrazyklin, wie z.B. Daunorubicin, Idarubicin, oder Mitoxantron. Zeitgleich erfolgt
eine kontinuierliche siebentägige Infusion von Cytarabin. Hiermit soll die Tumorlast
reduziert und eine komplette Remission erreicht werden. (6) In der Gruppe der 60-Jährigen erreichen
40-60% eine komplette Remission. (6)
Die Bedeutung des genetischen Risikoprofils wird bei verschiedenen genetischen
Subgruppen deutlich. (5) So werden AML-Patienten mit einer FLT3-Mutation
zusätzlich zur Induktionstherapie mit Midostaurin behandelt. (36) NPM1-mutierte
10Patienten profitieren möglicherweise von einer Therapie mit All-trans-Retinolsäure (ATRA). (37) Bei der Core-binding-factor (CBF)-AML ist zu erwägen Gentuzumab Ozogamicin der Induktionschemotherapie hinzuzufügen. (38) Nach Erreichen der kompletten Remission folgt die Konsolidierungstherapie an drei Tagen mit hochdosiertem Cytarabin in jeweils zwölfstündigem Abstand. (39) Eine andere Möglichkeit der Konsolidierung stellt die allogene Stammzelltransplantation (SCT) dar. Transplantiert werden vor allem Patienten mit schlechter Prognose und hoher Rezidivwahrscheinlichkeit, sowie bei Versagen der Chemotherapie, da sie von allen Therapien die stärkste antileukämische Wirkung hat. (40) Patienten, die nicht für eine intensive Chemotherapie in Frage kommen, bekommen eine anderweitige Behandlung. Eine Möglichkeit ist die Behandlung mit niedrigdosierten Cytarabinen, welche jedoch nicht bei Patienten der ungünstigen zytogenetischen Risikogruppe nach ELN angewendet werden soll. (6) Mittlerweile konnte gezeigt werden, dass die Behandlung mit hypomethylierenden Substanzen, wie Decitabin und Azacitidin zu einem besseren Gesamtüberleben führt. (41) In der AZA-AML-001 Studie stellte sich heraus, dass Patienten mit einem ungünstigen zytogenetischen Risikoprofil nach ELN besonders von der Behandlung mit Azacitidin profitierten. (42) Die wichtigsten Parameter zur Einschätzung der Prognose wurden bereits in Abschnitt 1.1.5 erklärt. Die 5-Jahres-Überlebensrate bei den jüngeren Patienten (
und Blasten im Knochenmark. Die Kriterien des Myelodysplastischen Syndroms
(MDS) und der AML werden somit nicht erfüllt. Eine paroxysmale nächtliche
Hämoglobinurie, eine monoklonale Gammopathie unklarer Signifikanz und eine
monoklonale B-Lymphozytose müssen ausgeschlossen werden. Der Nachweis der
klonalen Hämatopoese und damit der Klonalität muss durch eine somatische Mutation,
die mindestens eine variante Mutationsrate von >/=2% beträgt, erfolgen. (45) Am
häufigsten finden sich Mutationen in den Genen DNMT3A, TET2 und ASXL1. (46) Es
können aber auch andere Gene wie JAK2, TP53, PPM1D, BCORL1 und SF3B1
betroffen sein. (47) Mutationen in DNMT3A weisen die höchste Mutationsrate auf (26
bis 58%). (46-48)
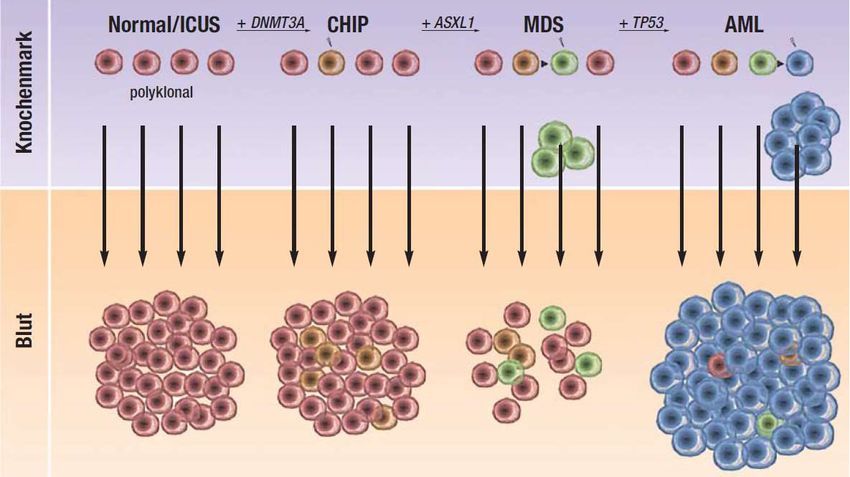
Abbildung 1. Schematische Darstellung der Entwicklung der AML über CHIP und MDS.
Schrittweise Entwicklung von polyklonaler (normaler, ohne Nachweis somatischer Mutationen) zu
klonaler Hämatopoese von unbestimmtem Potenzial (CHIP), myelodysplastischen Syndrom (MDS) und
akuter myeloischer Leukämie (AML). ICUS: idiopathische Zytopenie unbestimmter Signifikanz. Die
mutierten Gene (DNMT3A, ASXL1, TP53) sind nur exemplarisch dargestellt; es können andere Gene
und in anderer Reihenfolge betroffen sein. Mit Genehmigung des Deutschen Ärzteverlages GmbH. (44)
Daneben existiert der Begriff der altersassoziierten klonalen Hämatopoese (age-
related clonal hematopoiesis, ARCH). Diese ist definiert als klonale Expansion von
hämatopoetischen Stammzellen, die Mutationen im Genom aufweisen, ohne dass
zuvor eine hämatologische Neoplasie diagnostiziert war. (49) 2014 zeigten drei große
Studien, dass im Alter besonders Mutationen in den Genen DNMT3A, TET2 und
12ASXL1 auftreten, mit einer VAF von >2%, die im Median ungefähr 12% betrug.
(12,47,48) Die Begriffe CHIP und ARCH wurden von verschiedenen Arbeitsgruppen
geprägt, werden aber überwiegend synonym verwendet. Im Folgenden werde ich
CHIP verwenden.
Des Weiteren ist in der Literatur der Begriff der klonalen Hämatopoese mit erheblichem
onkogenen Potential (clonal hematopoiesis with substantial oncogenic potential,
CHOP) zu finden. Dieser wird für Patienten mit einem erhöhten Risiko für einen
Krankheitsübergang durch sogenannte Drivermutationen verwendet, ohne dass
andere Diagnoskriterien erfüllt sind. (50)
1.2.2 Epidemiologie
Die Prävalenz von CHIP steigt mit zunehmendem Alter an. (44) Die Ergebnisse von
drei großen Studien zu CHIP von Xie et al., Genovese et al. und Jaiswal et al. zeigen,
dass bei weniger als 1% aller 40-jährigen klonale Hämatopoese nachweisbar ist. In
der Altersgruppe der 70 bis 80-Jährigen fand sich bei 9,5 bis 13,9% klonale
Hämatopoese. Ab dem 80. Lebensjahr stieg die Zahl auf 16,4%. (46-48)
1.2.3 Bedeutung
Der Nachweis von CHIP geht mit einem gering erhöhten Risiko für eine
hämatologische Neoplasie einher. Dieses beträgt 0,5 bis 1% pro Jahr und korreliert
mit der Größe des somatischen Klons. Auch wenn die Progressionsrate
vergleichsweise niedrig ist, trägt die mutierte hämatologische Stammzelle stärker zur
Blutbildung bei als die Unmutierte und stellt somit eine mögliche Vorstufe
hämatologischer Neoplasien dar. (45) In verschiedenen Studien wurde belegt, dass die
Gesamtmortalität der Patienten mit CHIP erhöht ist. Dies ist zum einen auf eine
erhöhte Krebsmortalität zurückzuführen. (48) Zum anderen erhöht sich bei Patienten
mit somatischen Mutationen das Risiko für kardiovaskuläre Todesfälle durch koronare
Herzerkrankungen und Schlaganfälle. (46) Mit der Höhe der Mutationsrate der
somatischen Mutation steigt das Risiko eine Neoplasie zu entwickeln, sowie die
Gesamtmortalität. Daraus lässt sich ein kausaler Zusammenhang schließen, sodass
der Nachweis von CHIP neben seiner hämatologischen Bedeutung auch den
Alterungsprozess anzeigt. (44)
DNMT3A ist das am häufigsten mutierte Gen mit der höchsten Mutationsrate bei CHIP.
Vorarbeiten unserer Arbeitsgruppe haben gezeigt, dass DNMT3A Mutationen bei
13AML-Patienten mit einem kürzeren Gesamtüberleben einhergehen. Auswirkungen auf
das rezidivfreie Überleben (relapse free survival, RFS) oder das Erreichen der
kompletten Remission (complete remission, CR) ließen sich nicht feststellen. (17) Die
Höhe der DNMT3A Mutationsrate korreliert allerdings nicht mit dem Outcome.
Untersuchungen der Arbeitsgruppe Gaidzik et al. haben ergeben, dass die
Mutationsrate selbst nach der Chemotherapie konstant hoch bleiben kann und keine
Korrelation mit Remissionsraten oder dem Gesamtüberleben zeigt. Dies führt zu der
Annahme, dass klonale Hämatopoese auch noch in hämatologisch kompletter
Remission persistiert und keine ungünstige Wirkung auf die Prognose hat. (51)
1.3 Minimale Resterkrankung
1.3.1 Definition
Die Überwachung der minimalen Resterkrankung (minimal residual disease, MRD) hat
in den letzten Jahren durch die Entdeckung vieler molekularer Parameter erheblich an
Bedeutung gewonnen. In der European LeukemiaNet (ELN) Klassifikation findet sich
seit 2017 eine neue Kategorie hinsichtlich der Ansprechraten „CR ohne MRD“. (6) Das
Monitoring beruht auf genetische Marker, die spezifisch für leukämische Klone sind
und sowohl bei der Diagnose als auch beim Rezidiv vorliegen. (52) Ein großer Teil der
AML-Patienten entwickeln nach allogener Stammzelltransplantation ein Rezidiv. (53)
Das ist vor allem auf leukämische Klone zurückzuführen, die auch in morphologisch
kompletter Remission persistieren. Diese können erneut zu einer leukämischen
Zellpopulation heranwachsen und zu einem Rückfall führen. Die zytomorphologische
Methode zur Feststellung der kompletten Remission mit dem Mikroskop reicht nicht
aus, um diese Klone zu erfassen. (54) So sind bei der Diagnosestellung etwa 1012
leukämische Zellen nachweisbar. Nach der Induktionstherapie in morphologisch
kompletter Remission können noch bis zu 1010 der Zellklone nachgewiesen werden.
(55) Die Untersuchung mit dem Lichtmikroskop erlaubt nur begrenzt eine präzise
Quantifizierung der Myeloblasten, da diese je nach Untersucher variieren können.
Zudem kann man anhand der Zytomorphologie kaum normale von präleukämischen
Myeloblasten unterscheiden. Dazu kommt, dass die Verteilung der Myeloblasten sehr
unterschiedlich sein kann und die Lichtmikroskopie nur einen sehr kleinen Teil
abbildet. (56) Im Laufe der Jahre wurden deutlich sensitivere Methoden der MRD-
Messung entwickelt, die im Folgenden vorgestellt werden. Bei dem Krankheitsbild der
14chronischen myeloischen Leukämie (CML) ist die Überwachung der minimalen
Resterkrankung anhand des BCR/ABL1 Fusionsgens mit der qRT-PCR bereits
etabliert. (57)
Anhand des MRD-Levels können prognostische Aussagen über den voraussichtlichen
Krankheitsverlauf getroffen werden. Des Weiteren kann es als Anhaltspunkt zur
Intensivierung der Therapie bei einem erhöhten Rezidivrisiko dienen. (54) Bei etwa
60% aller AML-Patienten kann jedoch keine Überwachung der minimalen
Resterkrankung erfolgen, da es an geeigneten MRD-Markern fehlt. (30)
Die Detektion der minimalen Resterkrankung ist im Wesentlich abhängig von zwei
Faktoren: der Effizienz der Therapie und der Sensitivität der Detektionsmethode. Da
er als alleinstehender Begriff wenig Bedeutung hat, wird auch von „messbarer
Resterkrankung“ (measurable residual disease) gesprochen. Dabei sollte jedoch
immer die Messmethode und deren Sensitivität erwähnt werden. (58)
Derzeit werden verschiedene Messmethoden, wie die quantitative Real-Time-
Polymerase-Kettenreaktion (qRT-PCR), die Durchflusszytometrie (MFC), und das
Next Generation Sequencing (NGS), angewandt wobei je nach Patientenkollektiv und
angestrebtem Sensitivitätslevel unterschiedliche Methoden bevorzugt werden. (59)
Nach den aktuellen Empfehlungen der ELN sollte nach Möglichkeit eine Kombination
der verschiedenen Messverfahren erfolgen. (60)
15Abbildung 2. Schematische Darstellung des MRD-Messprinzipes.
Modifiziert nach Heuser et al. A zeigt den Verlauf der Leukämieerkankung ohne MRD-Monitoring, wobei
das molekulare Rezidiv im möglichen Handlungsfenster nicht erfasst wird. In B wird deutlich, dass mit
Hilfe der MRD-Analyse ein Rezidiv frühzeitig erkannt und therapiert werden kann.
1.3.2 MRD-Messmethoden
1.3.2.1 Quantitative Real-Time-Polymerase-Kettenreaktion (qRT-PCR)
Bei der qRT-PCR wird neben der Vervielfältigung der Nukleinsäuren gleichzeitig eine
Quantifizierung mittels Fluoreszenz-markierten Sonden am Ende jedes
Amplifikationszyklus vorgenommen. Bei der Messung der Mutationsraten wird ein
Sensitivitätslevel von 10-4 bis 10-6 gewährleistet. (61) Das Monitoring von BCR/ABL1
bei der CML mit der qRT-PCT ist ein fester Bestandteil der Behandlung, da sein Nutzen
als sensitiver MRD-Marker in zahlreichen Studien belegt werden konnte. (57) Eine
einzelne Mutation als MRD-Marker analog zu BCR/ABL1 existiert in der AML nicht. (62)
Dennoch haben sich einige Marker, die mit der qRT-PCR erfasst werden, etabliert. So
16wird bei der Promyelozytenleukämie das onkogene Fusionsprotein PML/RARA
nachgewiesen, dass durch die Translokation t(15;17) bedingt ist. Wenn dieses am
Ende der Therapie nachweisbar ist, spricht dies für ein bevorstehendes Rezidiv. (63)
Auch bei den Core-binding-factor Leukämien (CBF AML) sind MRD-Marker etabliert,
wie etwa inv(16) und t(8;21). Letzteres führt zu einer RUNX1/RUNX1T1 Fusion. (61)
Obwohl diese Tests sehr sensitiv sind und Anstiege der Marker mit einer erhöhten
Wahrscheinlichkeit für ein Rezidiv einhergehen, können besonders bei
Punktmutationen immer noch falsch-negative Ergebnisse auftreten. (62) Auch
Mutationen in NPM1 können effektiv mit dieser Methode erfasst wird. Seine Persistenz
in kompletter Remission deutet auf ein erhöhtes Rezidivrisiko hin (>80%). (64) NPM1
wird sowohl im peripheren Blut als auch im Knochenmark gemessen. Bei Anstieg des
Markers sollte die Einleitung einer Salvage-Therapie in Erwägung gezogen werden.
Ist der Marker negativ, sollte die Testung nach Beendigung der Therapie alle 3 Monate
für mindestens zwei Jahre wiederholt werden. (65) Zusammen decken diese MRD-
Messmethoden etwa 60% aller AML-Fälle bei den unter 60-Jährigen ab, aber deutlich
weniger bei älteren AML Patienten. (59)
1.3.2.2 Durchflusszytometrie (multi-parameter flow cytometry, MFC)
Die MRD-Messung mittels Durchflusszytometrie erfasst vor allem den Phänotyp der
leukämischen Zellen. Hierbei werden Zellpopulationen anhand veränderter
Antigenexpressionen unterschieden, an die mit Fluoreszenzfarbstoff markierte
Antikörper binden. Antigene können mit anderen Populationen gekreuzt, über- oder
unterexprimiert sein oder ganz fehlen. (59) Untersucht werden beispielsweise die
Clustermoleküle CD34, CD117, CD2, CD7, CD33 und CD56. Die Methode lässt sich
zwar auf die Mehrheit der AML-Patienten anwenden, ist jedoch schwer zu
standardisieren. (61) Es gibt zwei Arten sich der MRD-Messung mittels MFC zu
bedienen. Zum einen kann man anhand der Erstdiagnose einen Leukämie-
assoziierten Phänotyp definieren (leukemia-associated immunophenotype, LAIP), den
man in den nachfolgenden Proben verfolgt. Zum anderen kann man aberrante
Differenzierungen und Reifungen der Oberflächenmarker detektieren (different from
normal, DfN). Dies ist vor allem für die Identifizierungen von sogenannten „Shifts“ der
Immunphänotypen von Bedeutung, wobei neue Aberrationen bei gleichzeitigem
Verschwinden von Aberrationen bei Erstdiagnose auftreten. (66) Die relative
Sensitivität der Methode liegt bei 10-3, was einer von 1000 Zellen entspricht. (62)
171.3.2.3. Next Generation Sequencing (NGS)
Beim Next Generation Sequencing werden durch parallele Sequenzierung Millionen
von DNA-Strängen in nur einer Reaktion sequenziert. (67) So können bei >90% aller
AML-Patienten molekulare Aberrationen mittels NGS identifiziert werden. (30)
Theoretisch kann fast jede Mutation als MRD-Marker dienen. Vorarbeiten unserer
Arbeitsgruppe haben gezeigt, dass FLT3 und NPM1 mit der NGS-Technik geeignete
MRD-Marker darstellen. (52) FLT3 wurde bislang nicht als MRD-Marker verwendet,
weil es durch seine Instabilität nicht zwangsläufig in den leukämischen Blasten im
Rezidiv nachweisbar sein muss. (68) Weitere Limitationen waren, dass bei Diagnose
mehrere Klone vorliegen können und unklar ist, welcher im Rezidiv dominiert.
Außerdem variieren die FLT ITD-Mutationen stark zwischen den einzelnen Patienten.
Mit der NGS-Messung konnte die genaue Insertionsstelle bestimmt werden, sowie die
ITD-Länge und die Mutationsrate. Ebenso konnte der dominante Klon von Subklonen
identifiziert und in Remission und im Rezidiv erneut gemessen werden. Beim Vergleich
der NGS-MRD-Messung von NPM1 mit dem Goldstandard der qRT-PCR zeigte sich
eine Übereinstimmung von 95%. (52) Untersuchungen der Arbeitsgruppe Kohlmann et
al. ergänzten RUNX1 als Marker zur Quantifizierung der minimalen Resterkrankung
im Rezidiv mittels NGS. (69) Die größte Herausforderung bei der Messung mit NGS
liegt bei einem Sequenzierungsfehler von ungefähr 1% pro Nukleotid. (67) Dies macht
es schwierig minimale Resterkrankung mit sehr niedrigen Frequenzen von diesen zu
unterscheiden. Die Arbeitsgruppe Papaemmanuil et al. konnte mehr als 5000
Drivermutationen in 76 Genen bei 1540 AML-Patienten identifizieren. Bereits hier
ließen sich Effekte auf das Gesamtüberleben feststellen. (30) Bedenkt man, dass die
MRD-Messung auf fast jede Mutation übertragen werden kann, wird das Potential der
Next Generation Methode deutlich.
1.4 Zielsetzung der Arbeit
Die Überwachung der minimalen Resterkrankung ist mittlerweile ein etabliertes
Verfahren, um den Krankheitsverlauf der AML zu beurteilen. (6) Ein großer Teil der
Erkrankten erleidet nach allogener Stammzelltransplantation ein Rezidiv und hat damit
eine deutlich schlechtere Prognose. (53) Um dies frühzeitig zu erkennen eignen sich
Technologien wie Next Generation Sequencing, da sie Mutationen zuverlässig
detektieren und so ein Großteil der Patienten molekular überwacht werden kann. Die
18Mutationen geben einen möglichen Anhaltspunkt zur Erfassung der minimalen
Resterkrankung, welche möglicherweise in kompletter Remission persistieren. (30)
Während einige Mutationen als MRD-Marker bereits gezeigt haben, dass sie
zuverlässig auf ein erhöhtes Rezidivrisiko hinweisen, ist die Relevanz der meisten
Mutationen noch nicht ausreichend verstanden. Einige Mutationen treten im Rahmen
der klonalen Hämatopoese auf (CHIP) und können auch in morphologisch kompletter
Remission nach Chemotherapie persistieren.(45) Bislang ist für die meisten Gene noch
ungeklärt welche klinische als auch molekulare Bedeutung eine persistierende klonale
Hämatopoese bei Patienten, die in kompletter Remission transplantiert wurden, für den
Verlauf der AML haben. Nur für DNMT3A wurde gezeigt, dass unabhängig von einer
Transplantation eine persistierende klonale Hämatopoese keinen prognostischen
Effekt hat. (51) CHIP ist auch bei Gesunden nachweisbar und geht dann mit einem
erhöhten Risiko für eine hämatologische Neoplasie einher, sowie einer erhöhten
Mortalität. (45) Wie genau sich CHIP in kompletter Remission bei transplantierten
Patienten verhält und welche Schlüsse sich daraus auf das Rezidivrisiko schließen
lassen ist bislang ungeklärt.
Wir untersuchten eine Kohorte von AML-Patienten, die in kompletter Remission
stammzelltransplantiert wurden. Dabei bezogen wir nur Genmutationen ein, die in
anderen Genen als DNMT3A auftraten. Wir verwendeten ein neues MRD-Verfahren
mit NGS, um zu identifizieren, welche Patienten MRD negativ oder positiv waren, um
so Aussagen über die Rezidivwahrscheinlichkeit zu treffen. Daneben definierten wir
eine dritte Gruppe von Patienten, deren Mutationsfrequenz in CR >5% war. Bei diesen
Patienten gingen wir von persistierender klonaler Hämatopoese aus, da bei einer
morphologisch kompletten Remission ein Blastengehalt von 5% Blasten vorliegen. Dieser Sicherheitsabstand
gewährte uns eine sichere Identifizierung hoher Mutationslasten.
Ziel der Arbeit war es weitere Erkenntnisse über das Verhalten molekularer Marker in
kompletter Remission zu gewinnen, um durch das Monitoring in Zukunft ein Rezidiv
frühzeitig zu erkennen. Besonders mit Blick auf die demografischen Veränderungen
der Gesellschaft sollte auch die Bedeutung von altersassoziierten Mutationen, wie die
persistierende klonale Hämatopoese weiter untersucht werden, um diese in
Zusammenhang mit den Krankheitsverlauf besser zu verstehen.
192. Patienten, Material und Methoden
2.1 Patientenkohorte
Das Patientenkollektiv umfasste Patienten, die im Rahmen ihrer AML-Erkrankung in
kompletter Remission eine allogene Stammzelltransplantation erhalten haben. Diese
mussten mindestens 18 Jahre alt und zwischen 1996 und 2016 an der Medizinischen
Hochschule Hannover behandelt worden sein. Es war sowohl eine DNA-Probe zum
Zeitpunkt der Erstdiagnose nötig als auch eine weitere Probe in kompletter Remission
vor der Stammzelltransplantation.
Von den insgesamt 116 Patienten wurden acht nach der Untersuchung der
Erstdiagnose auf dem Myeloid Panel ausgeschlossen, da keine geeignete Mutation,
die man mittels NGS-MRD hätte verfolgen können, gefunden wurde. Das Screening
mit dem Myeloid Panel diente dem Detektieren von geeigneten MRD-Markern, die im
Verlauf gemessen werden sollten. Ausgewählt wurden dann bis zu drei verschiedene
Gene pro Patient. Ausgeschlossen wurden Mutationen in DNMT3A und NPM1.
Bei 96 Patienten wurde die minimale Resterkrankung mit NGS zum Zeitpunkt der
kompletten Remission gemessen. Diese Patienten hatten mindestens einen
geeigneten Marker, dessen Mutationsrate vor der Stammzelltransplantation 5% persistierte. Hier bestand die Annahme, dass es sich um persistierende
Mutationen in differenzierten Zellen handelte, die am ehesten mit klonaler
Hämatopoese im Zusammenhang standen. Bei den 12 Patienten stammten sechs
Proben aus dem Knochenmark und sechs aus peripherem Blut.
Die klinischen und hämatologischen Daten der Patienten wurden mit deren
Einverständnis in Übereinstimmung mit der Deklaration von Helsinki aufgezeichnet.
Die wissenschaftliche Analyse der Proben erfolgte nach Zustimmung der
Ethikkommission der Medizinischen Hochschule Hannover (2179-2014).
202.2 Material
Die verwendeten Materialien und Geräte sowie deren Hersteller wurden im Folgenden
aufgeführt.
Tabelle 3. Materialien.
Name Hersteller
Allgemeine 50x TAE Puffer Carl Roth, Karlsruhe,
Chemikalien Germany
Natriumhydroxyid 8mM Carl Roth, Karlsruhe,
Germany
PBS Dulbecco w/o Ca2+, w/o Biochrome GmbH,
Mg2+ Berlin, Germany
5x DNA Loading Dye GelPilot Qiagen, Hilden,
Germany
UltraPureTM Destilled Water Thermo Fisher
DNase/RNase-Free Scientific, Waltham,
USA
UltraPureTM Agarose Thermo Fisher
Scientific, Waltham,
USA
Etidiumbromid Thermo Fisher
Scientific, Waltham,
USA Carl Roth,
Karlsruhe, Germany
Ethanol absolute for molecular AppliChem Panreac
biology 96% ITW companies,
Gatersleben,
Germany
Isopropanol, 2-Propanol Sigma Aldrich, St.
Louis, USA
Kits und spezielle Agencourt AMPure XP Beads Beckman Coulter,
Chemikalien Krefeld, Germany
DNA/RNA AllPrep Purification Kit Qiagen, Hilden,
Germany
GeneRead Size Selection Kit Qiagen, Hilden,
Germany
QIAquick PCR Purification Kit Qiagen, Hilden,
Germany
Illumina MiseqTM Reagent Kit V3 Illumina, San Diego,
USA
PhiX Control V3 Illumina, San Diego,
USA
HiSeq Rapid SBS Kitv2 Illumina, San Diego,
USA
TruSight® Myeloid Sequencing Illumina, San Diego,
Panel0 USA
Kapa Library Quantification Kit Kapa Biosystems,
Wilmington, USA
21QubitTM dsDNA BR Assay Kit Thermo Fisher
Scientific, Waltham,
USA
QubitTM dsDNA HS Assay Kit Thermo Fisher
Scientific, Waltham,
USA
Kleinmaterialien Pipettenspitzen Bio Rad, München,
Germany
MicroAmp Optica 96 well Applied Biosystem,
Reaction plate Victoria, Australia
Biosphere Qualitätsspitzen Duran Group, Mainz,
Pipettenspitzen Germany
Adhesive PCR Film Sarstedt, Nümbrecht,
Germany
Multiply µ-Strip, 0,2ml Kette Sarstedt, Nümbrecht,
Germany
Multiplate PCR Plate, 96 well Sarstedt, Nümbrecht,
Germany
8er Deckelkette Sarstedt, Nümbrecht,
Germany
Reagiergefäße 1,5ml Sarstedt, Nümbrecht,
Germany
15 ml Falcon Tube, Sarstedt, Nümbrecht,
Zentrifugenröhrchen Germany
50 ml Falcon Tube, Sarstedt, Nümbrecht,
Zentrifugenröhrchen Germany
Enzyme und Q5® High-Fidelity DNA New England Biolabs,
biologische Polymerase Ipswich, USA
Substanzen Desoxyribonukleosidtriphosphate Thermo Fisher
(dNTPs) 10mM Scientific, Waltham,
USA
Q5®-Puffer New England Biolabs,
Ipswich, USA
Oligodesoxyribonukleotide Eurofins scientific,
(Primer) Luxemburg
PCR Puffer 10x (15nM MgCl2) Qiagen, Hilden,
Germany
HotStar Taq DNA Polymerase Qiagen, Hilden,
Germany
1kb DNA Leiter Qiagen, Hilden,
Germany
Apparate und Hiseq 2500 Sequencer Illumina, San Diego,
Utensilien USA
MiseqTM Illumina, San Diego,
USA
Heraeus Multifuge 3 S-R Thermo Fisher
Scientific, Waltham,
USA
DynaMagTM 2 Magnet Thermo Fisher
Scientific, Waltham,
22Microcentrifuge 5415 R USA Eppendorf AG,
Hamburg, Germany
Thermomixer® compact Eppendorf AG,
Hamburg, Germany
Galaxy Mini Centrifuge – 26 Joule VWR, Radnor, USA
Qubit® 2.0 Fluorometer Thermo Fisher
Scientific, Waltham,
USA
McCyclerTM Thermal Cycler und Bio-Rad Laboratories
T100TM Thermal Cycler GmbH, München,
Germany
Vortex-Genie® 2 Scientific Industries
Inc, Bohemia, USA
Pipetten – Eppendorf Research® Eppendorf AG,
0,5-10µl; 2-20µl; 10-100µl; 20- Hamburg, Germany
200µl; 100-1000µl; 8-Kanal 0,5-
10µl
Pipetten – PIPETMAN P2, P10, GLISON Inc,
P20, P200, P1000 Middleton, USA
2.3 Methoden
2.3.1 Grundschema
Abbildung 3. Arbeitsablauf der Probenaufbereitung.
Validierung der
Extraktion von Bestimmung der Untersuchung Bioinformatische
Mutationen
DNA aus ED Mutationen im der MRD in CR Analyse und
mittels Sanger
Proben MP mittels Miseq Bewertung
Sequenzierung
ED: Erstdiagnose; MP: Myeloid Panel; MRD: messbare Resterkrankung, CR: komplette Remission
2.3.2 DNA-Extraktion aus Blut und Knochenmark
Die DNA-Isolation aus Vollblut und Knochenmark wurde gemäß der Herstellerangaben
mit dem Allprep DNA/RNA purification kit (Qiagen, Hilden, Germany) durchgeführt.
Das Material wurde aus Zellen gewonnen, die bei -196°C in flüssigem Stickstoff
gelagert wurden. Zuerst wurden die Zellen mithilfe einer phosphatgepufferten
Salzlösung (PBS) gemischt, um diese zu lösen. Durch mehrmaliges Zentrifugieren und
Abpipettieren wurde der PBS-Puffer wieder entfernt, sodass nur noch Zellen im
Reagenz vorlagen. Anschließend wurde ein guanidiniumthiocyonathaltiger RLT plus
23Sie können auch lesen

















































