Charakterisierung von Tumorstammzellen eines Plattenepithelkarzinoms des Hypopharynx - eDiss
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Aus der Klinik für Hals-Nasen-Ohrenheilkunde
(Prof. Dr. med. D. Beutner)
der Medizinischen Fakultät der Universität Göttingen
Charakterisierung von Tumorstammzellen
eines Plattenepithelkarzinoms
des Hypopharynx
INAUGURAL-DISSERTATION
zur Erlangung des Doktorgrades
der Medizinischen Fakultät der
Georg-August-Universität zu Göttingen
vorgelegt von
Ronja Kristine Hähne, geb. Gratz
aus
Hannover
Göttingen 2020Dekan: Prof. Dr. med. W. Brück Referent: Prof. Dr. med. F. Ihler Koreferent/in: Priv.-Doz. Dr. med. dent. Dr. med. P. Brockmeyer Drittreferent/in: Prof. Dr. med. M. Oppermann Datum der mündlichen Prüfung: 20. April 2021
Hiermit erkläre ich, die Dissertation mit dem Titel „Charakterisierung von Tumorstamm-
zellen eines Plattenepithelkarzinoms des Hypopharynx“ eigenständig angefertigt und keine
anderen als die von mir angegebenen Quellen und Hilfsmittel verwendet zu haben.
Oldenburg, den ……………..………… …………….…………………
(Unterschrift)Die Daten, auf denen die vorliegende Arbeit basiert, wurden teilweise publiziert: Ihler F, Gratz R, Wolff HA, Weiss BG, Bertlich M, Kitz J, Salinas G, Rave-Fränk M, Canis M (2018): Epithelial-Mesenchymal Transition during Metastasis of HPV-Negative Pharyngeal Squamous Cell Carcinoma. Biomed Res Int 2018, 7929104
Inhalt
VERZEICHNIS DER ABBILDUNGEN..................................................................................III
VERZEICHNIS DER TABELLEN..........................................................................................III
VERZEICHNIS DES ANHANGS............................................................................................III
VERZEICHNIS DER ABKÜRZUNGEN.................................................................................IV
1 EINLEITUNG............................................................................................................................1
1.1 PLATTENEPITHELKARZINOME DES KOPF-HALS-BEREICHES....................................................................1
1.2 TUMORBIOLOGIE.............................................................................................................................3
1.2.1 Tumorbiologie und Metastasierung................................................................................3
1.2.2 Epithelial-mesenchymale Transition...............................................................................4
1.2.3 Tumorheterogenität und Tumorstammzellen...................................................................5
1.2.4 E-Cadherin als epithelialer Marker...............................................................................7
1.2.5 CD44 als Tumorstammzellmarker...................................................................................7
1.3 FRAGESTELLUNG............................................................................................................................9
2 MATERIAL UND METHODEN.............................................................................................10
2.1 ETHIK........................................................................................................................................10
2.2 GEWINNUNG UND KONSERVIERUNG VON NATIVEN GEWEBEPROBEN.......................................................11
2.2.1 Konservierung für die Genexpressionsanalyse..............................................................11
2.2.2 Konservierung für Immunhistochemie...........................................................................11
2.3 XENOGRAFT-MODELL...................................................................................................................12
2.3.1 Tierpflege......................................................................................................................12
2.3.2 Implantation des Tumors...............................................................................................12
2.3.3 Verlaufskontrolle...........................................................................................................12
2.3.4 Gewinnung von Xenotransplantaten.............................................................................13
2.3.5 Dissoziation in Einzelzellen..........................................................................................13
2.4 ZELLKULTUR................................................................................................................................15
2.4.1 Etablierung einer Zellkultur..........................................................................................15
2.4.2 Herstellen von Einzelzellsuspension.............................................................................15
2.4.3 Zählen der Zellen..........................................................................................................16
2.4.4 Trypanblaufärbung........................................................................................................16
2.4.5 Einfrieren und Auftauen von Zellen..............................................................................16
2.4.6 Sphäroidkultur..............................................................................................................17
2.5 IMMUNHISTOCHEMIE......................................................................................................................19
2.5.1 Kern-Plasma-Färbung von Zellen................................................................................19
2.5.2 Vorbereitung der Gewebeproben für die Immunhistochemie.........................................19
2.5.3 Kern-Plasma-Färbung der Gewebeproben..................................................................20
2.5.4 Färbungen der Gewebeproben mit E-Cadherin und CD44..........................................20
2.6 DURCHFLUSSZYTOMETRIE...............................................................................................................22
2.7 GENEXPRESSIONSANALYSE..............................................................................................................23
2.7.1 Herstellung der Proben aus Zellkulturen......................................................................23
2.7.2 Verarbeitung von Proben im Transkriptomanalyselabor...............................................23
2.7.3 Datenanalyse durch das Transkriptomanalyselabor.....................................................23
2.7.4 Auswertung Mithilfe ausgewählter Gensets..................................................................24
3 ERGEBNISSE..........................................................................................................................26
3.1 PATIENTENCHARAKTERISTIKA..........................................................................................................26
3.1.1 Patient H194.................................................................................................................26
3.1.2 Patient H196.................................................................................................................27
I3.1.3 Patient H200.................................................................................................................27
3.1.4 Patient H204.................................................................................................................28
3.1.5 Patient H208.................................................................................................................29
3.2 XENOGRAFT-MODELL...................................................................................................................30
3.3 ZELLKULTUR................................................................................................................................33
3.4 EXPRESSION VON E-CADHERIN.......................................................................................................35
3.4.1 Immunhistochemie........................................................................................................35
3.4.2 Durchflusszytometrie.....................................................................................................35
3.4.3 Genexpressionsanalyse.................................................................................................36
3.5 EXPRESSION VON CD44...............................................................................................................38
3.5.1 Immunhistochemie........................................................................................................38
3.5.2 Durchflusszytometrie.....................................................................................................38
3.5.3 Genexpressionsanalyse.................................................................................................38
3.6 GENEXPRESSIONSANALYSE..............................................................................................................41
3.6.1 Allgemeine Betrachtung................................................................................................41
3.6.2 Vergleich der Genexpression durch ausgewählte Gensets.............................................43
4 DISKUSSION...........................................................................................................................46
4.1 METHODEN.................................................................................................................................46
4.2 VERGLEICH DES PRIMÄRTUMORS MIT SEINER METASTASE....................................................................47
4.3 TUMORSTAMMZELLEIGENSCHAFTEN UND MESENCHYMALER PHÄNOTYP IN DER SPHÄROIDKULTUR................49
4.4 DER VERLUST VON E-CADHERIN ALS MARKER FÜR EMT................................................................51
4.5 CD44 ALS TUMORSTAMMZELLMARKER............................................................................................54
5 ZUSAMMENFASSUNG..........................................................................................................58
6 ANHANG..................................................................................................................................60
7 LITERATURVERZEICHNIS.................................................................................................71
DANKSAGUNG.........................................................................................................................83
IIVerzeichnis der Abbildungen
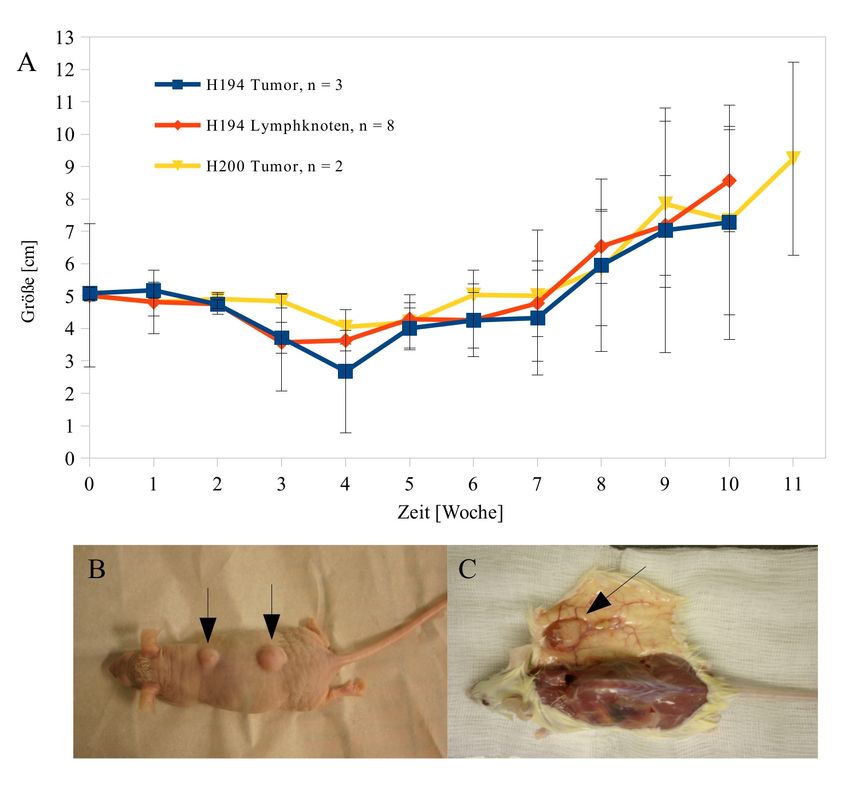
Abb. 1: Biometrie der Gewebeproben..................................................................................32
Abb. 2: Zellkulturen im Verlauf...........................................................................................34
Abb. 3: Expression von E-Cadherin.....................................................................................37
Abb. 4: Expression von CD44..............................................................................................40
Abb. 5: Varianz in der Genexpression der Gewebeproben und der Zellkulturen.................42
Abb. 6: Kombinationen der Probenvergleiche.....................................................................43
Abb. 7: Überschneidungen der verwendeten Gensets..........................................................44
Verzeichnis der Tabellen
Tab. 1: Gewebeproben der I. Generation.............................................................................30
Tab. 2: Gewebeproben der II. Generation...........................................................................31
Tab. 3: E-Cadherin-positive Zellen in der Durchflusszytometrie........................................35
Tab. 4: Genexpression von E-Cadherin (CDH1).................................................................36
Tab. 5: CD44-positive Zellen in der Durchflusszytometrie................................................38
Tab. 6: Genexpression von CD44........................................................................................39
Tab. 7: Expressionsanalyse der Gewebeproben und Zellkulturen mittels Gensets.............45
Verzeichnis des Anhangs
Anhang I: Liste der Chemikalien und Pharmazeutika..........................................................60
Anhang II: Liste des Zubehörs.............................................................................................62
Anhang III: Liste der Antikörper..........................................................................................63
Anhang IV: Liste der Versuchs- und Nachweiskits..............................................................63
Anhang V: Liste der Geräte..................................................................................................64
Anhang VI: Liste der Tiere...................................................................................................65
Anhang VII: Liste der Software...........................................................................................65
Anhang VIII: Zusammensetzung der verwendeten Lösungen und Medien.........................65
Anhang IX: Immunhistochemische Schnitte: Übersichtsbilder...........................................66
Anhang X: Auflistung der Gene der verwendeten Gensets..................................................67
IIIVerzeichnis der Abkürzungen
cDNA complementary DNA, komplementäre Desoxyribonukleinsäure
cRNA complementary RNA, komplementäre Ribonukleinsäure
CUP cancer of unknown primary, Karzinom mit unbekanntem Primärtumor
DMEM Dulbecco’s Modified Eagle Medium
DMSO Dimethylsulfoxid
DNA deoxyribonucleic acid, Desoxyribonukleinsäure
EDTA Ethylendiamintetraacetat
EGF epidermal growth factor, epidermaler Wachstumsfaktor
EMT epithelial-mesenchymale Transition
FDR false discovery rate, Rate der falsch-positiven Ergebnisse
FGF fibroblast growth factor, Fibroblasten-Wachstumsfaktor
FKS fetales Kälberserum
GSEA gene set enrichment analyses, Genanreichungsanalyse
HNO Hals-Nasen-Ohrenheilkunde
IVC individual-ventilated cages, einzeln belüftete Käfige
IVT In-vitro-Transkription
KHK koronare Herzkrankheit
kAECG-Medium komplettiertes Airway-Epithelial-Cell-Growth-Medium
LAVES Landesamt für Verbraucherschutz und Lebensmittelsicherheit
MET mesenchymal-epitheliale Transition
MSigDB Molecular Signatures Database
nom. nominal
ns nicht signifikant
PBS Phosphate-buffered Saline, phosphatgepufferte Salzlösung
PCA principal component analysis, Hauptkomponentenanalyse
PE Phycoerythrin
RNA ribonucleic acid, Ribonukleinsäure
TAL Transcriptome and Genome Analysis Laboratory, Transkriptom- und
Genomanalyselabor
TM Trademark
TNM-Klassifikation T = Tumor = Primärtumor, N = Node = Lymphknoten,
M = Metastasis = Fernmetastase
UICC Union for International Cancer Control
upm Umdrehungen pro Minute
ZTE Zentrale Tierexperimentelle Einrichtung
IV1. Einleitung
1 Einleitung
1.1 Plattenepithelkarzinome des Kopf-Hals-Bereiches
An bösartigen Tumore des Kopf-Hals-Bereichs erkranken jährlich weltweit 550 000 Men-
schen und 380 000 Todesfälle werden hierdurch registriert (Global Burden of Disease Can-
cer Collaboration et al. 2017). Bei Männern sind sie die sechsthäufigste Tumorentität welt-
weit (Moral und Paramio 2008). Bei Frauen sind sie weitaus seltener, nehmen allerdings
korrelierend zu dem vermehrten Auftreten von weiblichen Raucherinnen zu (Strutz und
Mann 2001). Kopf-Hals-Tumore unterscheiden sich in der Lokalisation, Histologie, Epide-
miologie und Ätiologie. Die häufigste Lokalisation für bösartige Tumore im Kopf-Hals-
Bereich ist die Mundhöhle (Simon und Plinkert 2008). Weiterhin treten maligne Ge-
schwulste im Larynx und im Pharynx auf. Letzterer lässt sich in Naso-, Oro- und Hypopha-
rynx unterteilen, wobei bösartige Tumore des Hypopharynx die schlechteste Prognose aller
Kopf-Hals-Malignitäten haben (Shen et al. 2013). Histologisch finden sich bei über 90 %
der Kopf-Hals-Tumore ein Plattenepithelkarzinom, aber auch Adenokarzinome, Sarkome,
lymphoepitheliale Tumore und Melanome (Riede et al. 2004; Kumar et al. 2009).
Plattenepithelien entwickeln sich aus embryonalem Bindegewebe, dem unpolaren Me-
senchym. Es besteht wie alle Epithelien aus Zellen mit apikaler-basaler Zellpolarität, die
durch Zell-Zell-Kontakte wie Zonula adhaerens, Zonula occludens und Desmosomen auf-
rechterhalten wird (Welsch und Deller 2014). Im Hypopharynx findet sich unverhorntes
mehrschichtiges Plattenepithel. Dabei befinden sich die mitotisch aktiven Stammzellen in
der Basalschicht auf der Basalmembran, welche eine mechanische Grenze zum darunter
liegenden Gewebe bildet (Lüllmann-Rauch 2015). Von der Basalschicht aus steigen die
Zellen über die Intermediärschicht zur Superfizialschicht am Lumen auf und differenzieren
sich dabei aus (Lüllmann-Rauch 2015). Ist die Ausdifferenzierung gestört, wird dies als
Dysplasie bezeichnet, welche je nach Ausprägung in drei Schweregrade eingeteilt werden
kann (Pschyrembel 2014). Entarten die Zellen weiter, entsteht ein Carcinoma in situ, ein
bösartiger Tumor aus epithelialen Zellen, welcher noch nicht die Basalmembran durch-
bricht. Bilden sich weitere Mutationen und Veränderungen aus, entwickelt sich ein Karzi-
nom mit invasivem und metastasierendem Verhalten (Lüllmann-Rauch 2015).
Als Risikofaktoren für Plattenepithelkarzinome gelten übermäßiger Alkoholkonsum,
Rauchen, Infektionen mit bestimmten Subtypen der humanen Papillomaviren, Ep-
stein-Barr-Virus-Infektionen und chronische Exposition mit einigen industriellen Substan-
zen (Sankaranarayanan et al. 1998; Strzelczyk et al. 2015). Der Altersgipfel für die Ausbil-
11. Einleitung
dung von HNO-Tumoren liegt bei Männern zwischen der fünften und siebten, bei Frauen
zwischen der sechsten und achten Lebensdekade (Mashberg und Samit 1995).
Als Hauptrisikofaktoren für die Entwicklung von Plattenepithelkarzinomen des Hypo-
pharynx gelten vor allem Alkohol- und Nikotinabusus. Sie metastasieren zunächst lympho-
gen und später auch hämatogen in Lunge, Leber und Knochen (Buckley und MacLennan
2000; Uwa et al. 2011; Lenarz und Boenninghaus 2012; Kulasinghe et al. 2015). Meist
werden Hypopharynxkarzinome erst im fortgeschrittenen Stadium erkannt. Dies liegt am
späten Auftreten von Symptomen wie Dysphagie, Odynophagie, Foetor ex ore, Lymphkno-
tenschwellungen, Heiserkeit, Hämoptysen, Hämatemesis oder Schmerzen (Grevers et al.
2008; Milisavljevic et al. 2009). Die prätherapeutische Einteilung wird nach klinischer Un-
tersuchung sowie Panendoskopie mit Probenentnahme anhand der TNM-Klassifikation
(Tumor, Node, Metastasis) und UICC-Stadium (Union for International Cancer Control)
festgelegt und ist bedeutend für die Behandlungsmöglichkeiten und die Prognose (Lenarz
und Boenninghaus 2012; Wittekind et al. 2015; Wittekind 2017). Günstigenfalls erfolgt die
Behandlung durch chirurgische Entfernung des Hypopharynxkarzinoms und gegebenen-
falls der regionalen Lymphknoten (Neck dissection). Bei fortgeschrittenen Stadien werden
adjuvant oder primär die Tumorregion und ihre Lymphabflusswege bestrahlt oder eine
kombinierte platinbasierte Radiochemotherapie durchgeführt. Die Prognose ist bei Hypo-
pharynxkarzinomen im Vergleich zu anderen Karzinomen im Kopf-Hals-Bereich sehr
schlecht. Bei Tumorprogress versterben die Patienten an den Komplikationen durch lokale
Rezidive, Zweittumoren und Fernmetastasen (Scanlon et al. 2013; Baxi et al. 2014).
21. Einleitung
1.2 Tumorbiologie
1.2.1 Tumorbiologie und Metastasierung
Im Allgemeinen beschreibt der Begriff Tumor ein Geschwulst beziehungsweise eine „ört-
lich umschriebene Zunahme des Gewebevolumens“ und kann benigne oder maligne sein
(Pschyrembel 2014). Um als bösartig zu gelten, muss er besondere Eigenschaften erlangen.
Hanahan und Weinberg (2011) beschrieben sechs Hauptcharakteristika, die einen bösarti-
gen Tumor von gesundem Gewebe unterscheiden. Diese beinhalten Aufrechterhaltung von
Signalen zur Proliferation, Entziehung vor Wachstumshemmern, Widerstand gegen den
Zelltod und Apoptosis, Induktion von Angiogenese, Möglichkeit zur unendlichen Zelltei-
lung sowie Invasivität und Metastasierung (Hanahan und Weinberg 2011). Durch diese
sechs Eigenschaften werden die natürlichen Alterungsprozesse einer Zelle und das Gleich-
gewicht im Zellverband zerstört.
Invasivität und Metastasierung haben eine besondere Bedeutung für die Prognose und
die therapeutischen Anforderungen, da die Metastasierung für über 90 % der tumorassozi-
ierten Mortalität verantwortlich ist (Weigelt et al. 2005; Jemal et al. 2006; Steeg 2006;
Patel und Chen 2012). Außerdem kommt es durch tumoröse Absiedlungen zu zusätzlichen
Krankheitssymptomen wie beispielsweise pathologischen Knochenbrüchen, Kompression
von Organen mit Lumen sowie Tumorthromben oder Tumorembolien (Böcker et al. 2012).
Damit eine Makrometastase entstehen kann, müssen die Zellen eine metastatische Kaskade
durchlaufen (Geiger und Peeper 2009). Zunächst findet das Durchbrechen der Basalmem-
bran und die Invasion des darunter gelegenen Gewebes statt. Anschließend löst sich eine
Zelle oder ein Zellverband los und migriert in der Extrazellulärmatrix zu den Gefäßen. Es
erfolgt die Intravasation in Blut- oder Lymphgefäße mit Hilfe geeigneter Oberflächenprote-
ine. Hier muss die Zelle außerhalb des Zellverbandes und ohne Kontakte zur Extrazellulär-
matrix überleben. Dies würde im gesunden Gewebe in der Anoikis, der eingeleiteten Apop-
tose bei Verlust von Zell-Zell-Kontakten, und damit im Zelltod enden (Frisch und Francis
1994). Widersteht die Zelle der Apoptoseeinleitung, zirkuliert die Tumorzelle bis sie im
Kapillarbett hängen bleibt oder mit Hilfe von Oberflächenproteinen das Gefäß wieder ver-
lässt (Extravasation). Dort erwartet die Zelle ein neues Milieu mit anderem Aufbau, ande-
ren Strukturen sowie Signalen als in ihrem Ursprungsgewebe und ihrer Ursprungsumge-
bung. Hier gibt es mehrere Möglichkeiten. Erstens kann es aufgrund fehlender wichtiger
Signale zum Zelltod kommen. Zweitens kann sich die Zelle in die Quieszenz begeben,
einen Status in der G0-Phase des Zellzyklus, in dem sie sich nicht teilt und Jahre überleben
31. Einleitung
kann (Scholzen und Gerdes 2000; Prince und Ailles 2008). Drittens kann es in einem pas-
senden neuen Milieu zur Kolonisation sowie Proliferation und somit zur Ausbildung einer
Mikrometastase kommen. Nicht jeder Ort im Körper bietet ein geeignetes Umfeld und so
metastasiert jeder Tumor nach einem charakteristischen Muster. Die Kopf-Hals-Karzinome
bilden bevorzugt Fernmetastasen in Lunge, Knochen und Leber aus (Kulasinghe et al.
2015). Dieses Phänomen wird in der Seed-and-Soil-Hypothese beschrieben, wonach die
Tumorzelle (Seed, Samen) und das Milieu des Metastasierungsortes (Soil, Erdboden) zu-
sammenpassen müssen (Paget 1889; Fidler 2003). Damit sich dort die Mikrometastase
weiter vergrößern kann, müssen die nutritiven Gegebenheiten verbessert werden (Gimbro-
ne et al. 1972) und es kommt zu einer regionalen Gefäßneubildung (Angiogenese). Es bil-
det sich eine Metastase mit starker Ähnlichkeit zum Primärtumor aus.
1.2.2 Epithelial-mesenchymale Transition
Anhand der metastatischen Kaskade kann der Vorgang der Metastasierung in zwei Haupt-
phasen aufgeteilt werden. Zuerst erfolgt der Weg samt Absiedlung an einem anatomisch
entfernten Ort und anschließend das Wachstum nach Vorbild des Primärtumors (Chaffer
und Weinberg 2011). Vor allem für die erste Phase braucht die Zelle Eigenschaften wie In-
vasivität, Migration und Überleben ohne Adhärenz. Deren Herausbildung lassen sich durch
die von Hay (1995) beschriebene epithelial-mesenchymale Transformation erklären. Hier-
bei wandelt sich eine durch Zell- und Basalmembrankontakte fest eingebundene und pola-
risierte Epithelzelle in eine Zelle mit mesenchymalen Phänotyp um. Da dieser Prozess auch
wieder umgekehrt werden kann, wird er auch als epithelial-mesenchymale Transition
(EMT) und mesenchymal-epitheliale Transition (MET) bezeichnet. Ursprünglich verwen-
det der Körper EMT bei der Fortpflanzung zur Implantation, Embryogenese und zur Or-
ganentwicklung (EMT Typ 1) (Kalluri und Weinberg 2009; Zeisberg und Neilson 2009).
Des Weiteren tritt EMT bei der Wundheilung von chronischen Entzündungen und Fibrosie-
rung zur Generierung von aktivierten Fibroblasten auf (EMT Typ 2) (Kalluri und Weinberg
2009; Zeisberg und Neilson 2009). Findet die EMT im Vorgang der Metastasierung statt,
so wird sie als EMT Typ 3 bezeichnet (Kalluri und Weinberg 2009; Zeisberg und Neilson
2009). Als Biomarker für die EMT gelten auf der einen Seite der Verlust von epithelialen
Markern wie E-Cadherin oder Zytokeratinen sowie der Abbau von Zonula adhaerens und
Desmosomen. Auf der anderen Seite erfolgt das Hervorbringen eines mesenchymalen Phä-
notyps mit einer spindelförmigen Morphologie sowie mit Expression von N-Cadherin,
41. Einleitung
α-smooth-muscle-Aktin, einem Vimentin-Zytoskelett und von EMT-vermittelnden Tran-
skriptionsfaktoren wie Snail, Slug und Twist (Hay 1995; Gavert und Ben-Ze’ev 2008; Kal-
luri und Weinberg 2009). Hervorzuheben ist der Verlust von E-Cadherin (CDH1-Gen) be-
ziehungsweise der Wechsel von E-Cadherin zu mesenchymalen Cadherinen wie
N-Cadherin (CDH2-Gen), welcher häufig bei Kopf-Hals-Tumoren zu finden ist (Birchmei-
er und Behrens 1994; Cavallaro und Christofori 2004; Hanahan und Weinberg 2011).
1.2.3 Tumorheterogenität und Tumorstammzellen
Ursprünglich wurde davon ausgegangen, dass ein Tumor homogen und ausschließlich aus
klonalen Zellen aufgebaut sei (Fialkow 1976; Nowell 1976). Allerdings stellte sich heraus,
dass sich die Tumorzellen nicht nur durch epigenetische Einflüsse im Phänotyp unterschei-
den, sondern sich zudem Subpopulationen mit unterschiedlichen genetischen Veränderun-
gen finden lassen (Heppner 1984; Hanahan und Weinberg 2011). Dies wird als Tumorhe-
terogenität bezeichnet (Heppner 1984; Hanahan und Weinberg 2011). Zum einen entwi-
ckeln sich Subpopulationen durch zusätzliche Mutationen, die durch eine genetische Insta-
bilität begünstigt werden (Nowell 1976; Prince et al. 2007). Auf der anderen Seite können
sich ganz unterschiedliche Zelllinien hierarchisch aus einer Mutterzelle entwickeln, wie es
physiologisch im hämatopoetischen Differenzierungsprozess stattfindet (Till und McCul-
loch 1980; Heppner 1984; Eaves 2015). Abgeleitet hiervon beschrieb Reya et al. (2001) die
Tumorstammzellhypothese. Diese besagt, dass sich im Tumor eine kleine Population mit
typischerweise einem Anteil von weniger als 10 % an Zellen des Gesamtgewebes befindet,
welche sich ähnlich wie Stammzellen im gesunden Gewebe verhalten (Reya et al. 2001;
Prince et al. 2007). Prince et al. (2007) konnte diese bei Plattenepithelkarzinomen im Kopf-
Hals-Bereich darstellen. Diese Tumorstammzellen teilen sich. Die Tochterzellen sind ent-
weder Tumorstammzellen oder differenzieren sich weiter (Clarke et al. 2006). Tumor-
stammzellen werden über vier Hauptcharakteristika definiert (Prince und Ailles 2008). Er-
stens ist die Selbsterneuerung essenziell. Eine Tumorstammzelle kann einen neuen Tumor
entstehen lassen und dessen Heterogenität mit tumorösen und nicht tumorösen Bestandtei-
len durch seine Tochterzellen wieder aufbauen (Clarke et al. 2006). Zweitens haben sie das
Potenzial, einen Tumor in einer immundefizienten Maus zu initiieren. Außerdem können
sie drittens über mehrere Generationen transplantiert und wieder angezüchtet werden. Vier-
tens haben sie besondere Marker auf ihrer Oberfläche, durch welche sie von anderen Tu-
morzellen separiert werden können (Pardal et al. 2003; Clarke und Fuller 2006; Wicha
51. Einleitung
et al. 2006; Dalerba et al. 2007; Prince und Ailles 2008). Mittlerweile wird oft noch eine
fünfte Eigenschaft genannt, welche eine Widerstandsfähigkeit gegenüber gängigen Chemo-
therapeutika beinhaltet (Lim et al. 2011). Grundlage hierfür ist, dass adulte Stammzellen
eine Resistenz gegen Giftstoffe besitzen. Dies geschieht unter anderem durch geringe Pro-
liferationsraten, Bereitstellung von Proteinen zur DNA-Reparatur oder den Transport von
schädlichen Stoffen aus der Zelle heraus, beispielsweise durch ATP-binding cassette trans-
porter (Dean et al. 2001; Shen et al. 2013). Zudem haben Tumorstammzellen und mesen-
chymale Zellen jeweils, im Gegensatz zu Epithelzellen, die Fähigkeit zur Migration ent-
lang der Extrazellulärmatrix (Hay 1995). Die Tumorstammzellhypothese hat eine enorme
Bedeutung für das Verständnis des Verlaufs einer Tumorerkrankung und damit für den Ein-
satz therapeutischer Strategien. Durch die Fähigkeit der Selbsterneuerung können Tumor-
stammzellen Metastasen bilden. Überlebt nur eine Zelle, so kann nach chirurgischer, Ra-
dio- oder Chemotherapie ein Rezidiv auftreten, aufgrund des Status der Quieszenz gegebe-
nenfalls erst Jahre nach der Therapie (Chen et al. 2011).
Es stellt sich die Frage, wie die Zellen mit Tumorstammzelleigenschaften in Karzino-
men entstehen. Eine Möglichkeit ist, dass adulte Stammzellen des Gewebes Mutationen
entwickeln, die Grundlage für einen bösartigen Tumor werden (Pardal et al. 2003; Brabletz
2012). Andererseits könnten sich Tumorstammzellen aus normalen epithelialen Tumorzel-
len entwickeln. Es gibt immer mehr Belege dafür, dass Letzteres mit Hilfe von epitheli-
al-mesenchymaler Transition geschieht. Der resultierende mesenchymale Phänotyp könnte
nicht nur die erste Hauptphase mit seinen neu gewonnenen Eigenschaften wie Invasivität
und Fähigkeit zur Migration absolvieren, sondern auch die zweite Phase der Kolonisation
(Morel et al. 2008; Singh und Settleman 2010; Chaffer und Weinberg 2011; Hanahan und
Weinberg 2011; da Silva et al. 2015). Durch Stimulation aus den umgebenen Zellen und
aus dem umliegenden Stroma wird der mesenchymale Phänotyp induziert (Chaffer und
Weinberg 2011).
Zur Selektion von Tumorstammzellen in vitro gibt es verschiedene Möglichkeiten, bei
denen die genannten Eigenschaften genutzt werden. Eine Option ist die Sphäroidkultur: In
einer serumfreien Zellkultur mit Böden ultraniedriger Adhäsion können epitheliale Zellen
aufgrund ihrer Eigenschaften nicht wachsen. Ohne Serum findet keine Differenzierung der
Zellen statt, aber die hinzugegebenen Wachstumsfaktoren fördern die Vermehrung (Satpute
et al. 2013). Es wird davon ausgegangen, dass durch klonales Wachstum aus jeweils einer
Tumorzelle ein Tumorsphäroid entstehen kann (Weiswald et al. 2015). Die resultierenden
61. Einleitung
Zellen besitzen nachgewiesenermaßen Tumorstammzelleigenschaften (Lim et al. 2011;
Weiswald et al. 2015; Shaheen et al. 2016). Eine Alternative zur Selektion von Tumor-
stammzellen ist die Erzeugung einer Untermenge von Zellen in der Durchflusszytometrie,
welche als Side Population bezeichnet wird. Diese Zellen besitzen an ihrer Oberfläche Pro-
teine der Familie der ATP-binding cassette transporter, die zuvor inkubierte Fluoreszenz-
farbstoffe wie Hoechst 33342 wieder aus der Zelle heraus transportieren können. Damit
würden sie in der Durchflusszytometrie als negativ selektiert werden (Goodell et al. 1996;
Behbod und Vivanco 2015).
1.2.4 E-Cadherin als epithelialer Marker
Cadherine sind eine große Gruppe von transmembranen Glykoproteinen, welche an
verschiedenen Zellverbindungen, wie zum Beispiel Zonula adhaerens, Desmosomen,
endothelialen und kardialen Junctions, beteiligt sind (Wheelock und Johnson 2003). Der
extrazelluläre Teil baut eine Verbindung mit dem auf der Nachbarzelle präsentierten
Cadherin auf, während der intrazelluläre Teil über weitere Proteine an das Zytoskelett
bindet oder Signale weiterleitet (Goodwin und Yap 2004; Jeanes et al. 2008).
E-Cadherin ist eines der Hauptproteine auf Epithelzellen zur Ausbildung einer
junktionalen Zonula adhaerens und ist über ß-Catenin mit dem Aktin-Zytoskelett der Zelle
verbunden (Bánkfalvi et al. 2002; Wheelock und Johnson 2003; Scanlon et al. 2013).
Durch diese calciumabhängigen Zell-Zell-Kontakte kann sich die Zellpolarität im Epithel
ausbilden (Cavallaro und Christofori 2004). Es scheint einen Zusammenhang zwischen der
Expression von E-Cadherin und Tumoreigenschaften zu geben. Wird der Verlust von
E-Cadherin induziert, kann dies zu Tumorinitiierung, Invasion und Metastasierung führen
(Derksen et al. 2006; Jeanes et al. 2008; Nijkamp et al. 2011). Wird dagegen E-Cadherin in
einer invasiven Zelllinie transfiziert, vermindert sich ihre Fähigkeit zur Invasion (Vle-
minckx et al. 1991).
1.2.5 CD44 als Tumorstammzellmarker
Ein Marker für Tumorstammzellen von Plattenepithelkarzinomen des Kopf-Hals-Bereichs
ist CD44 (Prince et al. 2007; Shen et al. 2013). CD44 ist ein Oberflächenglykoprotein, wel-
ches hauptsächlich als Ligand zur Hyaluronsäure in der Extrazellulärmatrix dient (Goodi-
son et al. 1999). Des Weiteren bindet es an Kollagen, Fibronectin, Laminin und Osteopon-
tin (Naor et al. 1997). Es treten zwei Isoformen auf: CD44s für die Standard- und CD44v
71. Einleitung
für die Variantisoform. Diese Formen entstehen durch alternatives Splicing und lassen sich
anhand der verwendeten Exone in weitere Untergruppen einteilen (Tölg et al. 1993; Todaro
et al. 2014). Exprimiert wird CD44 auf hämatopoetischen, mesenchymalen und einigen
epithelialen Zellen (Hanagiri et al. 2012; Shen et al. 2016). Es ist beteiligt an der Anregung
zur Aggregation, Zellproliferation, Migration und Angiogenese (Hanagiri et al. 2012; Shen
et al. 2016). Lymphozyten können CD44 zur Fortbewegung in der Extrazellulärmatrix ver-
wenden und aktivierte T-Lymphozyten können darüber an Endothelzellen von Gefäßen
binden und diese verlassen (DeGrendele et al. 1997; Estess et al. 1998; Goodison et al.
1999).
81. Einleitung
1.3 Fragestellung
Zielsetzung der Arbeit ist der Vergleich von Primärtumor und Metastase eines Plattenepi-
thelkarzinoms des Hypopharynx in Hinblick auf die Tumorstammzelleigenschaften sowie
die epithelial-mesenchymale Transition. Hierzu ergaben sich folgende Fragen:
1. Inwieweit unterscheiden sich Primärtumor, Lymphknotenmetastase, primäre Zellkultur
und Sphäroidkultur in der Ausprägung der EMT?
2. Eignet sich die Sphäroidkultur als Selektion für eine Zellpopulation mit
Tumorstammzelleigenschaften beziehungsweise mit einem mesenchymalen Phänotyp?
3. Wie sehr eignet sich der Verlust von E-Cadherin als Marker für EMT in Primärtumor,
Lymphknotenmetastase, primärer Zellkultur sowie Sphäroidkultur?
4. Ist CD44 dafür geeignet um im Primärtumor, in einer Lymphknotenmetastase sowie in
der Zellkultur und Sphäroidkultur des Primarius die Tumorstammzellen zu detektieren?
5. Welche Veränderungen in der Genexpression gehen mit CD44 einher?
92. Material und Methoden
2 Material und Methoden
Im folgenden Kapitel werden die verwendeten Methoden dargestellt. Eine genaue Auflis-
tung aller Materialien befindet sich im Anhang I - VIII.
2.1 Ethik
Die Genehmigung des Tierversuchantrages erfolgte am 01.02.2010 unter dem Geschäfts-
zeichen 33.14-42502-04-13/1105 durch das Niedersächsischen Landesamt für Verbraucher-
schutz und Lebensmittelsicherheit, LAVES, Postfach 3949, 26029 Oldenburg.
Die Entnahme von Gewebe wurde von der verantwortlichen Ethikkommission der
Universitätsmedizin Göttingen, Von-Siebold-Straße 3, 37075 Göttingen, unter dem Ge-
schäftszeichen 9/12/10 autorisiert.
102. Material und Methoden
2.2 Gewinnung und Konservierung von nativen Gewebeproben
Die in dieser Arbeit untersuchten Gewebeproben wurden im Rahmen einer kurativen Tu-
moroperation im Operationssaal entnommen und direkt in das Labor überführt. Nach dem
Transport erfolgte unter steriler Abluft in einer Petrischale zunächst die makroskopische
Reinigung der Probe von Bindegewebe, Nekrosen, Verbrennungen oder Ähnlichen. An-
schließend wurde die Probe je nach gewünschter Größe zerteilt und weiter verwendet.
2.2.1 Konservierung für die Genexpressionsanalyse
Für die Konservierung für die Genexpressionsanalyse erfolgte schnellstmöglich die Beför-
derung von drei 1 mm³ großen nativen Proben zusammen mit 1 ml RNAlater in ein
2-ml-Eppendorfgefäß. Diese wurde über Nacht bei 4 °C gekühlt und anschließend bei
-20 °C eingefroren, um sie später für Genexpressionsanalysen zu verwenden.
2.2.2 Konservierung für Immunhistochemie
Ein weiterer Block von 0,5 cm³ bis 1 cm³ großen Proben wurde für immunhistochemische
Färbungen über Nacht in 10 ml 4,5%iges Formalin eingelegt. Eine Weiterverarbeitung der
Probe erfolgte am nächsten Tag.
112. Material und Methoden
2.3 Xenograft-Modell
2.3.1 Tierpflege
Die in den vorliegenden Untersuchungen verwendeten Mäuse wurden in der Zentralen
Tierexperimentellen Einrichtung (ZTE) der Universitätsmedizin Göttingen gehalten. Alle
Tiere wurden in 1500 cm² großen IVC-Käfigen verwahrt. Die NOD-SCID-Mäuse erhielten
sowohl bestrahltes Futter als auch gefiltertes Wasser, wohingegen NMRI-nu-Mäuse norma-
les Futter und Wasser bekamen. Käfige, Futter und Wasser wurden wöchentlich
gewechselt. Die Gruppengrößen beinhalteten vier Mäuse pro Käfig.
2.3.2 Implantation des Tumors
Die Implantation von Gewebe als Xenograft in immundefiziente Mäuse erfolgte wie be-
reits von Morton und Houghten (2007) sowie Canis et al. (2013) beschrieben.
Nach der Gewinnung wurden Gewebeproben mit einen Durchmesser von bis zu 2 mm
in ein 15-ml-Röhrchen überführt und mit 3 ml Nährlösung (s. Anhang VIII: Medium der
primären Zellkultur) bedeckt. Anschließend erfolgte die Narkose der immunsupprimierten
Maus mit 3 ml/min Sauerstoff und 6 Volumen/% Sevofluran. Die Narkose wurde mit einer
Erhaltungsdosis von 3 ml/min Sauerstoff und 3 Volumen/% Sevofluran fortgesetzt.
Die Implantation erfolgte zwischen den Schulterblättern und auf dem kaudalen Teil des
Rückens in der Medianlinie. Zunächst wurden die Bereiche desinfiziert. Anschließend er-
folgte ein 2 mm langer horizontaler Schnitt im Operationsgebiet. Mit einer Präparations-
schere wurde die Subkutanschicht der Haut in kranialer Richtung aufgespalten. In die nun
entstandene Tasche wurde eine Gewebeprobe platziert und mit einem Tropfen ampicillin-
haltiges Medium (Anhang VIII: Medium der primären Zellkultur) bedeckt. Danach erfolgte
der Verschluss des Schnittes mit Einzelknopfnähten mittels resorbierbarem Nahtmaterial.
Anschließend wurde das Körpergewicht der Maus zum ersten Mal gemessen.
Als Schmerzmedikation bekamen die Mäuse 500 mg Metamizol in 500 ml Trinkwasser
ab zwei Tage vor bis drei Tage nach der Operation verabreicht.
2.3.3 Verlaufskontrolle
Als Verlaufskontrolle erfolgte wöchentlich die Messung des Gewichts der Tiere und der
Größe der Proben. Dabei wurde auch auf eventuelle Infektionen und weitere Auffälligkei-
ten geachtet. Der Durchmesser der Transplantate wurde in kranio-kaudaler sowie in hori-
122. Material und Methoden
zontaler Richtung mit einem Messschieber gemessen. Aus den beiden Werten erfolgte die
Berechnung des mittleren Durchmessers. Als angewachsen galt eine Probe, wenn sie sich
um mehr als 0,7 mm pro Woche über drei Wochen vergrößerte oder langfristig einen
mittleren Durchmesser größer als sieben Millimeter erreichte.
Die NOD-SCID-Mäuse wurden für die Messungen sediert. Im Gegensatz dazu konnten
die NMRI-nu-Mäuse unter steriler Abluft und wach gemessen werden.
2.3.4 Gewinnung von Xenotransplantaten
Jedes Transplantat mit einem mittleren Durchmesser über 9 - 10 mm wurde entnommen.
Dafür wurde die Maus zunächst durch Einleiten von 10 ml/min CO2 in einen Käfig eutha-
nasiert und zusätzlich durch einen Genickbruch sicher getötet. Anschließend erfolgten die
letzten Messungen des mittleren Durchmessers des Implantats und Fotografien der Maus
von der Seite und von oben. Schließlich wurden die Tumore unter sterilen Bedingungen
entnommen, vom restlichen Bindegewebe befreit und in eine Petrischale überführt. Es wur-
den wie bei den nativen Gewebeproben Anteile für Immunhistochemie, Genexpressions-
analyse und Xenotransplantation gesichert.
2.3.5 Dissoziation in Einzelzellen
Die Dissoziation der Xenotransplantate in Einzelzellen wurde mit dem gentleMACSTM-
Dissoziator (Miltenyi Biotec, Bergisch Gladbach, Deutschland) nach einem standardisier-
ten Protokoll des Tumor Dissociation Kit, human von Miltenyi Biotec (Bergisch Gladbach,
Deutschland) durchgeführt.
Zur Vorbereitung wurden die einzelnen Lösungen zubereitet. Hierfür wurde Lösung I
mit 3 ml komplettiertem Airway-Epithelial-Cell-Growth-Medium (kAECG-Medium, An-
hang VIII) gelöst und in 200 µl Portionen in Eppendorfgefäße gefüllt. Bei Lösung II er-
folgte die Lösung in 2,7 ml desselben Mediums und wurde in 100 µl Rationen portioniert.
Lösung III wurde mit 1 ml Reconstition Buffer zubereitet und jeweils 25 µl pro Eppendorf-
gefäß abgefüllt. Die so hergestellten Ansätze wurden bei -20 °C bis zu ihrer Verwendung
gelagert. Vor Dissoziationsbeginn wurde ein gentleMACSTM C-Tube (Miltenyi Biotec, Ber-
gisch Gladbach, Deutschland) pro Transplantat mit 4,7 ml kAECG-Medium (Anhang VIII)
und je einer Portion der Lösungen I, II, III befüllt.
Für die Herstellung der Zellsuspensionen erfolgte zunächst eine Desinfektion der noch
unzerschnittenen Gewebeprobe mit Schleimhautdesinfektionsmittel. Danach wurde sie mit
132. Material und Methoden
einem Skalpell auf eine Größe von 1 mm³ zerkleinert und anschließend in das vorbereitete
gentleMACSTM C-Tube überführt. Nun erfolgte die Durchführung der drei von Miltenyi
Biotec (Bergisch Gladbach, Deutschland) voreingestellten und standardisierten Programme
des gentleMACSTM-Dissoziator zur schrittweisen Herstellung einer Zellsuspension. Als Er-
stes wurde h_Tumor_01 verwendet, im Anschluss das C-Tube für 30 Minuten im Inkubator
aufbewahrt und alle 10 Minuten vorsichtig geschüttelt. Hinterher erfolgte die Ausführung
des Programms h_Tumor_02 und die Inkubation wie zuvor. Nachdem das Programm
h_Tumor_03 am Dissoziator durchgeführt wurde, ergab sich die gewünschte Zellsuspensi-
on. Diese wurde über einen 70-µm-Filter von noch vorhandenen größeren Zellverbänden
bereinigt und in ein 15-ml-Gefäß überführt. Anschließend erfolgte die Durchspülung des
Filters über einem zweiten Gefäß mit 10 ml Nährlösung. Die Zellsuspension wurde danach
10 Minuten lang bei 1200 upm zentrifugiert und der Überstand verworfen. Das Pellet
konnte nun zur Etablierung einer Zellkultur verwendet werden.
142. Material und Methoden
2.4 Zellkultur
Für die Zellkultur war der Inkubator auf eine Temperatur von 37 °C sowie 5 % CO2 einge-
stellt.
2.4.1 Etablierung einer Zellkultur
Die frisch dissoziierten Zellen im Pellet wurden in kAECG-Medium gelöst und auf 50-ml-
und 250-ml-Zellkulturflaschen verteilt. Im Anschluss erfolgte die Lagerung im Inkubator
für vier bis sieben Tage. Nach ein bis zwei Wochen wurden im kAECG-Medium noch
10 % FKS ergänzt. Anschließend erfolgte die Umstellung der primären Zellkultur in meh-
reren Verdünnungsschritten auf ein Medium mit DMEM/RPMI als Grundlage (An-
hang VIII: Medium der primären Zellkultur). Dies wurde je nach Verbrauch und Füllung
der Flaschen alle zwei bis drei Tage gewechselt.
Eine Passagierung der Zellen erfolgte entweder bei Platzmangel oder spätestens nach
vier- bis fünfmaligem Öffnen der Zellkulturflaschen. Hierfür wurde zunächst eine Einzel-
zellsuspension hergestellt, um die Zellen zählen zu können. Anschließend erfolgte die Pi-
pettierung der Zellen in neue Zellkulturflaschen und das Auffüllen mit Nährlösung.
2.4.2 Herstellen von Einzelzellsuspension
Einzelzellsuspensionen wurden auf verschiedenen Wegen gewonnen, je nachdem wie in-
tensiv die Zellen aneinander und am Flaschenboden hafteten.
Eine in dem Labor übliche Trypsinierung für weniger stark haftende Zellen erfolgte zu-
nächst durch Abgießen der Nährlösung. Anschließend wurden die Zellen gewaschen, in-
dem je nach Flaschengröße 1,5 - 3 ml PBS über den Flaschenboden gegeben und wieder
abpipettiert wurde. Hinterher wurde 1,5 - 3 ml 0,07%ige Trypsinlösung (Anhang VIII) in
die Zellkulturflasche gegeben und etwa fünf Minuten im Inkubator aufbewahrt. Nach
mehrmaligem Klopfen hatten sich die Zellen gelöst. Anschließend wurde die gewünschte
Menge mit dem jeweiligen Medium aufgefüllt.
Indessen erfolgte bei stark haftenden Zellen nach dem Abgießen des Mediums erst das
Waschen mit PBS und anschließend die fünfminütige Inkubation in 1,5 - 3 ml EDTA-
Lösung (Anhang VIII). Nachdem diese abpipettiert worden war, wurde 1,5 - 3 ml
0,05%ige Trypsinlösung hinzugegeben und die bedeckten Zellen fünf Minuten im Inkuba-
tor gelagert. Anschließend wurde die gewünschte Menge mit dem jeweiligen Medium auf-
gefüllt.
152. Material und Methoden
2.4.3 Zählen der Zellen
Zur Vorbereitung zum Zählen von Zellen wurden die Trägerstege einer Neubauer-Zählkam-
mer vorsichtig angefeuchtet, ein Deckglas auf sie aufgelegt und eine ausreichende Adhäsi-
on sichergestellt. Es war darauf zu achten, dass sich Newtonsche Interferenzringe zeigten
und das Deckglas mittig aufsaß, damit ein möglichst genaues Messvolumen gegeben war.
Anschließend wurde die Zellkultur in Einzelzellen dissoziiert und es erfolgte das Auf-
füllen mit Nährmedium bis zur 10-ml-Grenze. Nach gutem Durchmischen wurde mit einer
Pasteurpipette eine Probe entnommen und am Rand des Deckglases auf die Neubauer-
Zählkammer aufgetragen. Dieser Vorgang wurde mit einer neuen Pasteurpipette auf der
zweiten Seite der Zählkammer wiederholt.
Unter dem Mikroskop wurden alle Zellen in diesen Quadraten von oben nach unten ge-
zählt, es sei denn, sie befanden sich auf der unteren oder rechten Begrenzungslinie. Aus
beiden Zählungen der zwei Kammern wurde der Mittelwert gebildet und mit 10 5 multipli-
ziert. Die daraus resultierende Zahl wurde als Anzahl der Zellen in 10 ml betrachtet.
2.4.4 Trypanblaufärbung
Eine Typanblaufärbung diente dazu, lebende von toten Zellen zu unterscheiden, und erfolg-
te nach Angaben des Herstellers. Zunächst wurden die Zellen mit ihrer Nährlösung abpipet-
tiert und in ein 15-ml-Gefäß überführt. Anschließend erfolgte die zehnminütige Zentrifuga-
tion bei 1200 upm, um danach den Überstand abzugießen. Nun wurden die Zellen mit einer
Pipette gemischt und davon 200 µl in ein Gefäß befördert. Hinterher wurden noch 400 µl
der 0,04%ige Trypanblaulösung in das Gefäß hinzugefügt. Nach erneutem Mischen erfolg-
te das Zählen der Zellen.
Lebende Zellen konnten das Trypanblau nicht aufnehmen, leuchten dadurch weißlich,
maximal hellblau, auf blauem Grund und hatten eine runde Form. Abgestorbene Zellen wa-
ren blau.
2.4.5 Einfrieren und Auftauen von Zellen
Ein Einfrieren von Zellen ist eine Möglichkeit, um Überschüsse für einen späteren Zeit-
punkt zu konservieren. Hierfür wurde zunächst die Anzahl der Zellen zum Einfrieren be-
stimmt. Für ein 2-ml-Probengefäß erfolgte die Separation von einer Million Zellen und im
Anschluss die zehnminütige Zentrifugation bei 1200 upm. Der Überstand wurde verworfen
und die Zellen auf Eis in 1,8 ml kaltem Einfriermedium (Anhang VIII) aufgenommen. Die
162. Material und Methoden
Suspension wurde in ein Probengefäß überführt, mit -1 °C pro Minute in einer Einfrierbox
langsam gefroren und bei -80 °C dauerhaft gelagert.
Zum Auftauen wurden die Probengefäße bei Raumtemperatur gelagert, bis sich der ge-
frorene Inhalt von der Wand ablöste. Dieser wurde anschließend mit 10 ml Nährlösung für
die jeweilige Zellart verdünnt. Nun erfolgte die Abzentrifugation des Einfriermediums für
zehn Minuten bei 1200 upm. Nach der Resuspension der Zellen im gewünschten Medium
wurden diese auf Zellkulturflaschen verteilt und im Inkubator aufbewahrt. Nach drei bis
fünf Stunden wurde die Kultur kontrolliert. Hatten sich Zellen abgesetzt, wurde das über-
stehende Medium in eine neue 50-ml-Flasche überführt und die Kultur mit den abgesetzten
Zellen wieder aufgefüllt. Wenn sich keine Zellen abgesetzt hatten, stand die Kultur weitere
zwölf Stunden im Inkubator. Am Tag nach dem Auftauen wurde noch einmal die Nährlö-
sung gewechselt, um die verbliebenen Zelltrümmer zu beseitigen. Alle aufgetauten Zell-
kulturen, die sich nicht innerhalb von 48 Stunden abgesetzt hatten, wurden verworfen.
2.4.6 Sphäroidkultur
Eine Sphäroidkultur wurde nach Chen et al. (2011) und Lim et al. (2011) durchgeführt.
Das Tumorstammzellmedium bestand aus serumfreien DMEM/F12 im Verhältnis 1:1
mit je 1 ml/100 ml Medium und den Zusätzen N-2 sowie B-27. Außerdem wurden die
Wachstumsfaktoren EGF (recombinant human EGF, R&D Systems, Wiesbaden-Norden-
stadt, Deutschland) und FGF (recombinant human FGF basic 146 aa, R&D Systems, Wies-
baden-Nordenstadt, Deutschland) mit einer Konzentration von 20 ng/ml hinzugefügt (Lim
et al. 2011).
Zunächst erfolgte die Bestimmung der Zellzahl der jeweiligen Zelllinie. Anschließend
wurden 40 000 Zellen mit 3 ml Medium pro Kammer in 6-Kammer-Platten mit einer Ober-
fläche mit ultraniedriger Adhäsion angesetzt. Im weiteren Verlauf wurden die Platten im In-
kubator aufbewahrt. Alle zwei bis drei Tage wurde 0,5 ml Medium abpipettiert und 1 ml
frisches Tumorstammzellmedium hinzugefügt. Die Differenz zwischen hinzugefügten und
abpipettierten Medium glich sich dadurch aus, dass im Inkubator ein Teil der Nährlösung
verdampfte.
Die Stammzellkulturen wurden regelmäßig mikroskopisch auf Sphäroide kontrolliert.
Waren diese in einer großen Anzahl vorhanden, erfolgte die Abpipettierung des gesamten
Mediums einschließlich der Sphäroide aus der jeweiligen Kammer. Diese Suspension wur-
de zehn Minuten lang bei 1200 upm zentrifugiert, um anschließend das Medium abzupipet-
172. Material und Methoden
tieren. Das verbliebene Pellet wurde in EDTA-Lösung gelöst, acht Minuten inkubiert und
erneut wie zuvor zentrifugiert. Anschließend erfolgte das Lösen des Pellets in
0,07%iger Trypsinlösung und abermals eine Inkubation. Hierbei wurde die Zellsuspension
alle zwei Minuten geschüttelt. Daraufhin erfolgte die Neutralisation des Trypsins mittels
10 ml FKS/PBS = 1/100. Anschließend wurden die Zellen mit PBS gewaschen. Nachdem
das PBS nach zehnminütiger Zentrifugation bei 1200 upm abpipettiert wurde, erfolgte
diesmal die Lösung im gewünschten Medium, zum Beispiel Tumorstammzellmedium bei
Repassagierung in der Sphäroidkultur. In einer Zählkammer wurde mikroskopisch kontrol-
liert, ob alle Zellen voneinander dissoziiert waren. War dies nicht der Fall, erfolgte erneutes
Mischen mit einer Pipette bis zum gewünschten Ergebnis. Nun konnten die Einzelzellen je
nach Weiterverwendung übernommen werden.
182. Material und Methoden
2.5 Immunhistochemie
2.5.1 Kern-Plasma-Färbung von Zellen
Für eine Kern-Plasma-Färbung der Zellen wurde zunächst die Zellzahl der jeweiligen Zell-
kultur ermittelt und eine Konzentration von 20 000 Zellen/100 μl für insgesamt 1,5 ml an-
gestrebt. Dafür wurde die entsprechende Anzahl von Zellen entnommen, in ein
15-ml-Röhrchen überführt und zehn Minuten lang bei 1200 upm zentrifugiert. Im An-
schluss erfolgte die Resuspension des Pellets in 1,5 ml PBS.
Anschließend wurde eine Zytozentrifuge (Shandon Cytospin 4 Cytocentrifuge, Thermo
Scientific, Rockford, USA) zum punktförmigen auftragen von Zellen aus Zellsuspensionen
auf Objektträger verwendet. Zuvor wurden ein Objektträger, eine gelochte Filterkarte und
ein spezieller Trichter (Einfach-CytofunnelTM, Thermo Scientific, Rockford, USA) nach
Anleitung des Herstellers zusammengesetzt und mithilfe eines Clips (Cytoclip TM,
Thermo Scientific, Rockford, USA) aneinander befestigt. Nach dem Einsetzen der Kon-
struktion in den Rotorkopf wurden 100 µl (20.000 Zellen) der vorbereiteten Zellsuspension
in die Öffnung des Trichters gegeben. Es folgte die fünfminütige Zentrifugation bei
1500 upm. Anschließend wurden die Objektträger zum Trocknen aufgestellt.
Zur Kern-Plasma-Färbung wurden die Zellen zunächst für mindestens zehn Minuten in
Methanol auf dem Objektträger fixiert. Anschließend erfolgte das 15-malige Eintauchen
der haftenden Zellen in Eosin (Diff Quik II, Medion Diagnostics, Düdingen, Schweiz) und
danach in Methylenblau (Diff Quik I, Medion Diagnostics, Düdingen, Schweiz). Daraufhin
wurden die Objektträger zweimal in destilliertem Wasser gewaschen und bei Raumtempe-
ratur luftgetrocknet.
2.5.2 Vorbereitung der Gewebeproben für die Immunhistochemie
Für eine immunhistochemische Färbung von Gewebeproben wurden in Formalin eingeleg-
te native Tumorproben verwendet, welche durch Kooperationspartner paraffiniert und ge-
schnitten wurden.
Die Einbettung in Paraffin erfolgte in fünf Schritten durch einen Färbeautomaten
(Leica TP 1020, Leica, Nussloch, Deutschland). Jeder Schritt dauerte eine Stunde und sah
zwei Wechsel der jeweiligen Lösung vor. Die Lösungen orientierten sich an einer
aufsteigenden Alkoholreihe und bestanden aus (1) 70 % Ethanol, (2) 96 % Ethanol,
(3) 100 % Ethanol, (4) Xylol, (5) Paraffin. Die entstandenen Paraffinblöcke konnten bei
Raumtemperatur gelagert werden.
19Sie können auch lesen

















































