Die Rolle von Chemerin in Tiermodellen der hypertensiven Nephropathie und Glomerulonephritis - opus4.kobv.de
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Die Rolle von Chemerin in Tiermodellen der
hypertensiven Nephropathie und Glomerulonephritis
Kinder- und Jugendklinik des Universitätsklinikums Erlangen
Arbeitsgemeinschaft Prof. Dr. rer. nat. Hartner / PD Dr. med. Fahlbusch
Der Medizinischen Fakultät
der Friedrich-Alexander-Universität
Erlangen-Nürnberg
zur
Erlangung des Doktorgrades Dr. med.
vorgelegt von
Alexander Florian Claudius Mocker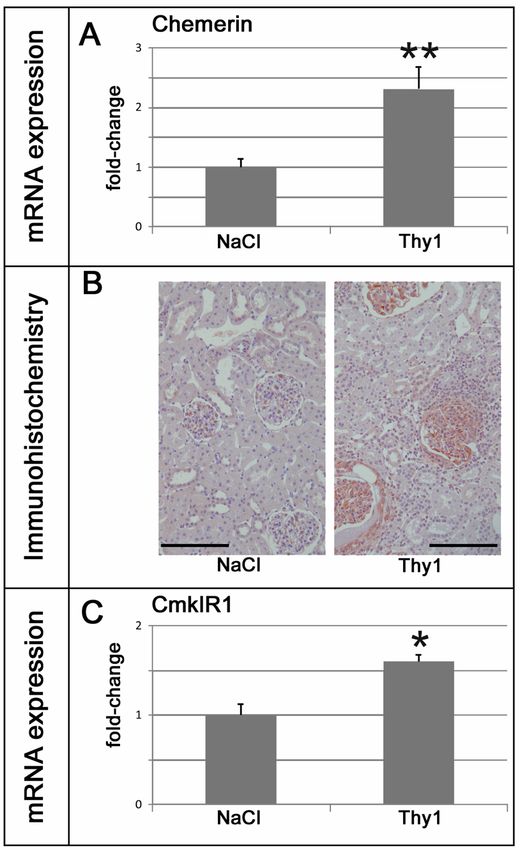
Als Dissertation genehmigt von der
Medizinischen Fakultät der Friedrich-Alexander-Universität Erlangen-
Nürnberg
Vorsitzender des Promotionsorgans: Prof. Dr. Markus F. Neurath
Gutachter: PD Dr. Fabian Fahlbusch
Gutachterin: Prof. Dr. Andrea Hartner
Gutachter: Prof. Dr. Carsten Willam
Gutachter: Prof. Dr. Jörg Dötsch
Tag der mündlichen Prüfung: 22. März 2022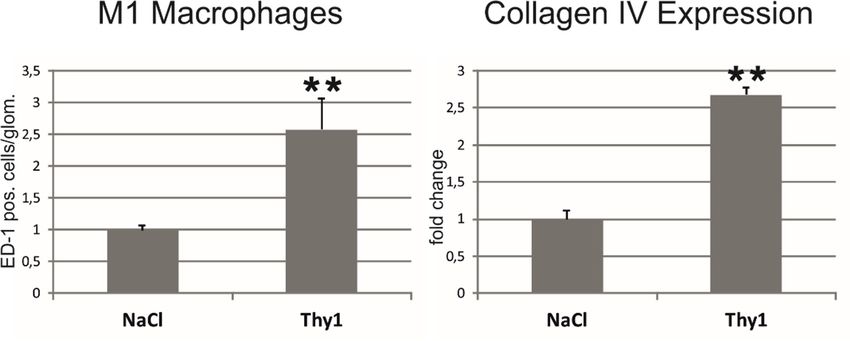
Inhaltsverzeichnis
1. Zusammenfassung 1
1.1. Deutsche Version 1
1.2. Englische Version 3
2. Einordnung der Publikationsdissertation in den wissenschaftlichen Hintergrund 4
2.1. Vitamin A 4
2.1.1. Struktur
2.1.2. Vorkommen, Mangel und Intoxikation
2.1.3. Intestinale und zelluläre Aufnahme
2.1.4. Intrazellulärer Retinoidsignalweg
2.1.5. Retinoide – Therapieansätze und Teratogenität
2.2. RARRES1 und Chemerin 7
2.2.1. RARRES1 – Lokalisierung und Funktion
2.2.2. Chemerin – Lokalisierung und Funktion
2.2.2.1. Einflüsse auf Stoffwechsel und Angiogenese
2.2.2.2. Einfluss auf Inflammation und Fibrose
2.2.2.3. Bezugspunkte zur chronischen Niereninsuffizienz
2.3. Chronische Nierenerkrankung 9
2.3.1. Definition und Einteilung
2.3.2. Epidemiologische Daten
2.3.3. Auswirkungen auf Gesellschaft und Gesundheitssystem
2.3.4. Physiologie
2.3.5. Häufige Ursachen und Pathophysiologie
2.3.6. Histologie der hypertensiven Nephropathie und Glomerulonephritis
2.4. Renale Inflammation und Fibrose 13
2.4.1. Das Renin-Angiotensin-Aldosteron-System (RAAS)
2.4.2. Überschneidungspunkte des RAAS mit Inflammation und Fibrose
2.4.3. Renale Entzündung – Akteure, Signalkaskaden, Effekte
2.4.4. Renale Fibrose – Gemeinsame Endstrecke multipler Nierenerkrankungen
2.5. Erläuterung der eingesetzten Tiermodelle 16
2.5.1. Orte der Nierenschädigung
2.5.2. Das „two-kidney, one-clip“ (2k1c) Tiermodell
2.5.3. Das „anti-Thy 1.1“ Tiermodell
3. Fragestellung 18
4. Quellenangaben 19
5. Publikation 24
6. Danksagung 40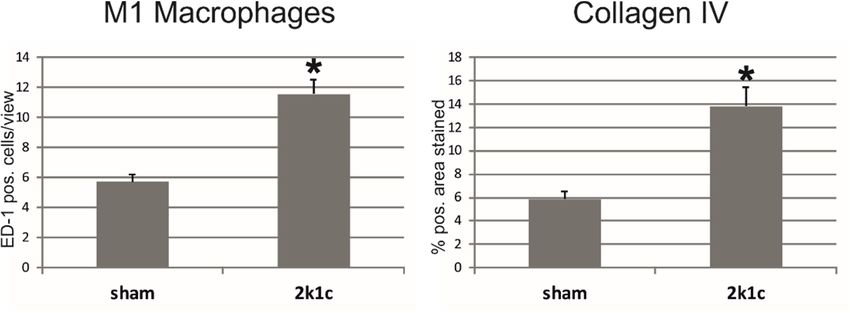
1. Zusammenfassung
1.1. Deutsche Version (adaptiert von Mocker et al. (1))
Hintergrund und Ziele
Chemerin und die Interaktion mit seinem Rezeptor, Chemokin-like Rezeptor 1 (CmklR1), sind
mit Volkskrankheiten, wie koronarer Herzkrankheit, Bluthochdruck oder dem metabolischen
Syndrom assoziiert. Diese Verknüpfung besteht insbesondere hinsichtlich der Prozesse
Chemotaxis und Inflammation, sowie über Endotheleffekte. Ferner konnte beim Menschen eine
inverse Korrelation zwischen Serum-Chemerinspiegeln und Nierenfunktion gezeigt werden.
Bisher ist nur wenig über die Rolle von Chemerin bei hypertensiver Nephropathie und renalen
Entzündungsvorgängen bekannt.
Methoden
In der Publikation von Mocker et al. (1) wurden in zwei unterschiedlichen Rattenmodellen
(two-kidney, one-clip-Modell und anti-Thy1.1-Modell) systemische und renale
Chemerinspiegel untersucht, um diese mit Markern renaler Entzündung und Fibrose zu
korrelieren. Ferner wurden in den Modellen arterieller Blutdruck, Serum- und Urinparameter
renaler Schädigung bestimmt. Die Plasmaspiegel von Chemerin wurden via Enzyme-linked
Immunosorbent Assay (ELISA) untersucht. Für immunhistochemische Färbungen von
Chemerin und proinflammatorischer beziehungsweise profibrotischer Marker wurden in
Methyl-Carnoy fixierte, renale Gewebeschnitte besagter Tiermodelle verwendet. Mittels
Echtzeit-Polymerase-Kettenreaktion (RT-PCR) wurde die Genexpression von Chemerin und
Fibrosemarkern untersucht. Die Proteinexpression von Chemerin wurde via Western Blot
dargestellt.
Ergebnisse und Beobachtungen
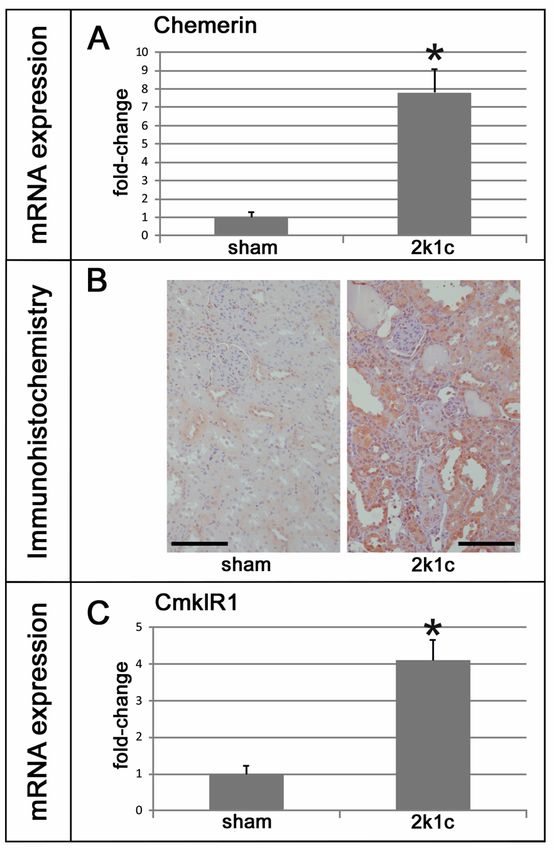
In den von Mocker et al. (1) verwendeten Tiermodellen konnten jeweils unterschiedliche
Formen der Nierenschädigung festgestellt werden. So waren im two-kidney, one-clip-Modell
(2k1c) ein Hypertonus mit erhöhten Serumkreatinin- und Harnstoffspiegeln nachweisbar,
während sich im anti-Thy1.1-Modell eine Normotonie und Anzeichen einer
Glomerulonephritis zeigten. Renale immunhistochemische Färbungen ergaben ein
Verteilungsmuster der Chemerin- beziehungsweise CmklR1-Expression, welches der
Lokalisation der Nierenschädigung im jeweiligen Modell entspricht (tubulär in 2k1c versus
glomerulär in anti-Thy1.1). In beiden Modellen waren die renale Expression von Chemerin und
CmklR1 (RT-PCR, Western Blot) im Vergleich zu Kontrollen erhöht und die
Chemerinexpression zeigte eine positive Korrelation verglichen mit Markern renaler
Entzündung und Fibrose. Im 2k1c-Modell zeigte sich eine gesteigerte Gewebsinfiltration von
1M1-Makrophagen und neutrophilen Granulozyten, Fibroblastenaktivierung, erhöhte TGFβ-1-
Expression und vermehrte Bildung von Fibronektin und Kollagenen. Im anti-Thy1.1-Modell
korrelierte die renale Chemerinexpression mit der glomerulären Infiltration von M1-
Makrophagen und der Expression von renalem Kollagen IV. Beide Modelle zeigten eine
Assoziation von Chemerin mit Serummarkern für Nierenschädigung, allerdings unabhängig
vom Hypertonus.
Schlussfolgerungen und Diskussion
Mocker et al. (1) konnten eine Verbindung zwischen der renalen Chemerinexpression und dem
Ausmaß lokaler Entzündungsprozesse und Fibrose in Modellen der Nierenschädigung
aufzeigen. Frei zirkulierendes Chemerin schien nur eine geringe Eignung als Serummarker der
Nierenschädigung in den verwendeten Modellen zu besitzen.
21.2 English version (adapted from Mocker et al. (1))
Introduction
An association of Chemerin and its receptor, chemokine-like receptor 1 (CmklR1), with
chemotaxis, inflammation, and endothelial function is especially seen in metabolic syndrome,
coronary heart disease, and hypertension. Circulating chemerin levels and renal function show
an inverse relation in humans. So far, little is known about the potential role of chemerin in
hypertensive nephropathy and renal inflammation.
Methods
Mocker et al. (1) determined systemic and renal chemerin levels in 2-kidney-1-clip (2k1c)
hypertensive and anti-Thy1.1 nephritic rats, respectively, to explore the correlation between
chemerin and markers of renal inflammation and fibrosis. They measured blood pressure, serum
and urine parameters. Plasma chemerin was determined via enzyme-linked immunosorbent
assay (ELISA). Immunohistochemical detection of chemerin and proinflammatory / profibrotic
markers was performed in methyl Carnoy-fixed sections of rat kidneys. Gene expression of
chemerin and markers of fibrosis was investigated via real-time polymerase chain reaction
(PCR). Protein expression of chemerin was examined via western blot.
Results
Mocker et al. (1) were able to show that 2k1c rats were hypertensive with increased serum urea
and creatinine, while Thy1.1 rats were normotensive with signs of glomerulonephritis.
Immunohistochemistry revealed a model-specific induction of chemerin and CmklR1
expression at the corresponding site of renal damage (tubular in 2k1c vs. glomerular in anti-
Thy1.1). In both models, renal expression of chemerin and its receptor CmklR1 (RT-PCR,
western blot) were increased and chemerin correlated positively with markers of inflammation
and fibrosis. In 2k1c rats, infiltration of M1 macrophages and neutrophil granulocytes, as well
as fibroblast activation, TGFβ-1 expression and the expression of fibronectin and collagens had
a strong correlation with chemerin expression. In anti-Thy1.1, chemerin was correlated with
glomerular infiltration of M1 macrophages and renal collagen IV expression. In both models
chemerin correlated with serum markers of renal damage, but not with blood pressure levels.
Conclusion
The findings of Mocker et al. (1) demonstrated an association between renal chemerin
expression and the degree of local inflammation and fibrosis related to renal damage. However,
its use as circulating biomarker of renal inflammation seemed to be limited in their rat models.
32. Einordnung der Publikationsdissertation in den wissenschaftlichen Hintergrund
2.1. Vitamin A
2.1.1. Struktur
Vitamin A ist für den Menschen und andere Säugetiere essenziell, da es wie alle Vitamine nicht
bedarfsgerecht hergestellt werden kann und über die Nahrung aufgenommen werden muss.
Relevante Nahrungsquellen ergeben sich aus der Aufnahme von Provitamin A, beispielsweise
in Form des Pflanzenfarbstoffs beta-Carotin oder von Retinsäurederivaten aus Geweben
tierischen Ursprungs (2). Der Begriff Vitamin A bezeichnet in erster Linie all-trans-Retinol,
wird aber gemeinhin als Sammelbezeichnung für Retinale, Retinole, Retinsäuren und
Retinylpalmitat verwendet (2, 3). Diese Unterformen werden treffender unter dem
Sammelbegriff der Retinoide zusammengefasst. Wichtige Gemeinsamkeiten in der
Molekularstruktur der Retinoide sind der beta-Jonon-Ring, eine Polyen-Seitenkette und daran
anschließende polare Endgruppen, welche sich je nach Retinoid unterscheiden (4).
2.12. Vorkommen, Mangel und Intoxikation
Besonders hohe Konzentrationen des fettlöslichen Vitamin A finden sich in Leber, Lunge oder
anderen Organgeweben, gefolgt von Milchprodukten (2). Relevante Quellen für Provitamin A
stellen Obst und Gemüse dar, insbesondere Möhren, Orangen oder Mangos (2).
Initial führt ein Mangel an Vitamin A zu reversibler Nachtblindheit und Blutbildungsstörungen
wie einer Leukozytopenie oder Thrombopenie (5). Da insbesondere die Fotorezeptoren des
Auges auf Vitamin A zur Aufrechterhaltung des Visus angewiesen sind, kommt es im Verlauf
zu Epithelveränderungen wie Netzhautflecken (Bitot-Flecken), Xerosis corneae und
Keratomalazie (6, 7). Auch andere Gewebe benötigen Vitamin A zur Zellproliferation.
Klassische Beispiele sind eine verminderte Tränensekretion und Xerosis cutis (7). Ferner führt
ein mittel- bis langfristiger Mangel an Vitamin A zu Wachstumshemmung, Gewichtsverlust
und schlimmstenfalls zum Tod (5, 8). Außerdem kommt es zu einer erhöhten Infektanfälligkeit,
erklärt durch die verminderte Produktion dendritischer Zellen, Makrophagen und T-
Lymphozyten, wobei insbesondere regulatorische T-Zellen betroffen sind (9). Auffallend sind
starke Schwankungsbreiten zwischen der Symptomintensität und dem Symptombeginn
aufgrund hoher Speicherkapazitäten für Vitamin A in Form von Retinylester in der Leber und
anderen Geweben (5).
4Ein Vitamin-A-Mangel während der Schwangerschaft führt beim Neugeborenen zu einer
erhöhten Rate an kongenitaler Blindheit und zu Fehlbildungen des Herz-Kreislaufsystems, des
ZNS, des Urogenitaltrakts, des Bewegungsapparats und der Lunge, welche bis zum
intrauterinen Fruchttod reichen können (10, 11).
Die erwähnte Speicherfunktion der Leber ist bei dem Verzehr Vitamin A-reicher Gewebe
ursächlich für akute Vergiftungen (12). Ähnlich anderer Intoxikationen zählen Übelkeit,
Erbrechen, Abgeschlagenheit und starker Kopfschmerz zu den häufigsten Symptomen (13).
Typisch ist eine perioral begrenzte oder das gesamte Integument betreffende Epidermolyse
(12). Eine chronisch gesteigerte Aufnahme von Vitamin A führt zu Anorexie, Haarverlust oder
trockener juckender Haut (13).
Im Kindesalter kann es durch eine übermäßige Aufnahme von Vitamin A zu einem vorzeitigen
Schluss der Epiphysenfugen und subperiostalen Knochenwucherungen kommen (14). Während
der Schwangerschaft kann eine erhöhte Aufnahme von Vitamin A insbesondere in den ersten
sechs Wochen post conceptionem zu fetalen Fehlbildungen führen und das Risiko einer
Totgeburt steigern (15). Eine Tagesmenge von über 25.000 IU sollte daher aufgrund der
Teratogenität nicht überschritten werden (16).
2.1.3. Intestinale und zelluläre Aufnahme
Retinsäuren werden nach Aufnahme über den Darm frei flottierend im Blutkreislauf verteilt
oder an Retinsäure-Bindeproteine, wie beispielsweise Retinol-Bindeprotein 4 (RBP4),
Interphotorezeptormatrix Retinoidbindeprotein (IRBP), Epididymales Retinoidbindeprotein
(ERBP) oder beta-trace, gekoppelt (3). Diese Bindeproteine sind für die Retinsäureaufnahme
nicht zwingend notwendig, da in Knock-out Tiermodellen nur unter gleichzeitiger Vitamin A-
Minderversorgung Mangelzustände auftreten (3). Die Serumspiegel der Bindeproteine ergeben
ferner keinen spezifischen Hinweis auf die Vitamin A-Versorgung des Organismus, da
beispielsweise RBP als negatives akute Phase Protein auf Entzündungszustände reagiert (17).
Die intrazelluläre Aufnahme von Vitamin A und Retinoiden erfolgt über den von Retinsäure 6
stimulierten Vitamin A-Rezeptor (STRA6) (18). Weitere zelluläre Aufnahmemöglichkeiten für
Retinoide sind die direkte Membrandiffusion oder die Aufnahme über Lipoproteine, hier
insbesondere durch Chylomikronen oder VLDL / LDL (3, 19).
52.1.4. Intrazellulärer Retinoidsignalweg
Sobald Vitamin A oder andere Retinoide über die zuvor erwähnten Aufnahmemechanismen in
die Zellen gelangt sind, binden sie an zelluläre Retinolbindeproteine (CRBP), wodurch ein
sofortiger Verlust der Retinoide via Rückdiffusion über Zellmembranen verhindert wird (20).
Nach Bindung können Retinoide über die Lecithin-Retinol-Acyltransferase (LRAT) mittels
Esterbildung in Retinylester überführt werden, welche die Speicherform für Vitamin A
darstellen (3). Einen regulatorisch bedeutenden Stellenwert nehmen Retinsäuren ein, welche
mittels zweifacher Dehydrierung über Retinal hergestellt werden (3). Als Leitsubstanz gilt hier
die all-trans Retinsäure (ATRA) (3). Intrazellulär binden Retinsäuren wiederum an zelluläre
Retinsäurebindeproteine (bspw. ATRA an CRABP-I oder -II), welche einen Transport in den
Zellkern ermöglichen (3).
Im Zellkern nehmen Retinsäuren über spezifische Rezeptoren (Retinoic Acid Receptor /
Retinoic X Receptor) Einfluss auf die Expression regulatorischer Gene von
Wachstumsprozessen, Zelldifferenzierung und Apoptose (21). Diese Rezeptoren bestehen aus
je einem variablen RAR (alpha, beta, oder gamma) und einem konstanten RXR-Anteil (21).
Nach Bindung von Retinsäure an den RAR-Anteil und daraus resultierender
Heterodimerisierung mit RXR werden in bestimmten Promotorregionen Retinsäure-
Responsive Elemente (RAREs) aktiviert (vgl. Abb. 1) (21). Diese führen in Folge zur
vermehrten Produktion der Retinsäurerezeptor Responderproteine 1 und 2 (RARRES1 /
RARRES2) (22-24).
RA
RARRES 1 / RARRES 2
RAR RXR
Genexpression
Promoter
Abb. 1: Schema der Promoteraktivierung durch Retinsäuren (RA) an einem Retinsäurerezeptor (RAR & RXR)
nach Hübner (24).
2.1.5. Retinoide – Therapieansätze und Teratogenität
Ein bekanntes Beispiel für Retinoide als systemischer Therapieansatz stellt die akute
Promyelozytenleukämie (APL) dar. Bei dieser Unterform der akuten myeloischen Leukämie
(AML) kommt es aufgrund einer Translokationsmutation zur Bildung eines Fusionsproteins im
Retinsäurerezeptor alpha mit daraus resultierender Funktionseinschränkung des Rezeptors (25).
6Die Gabe von all-trans Retinsäure (ATRA) fördert die Ausdifferenzierung von Tumorzellen zu
funktionell reifen Leukozyten und führt daher in vielen Fällen zu einer Komplettremission der
Erkrankung (25).
Häufiger wird ATRA zur dermatologischen Lokaltherapie von Leukoplakien, Krebsvorstufen
der Cervix uteri oder der aktinischen Keratose genutzt (26). Hier führt ATRA zum einen zu
einer verbesserten Ausdifferenzierung der Zellen, zum anderen kommen immunmodulatorische
Eigenschaften zum Tragen (26). Diese Immunmodulation lässt sich am Beispiel der
Aknetherapie gut veranschaulichen (27). Hier konnte gezeigt werden, dass die topische
Applikation von ATRA die durch Proprionibacterium acnes ausgelöste und durch Matrix-
Metalloproteasen (insbesondere MMP-9) und Toll-Like-Rezeptor 2 vermittelte Immunreaktion
unterbrechen kann (27). Des Weiteren kann die Immunantwort durch eine verminderte
Expression von Interleukinen (z.B. IL-12) verändert werden (9).
Eine Therapie mittels ATRA während der Schwangerschaft stellt eine Kontraindikation dar.
Sie kann fetale Fehlbildungen des ZNS, Thymus, des kardiovaskulären Systems und
kraniofaziale Fehlbindungen verursachen (28). Typisch sind eine Mikrotie oder Anotie mit
meist fehlendem äußerem und innerem Gehörgang (28).
2.2. RARRES1 und Chemerin
2.2.1. RARRES1 – Lokalisierung und Funktion
Retinsäuren nehmen über RAR/RXR Einfluss auf die Expression von Retinsäurerezeptor-
responder kodierender Gene (21). Diese Gene führen unter anderem zur Produktion der
Retinsäurerezeptor Responderproteine 1 und 2 (RARRES1 / RARRES2) (22, 23). RARRES1
wurde erstmals in Hautgewebe beschrieben, welches mit ATRA behandelt wurde und ist auch
unter den Begriffen Tazaroten-induziertes Gen 1 (TIG1), Latexin-like (LXNL) oder Phorbol
Ester-induziertes Gen 1 (PERG-1) bekannt (22). Es kommt ausschließlich membranständig vor
und wird vermehrt von Epithelzellen exprimiert (22).
RARRES1 hat in den meisten Tumoren wachstumshemmende und Apoptose-induzierende
Eigenschaften (29, 30), kann aber je nach Entität und epigenetischer Methylierung auch zu
einem Tumorprogress führen, wie beispielsweise im inflammatorischen Mammakarzinom (31).
Der negative Einfluss auf das Tumorwachstum wird von RARRES1 durch Angiogenese-
inhibition und gleichzeitiger Induktion von Autophagie erzielt (30). Außerdem sind Einflüsse
auf den Fettgewebsmetabolismus bekannt, wobei RARRES1 hier vor allem bei plötzlicher
Gewichtsabnahme und der Entdifferenzierung von Adipozyten eine Rolle spielt (32).
7Die Funktion von RARRES1 im Rahmen chronischer Nierenerkrankungen bleibt derzeit noch
unklar, wobei eine positive Korrelation zwischen der RARRES1-Expression und dem Ausmaß
glomerulärer Nierenschädigung bei diabetischer Nephropathie und fokal segmentaler
Glomerulosklerose nachgewiesen werden konnte (33).
2.2.2. Chemerin – Lokalisierung und Funktion
RARRES2 wurde wie RARRES1 zuerst in der Haut beschrieben und ist unter den Begriffen
Chemerin, HP10433 und Tazaroten-induziertes Gen 2 (TIG2) bekannt (23). Es wird
hauptsächlich in der Leber und in weißem Fettgewebe exprimiert, kommt allerdings auch in
Lungengewebe, Gefäßen, dem Ovar, den Nieren und der Plazenta vor (34). In diesen Geweben
existiert es als metabolisch inaktive Speicherform Pro-Chemerin, welche durch Proteolyse in
aktives Chemerin überführt wird (35). Im Gegensatz zu RARRES1 wird es sezerniert und wirkt
im Zielgewebe als Chemoattraktans für Makrophagen und unreife dendritische Zellen (35).
Seine Gewebewirkung wird größtenteils durch einen G-Protein-gekoppelten Rezeptor namens
Chemokin-like Rezeptor 1 (CmklR1, oder Chemerin Rezeptor 23) vermittelt, wobei weitere
Signalwege über G-Protein-gekoppelten Rezeptor 1 (GPR1) und C-C Motiv Rezeptor-like 2
(CCRL2) bekannt sind (35).
2.2.2.1. Einflüsse auf Stoffwechsel und Angiogenese
Chemerin verursacht als Adipozytokin eine vermehrte Bildung von weißem Fettgewebe und
führt zu einer vermehrten Glukoseaufnahme in Zellen (36, 37). Dadurch liegt eine Assoziation
zu Übergewicht und Typ-2-Diabetes nahe (36, 38). Erhöhte Serumspiegel von Chemerin sind
ferner mit dem metabolischen Syndrom und den Volkskrankheiten Bluthochdruck, koronarer
Herzerkrankung und Atherosklerose assoziiert (38, 39). Es konnte zudem gezeigt werden, dass
Chemerin durch Leukozytenaktivierung und Stimulation der Chemotaxis von dendritischen
Zellen und Makrophagen eine Rolle bei Immun- und Entzündungsreaktionen spielt (35, 39).
Diese migrationsfördernden Eigenschaften spiegeln sich im angiogenen Potential von
Chemerin wider (40). So wird einerseits die Adhäsion und Proliferation von Endothelzellen
gesteigert, andererseits eine Atherosklerose-typische Entzündungsreaktion in der Gefäßwand
ausgelöst (41, 42). Im Gegensatz dazu kann Chemerin über Reduktion der Proliferation und
Invasion bestimmter Tumorzellreihen und über Leukozytenrekrutierung tumorsuppressive
Eigenschaften ausüben (43).
In der Schwangerschaft beeinflusst Chemerin den maternofetalen Nährstoffaustausch und ist
mit erhöhter maternaler Insulinresistenz und Gestationsdiabetes assoziiert (44).
82.2.2.2. Einfluss auf Inflammation und Fibrose
Der Einfluss von Chemerin auf das Immunsystem wird größtenteils über CmklR1 ausgeübt,
welcher als einziger Chemerin-Rezeptor in signifikantem Ausmaß in die Expression
proinflammatorischer Gene involviert ist (41). Neben hämatopoietischen Zelllinien,
Endothelzellen und Fettgewebe kommt Chemerin daher insbesondere in Zellen des
Immunsystems vor, wie beispielsweise in Monozyten, Makrophagen, unreifen dendritischen
Zellen oder Lymphozyten (41, 45). Ferner kann über CmklR1 die Migration von Lymphozyten
in entzündetes Gewebe und Lymphorgane gesteuert werden, was im Falle von Makrophagen
über den transformierenden Wachstumsfaktor-beta (TGF-beta), in Monozyten via
Tumornekrosefaktoren (TNF) und Interferon-gamma (IFN-gamma) und in NK-Zellen mittels
Interleukin 2 und 15 reguliert wird (45).
2.2.2.3. Bezugspunkte zur chronischen Niereninsuffizienz
Bei chronischer Niereninsuffizienz (CKD) konnte in mehreren Versuchen eine inverse
Korrelation zwischen dem Serumchemerinspiegel und der glomerulären Filtrationsrate (GFR)
gezeigt werden (46). So steigen im Rahmen der Niereninsuffizienz Serumchemerin und
Serumkreatinin konkordant an (46). Bei dialysepflichtigen Patienten sind die
Serumchemerinspiegel chronisch erhöht und fallen drei Monate nach Nierentransplantation auf
Normalwerte ab (46). Ob diese Erhöhung der Serumchemerinspiegel auf eine Überexpression
im Rahmen metabolischer Erkrankungen, die verminderte renale Funktionsfähigkeit selbst,
oder auf eine Eigenproduktion der Niere zurückzuführen ist, konnte bisher nicht klar
differenziert werden (34, 46).
2.3. Chronische Nierenerkrankung
2.3.3. Definition und Einteilung
Definiert wird die chronische Niereninsuffizienz nach den Kidney Disease: Improving Global
Outcome (KDIGO) Leitlinien von 2012 (siehe Tabelle 1) als Strukturveränderung oder
Funktionsstörung der Niere, die länger als 3 Monate besteht (47). Hierunter fallen
beispielsweise eine auf < 60 mL/min/1,73m2 verminderte GFR oder eine Albuminurie ≥ 30
mg/24h (47). Je nach Ursache, GFR-Verlust oder Schwere der Albuminurie ergeben sich
unterschiedliche Abstufungen (Tabelle 1) (47). So wird die Minderung der GFR in 5 Stadien
unterteilt, wobei ohne Vorschädigung erst ab Stadium 3 von einer Niereninsuffizienz
gesprochen wird (47). Eine terminale Niereninsuffizienz wird als GFR unter 15 mL/min/1,73m2
9definiert (47). Nach Schwere der Albuminurie kann die CKD in 3 Stadien von unter 30 mg/g,
zwischen 30 - 300 mg/g und über 300 mg/g differenziert werden (47).
Risikofaktoren für die Progression einer CKD sind neben deren Ursache und des
Krankheitsstadiums insbesondere Diabetes, arterielle Hypertonie, kardiovaskuläre
Begleiterkrankungen, Dyslipidämie, Adipositas, Nikotinkonsum und nephrotoxische
Medikamente (47, 48). Diese Risikofaktoren sind von besonderer klinischer Bedeutung, da sie
durch entsprechende Individualtherapie und Lebensstiländerung modifizierbar sind (47, 48).
Albuminurie
(in mg/mmol)
A1 A2 A3
Normal bis Moderat Stark erhöht
leicht erhöht erhöht
30
G1 Normal bis hoch ≥ 90
G2 Leicht vermindert 60 - 89
G 3a Leicht bis moderat 45 - 59
GFR
vermindert
(in
G 3b Moderat bis stark 30 - 44
ml/min/
vermindert
1.73 m2)
G4 Stark vermindert 15 - 29
G5 Terminale ≤ 15
Niereninsuffizienz
Tab. 1: Risiko für terminale Niereninsuffizienz und kardiovaskuläre Komplikationen bei CKD in Abhängigkeit
von GFR und Albuminurie nach KDIGO 2012: A = Albuminurie, G = GFR, Grün = kein erhöhtes Risiko bzw.
keine CKD, gelb = mäßig erhöhtes Risiko, orange = hohes Risiko, rot = sehr hohes Risiko (47).
2.3.1. Fallzahlen in Deutschland und weltweit
Etwa 12,7% der deutschen Bevölkerung zeigten in einer Stichprobe des Robert-Koch-Instituts
aus dem Jahr 2011 Anzeichen einer eingeschränkten Nierenfunktion (49). In dieser Stichprobe
wurde eine Einschränkung der Nierenfunktion als eine Albuminurie ≥ 30 mg/L oder eine
Einschränkung der eGFR < 60 mL/min/1,73m2 (49) definiert. Weltweite Schätzungen über die
Prävalenz chronischer Niereninsuffizienz gehen von etwa 10-15% der Weltbevölkerung aus
(50, 51). Interessant ist hier ein höherer Anteil von Betroffenen in westlich geprägten Regionen
10wie Nordamerika, Europa oder Australien (50, 51). Dies legt eine Assoziation mit
Wohlstandserkrankungen wie Bluthochdruck, Übergewicht oder Diabetes mellitus nahe, die in
diesen Bevölkerungsgruppen eine ähnliche oder höhere Verbreitung haben (52, 53). Besonders
auffallend ist eine Dynamik in Schwellenländern, welche durch die steigende Industrialisierung
nicht nur bei Bluthochdruck, Diabetes mellitus und Übergewicht einen Anstieg der Fallzahlen
verzeichnen, sondern auch bei chronischer Niereninsuffizienz (51-53).
2.3.2. Epidemiologische Daten
Trotz der bereits hohen Prävalenz der CKD und angesichts steigender Fallzahlen besteht nur
ein geringes Krankheitsbewusstsein in der Bevölkerung. So wussten in der oben erwähnten
Kohorte des RKI nur 28% der Probanden von ihrer eingeschränkten Nierenfunktion und nur
etwa zwei Drittel der Erkrankten waren aufgrund ihrer CKD bei einem Arzt vorstellig (49). Aus
den Vereinigten Staaten sind ähnliche Schätzungen aus dem Jahr 2008 vorhanden, wobei in
dieser Gruppe nur 10% der Probanden über ihre eingeschränkte Nierenfunktion Bescheid
wussten (54). Gründe hierfür sind in der Symptomarmut und dem Mangel an spezifischen
Symptomen in Frühstadien der CKD zu suchen (51, 55).
In Kontrast dazu stehen die hohen gesundheitsökonomischen Kosten, die im Spätstadium der
Niereninsuffizienz verursacht werden (56, 57). Beispielsweise können 10,2% der Ausgaben der
gesetzlichen Krankenversicherung auf Patienten mit chronischer Niereninsuffizienz in den
Stadien 3 und 4 zurückgeführt werden (57). Weitere 1,6% werden durch etwa 92.000
Dialysepatienten deutschlandweit verursacht (57, 58).
Diese weltweite Entwicklung erhält durch den Mangel an präventivtherapeutischen
Maßnahmen in Frühstadien der chronischen Niereninsuffizienz und damit verbundener hoher
Morbidität und Mortalität eine besondere Brisanz (59).
2.3.4. Physiologie
Um die renalen Schädigungsprozesse der verschiedenen Ursachen von CKD besser zu
verdeutlichen, lohnt es an dieser Stelle kurz die Physiologie einer gesunden Niere zu
rekapitulieren. Der Filtrierapparat einer gesunden humanen Niere besteht aus etwa einer Million
Nephronen, bei Ratten beläuft sich deren Anzahl auf etwa 30.000 (60, 61). Diese können grob
in ein rindenständiges Nierenkörperchen und das Tubulussystem untergliedert werden (62). Das
Nierenkörperchen selbst besteht aus zu- bzw. abführenden Gefäßen (Glomerulus) und deren
Endothel, einer glomerulären Basalmembran und den Podozyten (62). Die Podozyten bilden
gemeinsam mit Plattenepithelzellen und der Glomeruluskammer die Bowman-Kapsel (62).
11Hier wird ein serumähnliches Ultrafiltrat abgepresst, welches im Tubulussystem zu Restharn
konzentriert wird (63). Das an die Glomeruluskapsel anschließende Tubulussystem teilt sich in
einen proximalen und distalen Tubulus auf, deren gerade Anteile als Henle-Schleife in das
Nierenmark ziehen (62). Der distale Tubulus zieht im Verlauf am Glomerulus vorbei und bildet
mit der Macula densa den juxtaglomerulären Apparat (JGA) als Nephron-internen
Rückkopplungsmechanismus (62, 63). Im Anschluss wird der Harn über das
Sammelrohrsystem weiter konzentriert und über die ableitenden Harnwege ausgeschieden (63).
Die renale Gefäßversorgungsarchitektur lässt sich grob in zwei Abschnitte zwischen Rinde und
Mark einteilen, die durch die Aa. arcuatae voneinander getrennt werden (62). Das arterielle Blut
gelangt über die A. renalis nach Aufspaltung in die Segmentarterien über die Aa. interlobares
an den Markpyramiden vorbei und mündet in den Aa. arcuatae (64). Von dort werden zuerst
die Glomeruli versorgt (62). Das danach vom Ultrafiltrat befreite Blut fließt bei marknahen
Glomeruli über die Arteriola recta in das Nierenmark, während abführende Gefäße
oberflächennaher Glomeruli die Rinde mit Nährstoffen versorgen. Gemeinsame Endstrecke
sind die Vv. arcuatae und die abführende V. renalis (62).
2.3.5. Häufige Ursachen und Pathophysiologie
Die Ursachen chronischer Niereninsuffizienz sind vielfältig und regional unterschiedlich. In
westlichen Ländern führen vor allem metabolische Systemerkrankungen wie Diabetes mellitus,
Übergewicht oder arterielle Hypertonie zu einer Nierenschädigung (65, 66). Weitere
ursächliche Grunderkrankungen sind in Abstufung ihrer Häufigkeit chronisch verlaufende
Glomerulonephritiden und polyzystische Nierenerkrankungen, gefolgt von seltenen hereditären
Syndromen (66, 67).
Ferner kann man anhand der Klinik und laborchemischer Ergebnisse Rückschlüsse auf den
Schädigungsort und die Grunderkrankung ziehen. Defekte der glomerulären Basalmembran
führen zu einer erhöhten Permeabilität von Proteinen, während zelluläre Bestandteile noch
zurückgehalten werden können (55, 63). Da suffiziente Rückresorptionsmechanismen für
Makroproteine fehlen, zeigen sich ein erhöhter Albumingehalt im Urin oder ein erhöhter
Albuminquotient zwischen Blut und Urin (47, 55). Die isolierte Makroproteinurie tritt regelhaft
bei der diabetischen Glomerulosklerose und bei Glomerulonephritiden auf (47, 55, 68). Auch
bei Vaskulitiden, Pyelonephritiden oder im Verlauf der hypertensiven Nephropathie kann sich
eine Albuminurie entwickeln (47, 55, 68). Sollten die Rückresorptionsmechanismen des
Tubulussystems geschädigt sein, kommt es in der Folge zu einer fehlenden Wiederaufnahme
von Mikroproteinen wie alpha1- oder beta2-Mikroglobulin (69). Weitere Hinweise auf die
12renale Grunderkrankung gibt das Urinsediment. Erythrozyten, insbesondere Akanthozyten und
Erythrozytenzylinder, weisen meist auf eine Schädigung des glomerulären Endothels hin,
wodurch Zellen durch die geschädigte Basalmembran und die vergrößerten Zwischenräume
zwischen den Podozytenschlitzen in den Harn übertreten können (47, 55, 68). Leukozyten und
Leukozytenzylinder weisen hingegen auf ein inflammatorisches Geschehen im Tubulussystem
und Interstitium hin und treten gehäuft im Rahmen der interstitiellen Nephritis auf (47, 55, 68).
2.3.6. Histologie der hypertensiven Nephropathie und Glomerulonephritis
Endstrecke der meisten länger andauernden Nierenschädigungen sind fibrosierende
Umbauprozesse im Nierengewebe, welche zu Glomerulosklerose, Tubulusatrophie und
interstitieller Fibrose führen (55, 65). Diese Fibrose entsteht durch die langfristige
Gewebsschädigung im Rahmen der zugrundeliegenden Erkrankung (beispielsweise renale
Hypertonie oder Diabetes mellitus) und führt zu einer sich selbsterhaltenden
Entzündungsreaktion mit Verdickung der Basalmembran, Hypoxie und Ausschüttung
proinflammatorischer Produkte (55, 70).
Bei der hypertensiven Nephropatie kommt es durch den gesteigerten intravasalen Druck und
der konsekutiven Arteriosklerose zu Umbauprozessen der Gefäßwand und durch rezidivierende
Ischämien im mäßig oxygenierten Tubulussystem zu fibrotischem Umbau in diesen Bereichen
(71).
Im Gegensatz dazu sind viele chronische Glomerulonephritiden von glomerulär-mesangialen
Veränderungen geprägt. Als typisches Beispiel ist vor allem die mesangioproliferative
Glomerulonephritis als Folge von Medikamentenintoxikationen zu nennen (72).
2.4. Renale Entzündung und Fibrose
2.4.1. Das Renin-Angiotensin-Aldosteron-System (RAAS)
Bei den drei Hauptursachen der CKD, hypertensiver Nephropathie, diabetischer Nephropathie
und Glomerulonephritis, sind inflammatorische Mechanismen bei der Entstehung oder
Aufrechterhaltung der Nierenschädigung involviert (65).
Ursächlich hierfür ist im Falle der hypertensiven Nephropathie die renale Minderperfusion bzw.
Ischämie-Reperfusion und die damit verbundene Aktivierung des Renin-Angiotensin-
Aldosteron-Systems (RAAS) (73). Durch Sympathikusaktivierung, Verminderung des renalen
Perfusionsdrucks oder des Serum-Chloridgehalts wird vermehrt Renin aus Prorenin durch
Zellen des juxtaglomerulären Apparats (JGA) mittels Proteolyse abgespalten (73). Das Prä-
13Prohormon Angiotensinogen wird hauptsächlich in der Leber gebildet (73). Die Bindung von
Renin an Angiotensinogen bewirkt die Abspaltung des Dekapeptids Angiotensin I von
Angiotensinogen (73, 74). Angiotensin I wird von dem hauptsächlich membrangebundenen
Angiotensin-Converting-Enzym (ACE) in das metabolisch aktive Oktapeptid Angiotensin II
überführt (73, 74). Dies geschieht vor allem an Gefäßendothelien, am Bürstensaumepithel oder
an Neuroendothelzellen (73). Angiotensin II führt unter anderem zu Vasokonstriktion,
Natriumretention, Gewebefibrose, oxidativen Stress und Aldosteronfreisetzung (73, 74).
Aldosteron wird als Mineralocorticoid von der Nebennierenrinde hergestellt und sorgt für eine
vermehrte renale Natrium- bzw. Wasserretention in Verbindung mit einer vermehrten
Ausscheidung von Kalium und Protonen (75).
2.4.2. Überschneidungspunkte des RAAS mit Inflammation und Fibrose
Die Komponenten des RAAS haben mehrere Überschneidungspunkte zu inflammatorischen
Prozessen. Renin und dessen Propeptid Prorenin induzieren über einen gemeinsamen Rezeptor
unabhängig von Angiotensin II die Bildung von Transforming-Growth-Factor-beta (TGF-beta),
Plasminogen-Aktivator-Inhibitor-1 (PAI-1) und die Bildung von Bestandteilen der
Extrazellulärmatrix wie Fibronektin oder Kollagen-1 (74).
Eine weitere Verbindung von RAAS und Inflammation besteht über Angiotensin II selbst.
Dessen Expression führt über den AT1-Rezeptor zur vermehrten Produktion profibrotischer
Wachstumsfaktoren wie TGF-beta und dadurch zur Proliferation der Extrazellulärmatrix (76,
77). Hierdurch werden in Mesangiumszellen vermehrt Typ 1 Prokollagen und Fibronektin
gebildet (78). Ferner werden via AT1 und AT2 vermehrt proinflammatorische Signalkaskaden
angestoßen, wie Nuclear-Factor-kappa-B (NF-kappa B), der Mitogen-aktivierte Proteinkinase
(MAPK) Signalweg, Rho- oder Redox-Signalwege (77). Durch Aktivierung dieser
Signalkaskaden wird die Produktion von Interleukin 6 und Monozyten-Chemoattraktant-
Protein-1 (MCP-1) angeregt (77). Über AT2 wird außerdem die endogene Synthese von TGF-
beta angeregt, welches zu vermehrter renaler Fibrose führt (76).
Den dritten Überschneidungspunkt stellt Aldosteron dar, welches mittels Bildung von
Sauerstoffradikalen zu einer verstärkten NF-kappa B-Antwort führt und eine vermehrte
Genexpression von TGF-beta, PAI-1, Endothelin 1 (ET-1) und weiterer Wachstumsfaktoren
anregt (79). Ferner verstärkt Aldosteron die Effekte von Angiotensin II auf den systolischen
Blutdruck durch Aktivierung des MAP-Kinase Signalwegs in Gefäßmuskelzellen (80).
142.4.3. Renale Entzündung – Akteure, Signalkaskaden, Effekte
Die zugrundeliegenden Ursachen chronischer Nierenschädigung sind vielfältig, führen jedoch
meist zu chronischer Entzündung und Fibrose (55, 70). Im Zentrum der Entzündungsreaktion
steht die Einwanderung von Leukozyten in das Interstitium, welche durch Zytokine und
Chemokine initiiert und moduliert wird (65). Diese Zyto- und Chemokine können von beinahe
allen Zellen der Niere hergestellt werden (70). Von besonderem Interesse als Bestandteil der
ausgeschütteten Entzündungsmediatoren sind die Chemokine, zu denen auch Chemerin zählt
(35). Die eingewanderten Leukozyten kann man grob in Regulator- und Effektorzellen
einteilen, wobei letztere sich in M1 & M2 Makrophagen, neutrophile Granulozyten, CD4
positive T-Helferzellen, zytotoxische CD8 positive T-Zellen, B-Zellen und dendritische Zellen
unterteilen lassen (65). Je nach aktivierendem Chemokin werden andere Zellreihen an den Ort
der Entzündung gelockt (70). Im Falle von Chemerin werden besonders
Haupthistokompabilitätskomplex (MHC) II positive antigen-präsentierende Zellen wie
Monozyten, Makrophagen und dendritische Zellen rekrutiert (81).
Diese eingewanderten Makrophagen können selbst Entzündungsmediatoren wie verschiedene
TNF oder reaktive Sauerstoffspezies herstellen und auf diesem Weg die Entzündung
aufrechterhalten (65, 70). Durch die ausgeschütteten TNF werden auf auto- und parakrinem
Weg weitere Leukozyten aktiviert und die Apoptose von Zellen des Nierenparenchyms wie
Gefäßendothelien, Tubulusepithelien oder Podozyten induziert (82). Diese
Leukozytenaktivierung geschieht durch die Aktivierung proinflammatorischer Signalkaskaden
wie NF-kappa B (83). Dies regt die nukleäre Expression einer breiten Palette von Genen an,
welche für Inflammation, Immunsystem, Apoptose, Zellproliferation und Zelldifferenzierung
kodieren (83). Zu den in der Frühphase der Entzündungsreaktion vermehrt exprimierten
Proteinen zählen Zytokine wie IL-6, IL-8 und MCP-1, wobei in der Spätphase
Oberflächenrezeptoren, Adhäsionsmoleküle und Chemokine eine größere Rolle spielen (83).
2.4.4. Renale Fibrose – Gemeinsame Endstrecke multipler Nierenerkrankungen
Wenn die renale Entzündung lange genug besteht, werden im weiteren Verlauf von
Makrophagen vermehrt profibrotische Wachstumsfaktoren wie TGF-beta ausgeschüttet (82).
Dies führt über die vermehrte Bildung von Myofibroblasten, Fibronektin und Kollagenen
(insbesondere Kollagen Typ III) zu einer Verdickung der Extrazellulärmatrix (EZM) (84).
Ferner werden durch TGF-beta über verschiedene Smad-Proteine profibrotische Gene und
proteolytische Gewebsfaktoren wie Matrix-Metalloprotease-1 (MMP-1) aktiviert (85).
Außerdem können durch TGF-beta selbst via MCP-1 Makrophagen in das Gewebe einwandern
15und Fibroblasten in Myofibroblasten differenziert werden (85). Bei dieser Umwandlung wird
Smooth Muscle Aktin (SMA) gebildet, welches als Marker für Gewebsfibrose verwendet
werden kann (86). Allerdings bilden nicht nur Fibroblasten und Myofibroblasten Bestandteile
der Extrazellulärmatrix. Auch renales Tubulusepithel, glatte Gefäßmuskulatur und einige
Makrophagen können zu einer Verdickung beitragen, welche durch die Produktion und
Freisetzung von Fibronektin und Kollagenen wie Typ I und Typ III Kollagen verursacht wird
(87, 88).
Durch die Verdickung der Extrazellulärmatrix kommt es zu einer Tubulusatrophie und zur
Rarefizierung kleiner Gefäße, wodurch Gewebshypoxie begünstigt wird und somit ein renaler
Teufelskreis aus selbstverstärkender Inflammation und Fibrose entsteht (87, 88). Dadurch
bildet Fibrose die Endstrecke der meisten chronischen Nierenerkrankungen (55, 70, 86).
2.5. Erläuterung der eingesetzten Tiermodelle
2.5.1. Orte der Nierenschädigung
Der Publikation von Mocker et. al (1) lagen methodisch zwei etablierte Tiermodelle renaler
Schädigung zugrunde, welche häufige Ursachen humaner CKD abbilden. Im Folgenden wird
daher kurz auf diese Modelle eingegangen.
Das 2k1c-Modell (two-kidney, one-clip, Details siehe 2.5.2.) führt durch Verschluss einer
Nierenarterie mittels Metallclip zu einer hypertensiven Nephropathie mit konsekutiven
Schäden am Tubulussystem und renoparenchymatösen Gefäßen (89). Im Gegensatz dazu ist die
durch Antikörper gegen Thymozyten-Antigen 1.1 ausgelöste mesangioproliferative
Glomerulonephritis mit Glomerulosklerose des anti-Thy1.1-Modells (Details siehe 2.5.3.)
durch Schädigung am Nierenkörperchen geprägt (72). Damit ergeben sich bei gleichzeitig
hoher klinischer Relevanz der abgebildeten Krankheitsmuster nur geringe
Überschneidungspunkte für den Hauptort der renalen Schädigung zwischen beiden Modellen.
2.5.2. Das „two-kidney, one-clip“ (2k1c) Tiermodell
Das 2k1c Tiermodell wurde erstmals 1934 zur Investigation der renalen Hypertonie in
Haushunden beschrieben (90). Im Gegensatz zum Hund neigen Nagetiere seltener zu renalen
Kollateralkreisläufen und bilden daher eine dem Menschen ähnlichere Gefäßversorgung ab
(64). Die Grundzüge des Modells sind seit seiner Etablierung beibehalten worden und besitzen
daher eine entsprechend gute Datenlage in der Literatur. Der Ablauf der renalen Schädigung
unterteilt sich in eine akute und eine chronische Phase (91).
16Direkt nach Clipping einer Nierenarterie kommt es durch die Minderperfusion zur Aktivierung
des RAAS der betroffenen Niere mit Anstieg des Plasmarenins und Aktivitätssteigerung des
ACE mit in der Folge erhöhtem AT2 (91). Durch diese Mechanismen und dem daraus
resultierenden Hypertonus versucht der Körper eine Gewebeperfusion in der geclippten Niere
aufrechtzuerhalten. Im Anschluss pendelt sich der Blutdruck bei erhöhten Werten ein und die
Plasmaspiegel der RAAS-Hormone normalisieren sich, während die Expression von Renin und
AT1/AT2 in der geclippten Niere konstant erhöht bleibt (91, 92).
Morphologisch führt die Stenose nach etwa zwei bis vier Wochen zu einer Schrumpfniere auf
der geclippten Seite und kontralateral zu reaktiver Hypertrophie (92). Ferner werden nach
dieser Zeit Umbauprozesse durch den erhöhten renalen Gefäßwiderstand und den verminderten
Ultrafiltrationskoeffizienten in der ungeclippten Niere sichtbar. Dadurch sinkt die Gesamt-GFR
und es kommt zu einer Salz- und Wasserretention (93). Ursächlich ist dabei die erhöhte lokale
Aktivität von ACE und AT2 in der ungeclippten Niere (91).
Charakteristisch sind die unterschiedlichen histopathologischen Veränderungen beider Nieren
nach 5 Wochen. Während die geclippte Niere eine globale Nierenschädigung mit
Glomerulosklerose, Tubulusatrophie und interstitieller Fibrose zeigt, sind die Schäden bei der
ungeclippten Niere diskreter und betreffen primär das Tubulussystem und Interstitium (94).
2.5.3. Das „anti-Thy1.1“ Tiermodell
Im anti-Thy1.1-Modell nutzt man das auf Mesangiumzellen der Ratte vorkommende
Thymozyten-Antigen 1.1 zum Auslösen einer Typ 2 Immunreaktion mit konsekutiver Zelllyse
und Apoptose durch Komplementaktivierung (72). Die Injektion von aufgereinigtem
Antithymozytenserum oder monoklonalen anti-Thy1.1-Antikörpern in den Blutkreislauf der
Ratten führt initial zu einer renalen Mesangiolyse und Bildung von Mikroaneurysmen mit
darauffolgender Leukozyteninvasion (72, 95).
In Verbindung mit den durch Nekrose der Mesangiumszellen ausgeschütteten
Entzündungsmediatoren bildet sich ein inflammatorisches Milieu, das zu einer Verdickung des
Mesangiums führt (72, 96). Diese Verdickung ist zum einen auf die Proliferation der
Mesangiumszellen, zum anderen auf eine verstärkte Bildung von Proteoglykanen, Kollagen
Typ I & Typ IV und Laminin zurückzuführen (96). Ihr Maximum erreicht diese
Mesangiumsproliferation etwa 14 Tage nach Injektion, bevor Abräumprozesse einsetzen (97).
Interessanterweise bleiben die Hyperzellularität und die Folgen der Mikroaneurysmen bis weit
über 6 Wochen nach Injektion histologisch nachweisbar (96, 97).
17Charakteristisch für die Klinik der anti-Thy-1.1-mesangioproliferativen Glomerulonephritis ist
eine sofort beginnende massive Proteinurie, welche nach 2-4 Tagen ihr Maximum erreicht und
nach etwa 3 Wochen auf Normalwerte absinkt (97).
Zusammenfassend sind durch den kombinierten Einsatz obiger Tiermodelle im Rahmen der
hier vorgestellten Studie (Mocker et al.) Rückschlüsse über die renale Expression und
Lokalisation von Markergenen und Markerproteinen möglich, um deren Expressionsverhalten
in Zusammenhang mit verschiedenen renalen Schädigungsmustern zu setzen (1).
3. Fragestellung von Mocker et al. 2020
Im Rahmen der Publikation von Mocker et. al. wurde die Rolle von Chemerin als potentielles
renales Markergen und Markerprotein für Gewebeschädigung im 2k1c und anti-Thy1.1.
Tiermodell untersucht. Besonderer Fokus lag dabei auf der Korrelation von Chemerin und
dessen Rezeptor CmklR1 mit charakteristischen Inflammations- und Fibroseprozessen dieser
CKD Modelle (1). Ferner wurde hierbei die renale Lokalisation von Chemerin analysiert (1).
In Hinblick auf humane Studien zu erhöhten Chemerinspiegeln im Serum von Dialysepatienten
soll untersucht werden, ob Chemerin in den verwendeten Tiermodellen als Serummarker für
Nierenschädigung fungieren kann (1, 46).
Da insbesondere Angiotensin II und Chemerin über NF-kappa-B und den MAPK Signalweg
auf ähnliche Weise Einfluss auf Fibrose nehmen können (42, 77), sollten Schnittpunkte
zwischen RAAS und Chemerin in den verwendeten Tiermodellen identifiziert werden (1).
Hinsichtlich der zahlreichen Interaktionspunkte zwischen chronischer Nierenschädigung, dem
RAAS, renaler Gewebsinflammation und -fibrose sollte daher untersucht werden, ob Chemerin
hier als Schlüsselmediator gesehen werden kann (1). Da Chemerin mittels
Leukozytenaktivierung und Stimulation der Chemotaxis von dendritischen Zellen und
Makrophagen Immun- und Entzündungsreaktionen beeinflusst (35, 39), sollte im Speziellen
evaluiert werden, welche Leukozytensubpopulationen am ehesten mit der renalen
Chemerinexpression korrelieren (1).
184. Quellenangaben
1. Mocker A, Hilgers KF, Cordasic N, Wachtveitl R, Menendez-Castro C, Woelfle J, et
al. Renal Chemerin Expression is Induced in Models of Hypertensive Nephropathy and
Glomerulonephritis and Correlates with Markers of Inflammation and Fibrosis. International
Journal of Molecular Sciences. 2019;20(24):6240.
2. Ross C. Vitamin A. In: Coates P, Betz J, Blackman M, editors. Encyclopedia of
Dietary Supplements. 2nd ed. London and New York: Informa Healthcare; 2010. p. 778-91.
3. O'Byrne SM, Blaner WS. Retinol and retinyl esters: biochemistry and physiology. J
Lipid Res. 2013;54(7):1731-43.
4. Tang XH, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol.
2011;6:345-64.
5. Wolf G. The Experimental Induction of Vitamin A Deficiency in Humans. The
Journal of nutrition. 2002;132(7):1805-11.
6. Bitot. Lésion conjonctivale non encore décrite: coïncidant avec l'héméralopie: Imp. G
Gounouilhou; 1863.
7. Dowling JE. Vitamin A: its many roles-from vision and synaptic plasticity to infant
mortality. J Comp Physiol A Neuroethol Sens Neural Behav Physiol. 2020.
8. Sommer A, Tarwotjo I, Djunaedi E, West KP, Jr., Loeden AA, Tilden R, et al. Impact
of vitamin A supplementation on childhood mortality. A randomised controlled community
trial. Lancet. 1986;1(8491):1169-73.
9. Pino-Lagos K, Guo Y, Noelle RJ. Retinoic acid: A key player in immunity.
BioFactors. 2010;36(6):430-6.
10. Bastos Maia S, Rolland Souza AS, Costa Caminha MF, Lins da Silva S, Callou Cruz
R, Carvalho Dos Santos C, et al. Vitamin A and Pregnancy: A Narrative Review. Nutrients.
2019;11(3).
11. Biesalski HK, Nohr D. Importance of vitamin-A for lung function and development.
Mol Aspects Med. 2003;24(6):431-40.
12. Rodahl K, Moore T. The vitamin A content and toxicity of bear and seal liver.
Biochem J. 1943;37(2):166-8.
13. Bendich A, Langseth L. Safety of vitamin A. The American Journal of Clinical
Nutrition. 1989;49(2):358-71.
14. Rothenberg AB, Berdon WE, Woodard JC, Cowles RA. Hypervitaminosis A-induced
premature closure of epiphyses (physeal obliteration) in humans and calves (hyena disease): a
historical review of the human and veterinary literature. Pediatr Radiol. 2007;37(12):1264-7.
15. Gutierrez-Mazariegos J, Theodosiou M, Campo-Paysaa F, Schubert M. Vitamin A: a
multifunctional tool for development. Semin Cell Dev Biol. 2011;22(6):603-10.
16. WHO. Guideline: Vitamin A supplementation in pregnant women. Geneva: World
Health Organization; 2011.
17. Ingenbleek Y, Young V. Transthyretin (prealbumin) in health and disease: nutritional
implications. Annu Rev Nutr. 1994;14:495-533.
18. Amengual J, Golczak M, Palczewski K, von Lintig J. Lecithin:retinol acyltransferase
is critical for cellular uptake of vitamin A from serum retinol-binding protein. The Journal of
biological chemistry. 2012;287(29):24216-27.
19. Noy N. The ionization behavior of retinoic acid in lipid bilayers and in membranes.
Biochim Biophys Acta. 1992;1106(1):159-64.
20. Harrison EH, Hussain MM. Mechanisms involved in the intestinal digestion and
absorption of dietary vitamin A. The Journal of nutrition. 2001;131(5):1405-8.
21. le Maire A, Teyssier C, Balaguer P, Bourguet W, Germain P. Regulation of RXR-
RAR Heterodimers by RXR- and RAR-Specific Ligands and Their Combinations. Cells.
2019;8(11):1392.
1922. Nagpal S, Patel S, Asano AT, Johnson AT, Duvic M, Chandraratna RA. Tazarotene-
induced gene 1 (TIG1), a novel retinoic acid receptor-responsive gene in skin. The Journal of
investigative dermatology. 1996;106(2):269-74.
23. Nagpal S, Patel S, Jacobe H, DiSepio D, Ghosn C, Malhotra M, et al. Tazarotene-
induced gene 2 (TIG2), a novel retinoid-responsive gene in skin. The Journal of investigative
dermatology. 1997;109(1):91-5.
24. Huebner H, Strick R, Wachter DL, Kehl S, Strissel PL, Schneider-Stock R, et al.
Hypermethylation and loss of retinoic acid receptor responder 1 expression in human
choriocarcinoma. Journal of experimental & clinical cancer research : CR. 2017;36(1):165.
25. Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Ogden A, et al.
All-trans-Retinoic Acid in Acute Promyelocytic Leukemia. New England Journal of
Medicine. 1997;337(15):1021-8.
26. Schenk T, Stengel S, Zelent A. Unlocking the potential of retinoic acid in anticancer
therapy. British Journal of Cancer. 2014;111(11):2039-45.
27. Leyden J, Stein-Gold L, Weiss J. Why Topical Retinoids Are Mainstay of Therapy for
Acne. Dermatol Ther (Heidelb). 2017;7(3):293-304.
28. Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, Braun JT, et al. Retinoic
Acid Embryopathy. New England Journal of Medicine. 1985;313(14):837-41.
29. Coyle KM, Murphy JP, Vidovic D, Vaghar-Kashani A, Dean CA, Sultan M, et al.
Breast cancer subtype dictates DNA methylation and ALDH1A3-mediated expression of
tumor suppressor RARRES1. Oncotarget. 2016;7(28):44096-112.
30. Roy A, Ramalinga M, Kim OJ, Chijioke J, Lynch S, Byers S, et al. Multiple roles of
RARRES1 in prostate cancer: Autophagy induction and angiogenesis inhibition. PloS one.
2017;12(7):e0180344.
31. Wang X, Saso H, Iwamoto T, Xia W, Gong Y, Pusztai L, et al. TIG1 promotes the
development and progression of inflammatory breast cancer through activation of Axl kinase.
Cancer research. 2013;73(21):6516-25.
32. Maimouni S, Issa N, Cheng S, Ouaari C, Cheema A, Kumar D, et al. Tumor
suppressor RARRES1- A novel regulator of fatty acid metabolism in epithelial cells. PloS
one. 2018;13(12):e0208756.
33. Chen A, Feng Y, Lai H, Ju W, Li Z, Li Y, et al. Soluble RARRES1 induces podocyte
apoptosis to promote glomerular disease progression. J Clin Invest. 2020;130(10):5523-35.
34. Bozaoglu K, Bolton K, McMillan J, Zimmet P, Jowett J, Collier G, et al. Chemerin is
a novel adipokine associated with obesity and metabolic syndrome. Endocrinology.
2007;148(10):4687-94.
35. Wittamer V, Franssen JD, Vulcano M, Mirjolet JF, Le Poul E, Migeotte I, et al.
Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from
human inflammatory fluids. The Journal of experimental medicine. 2003;198(7):977-85.
36. Roh SG, Song SH, Choi KC, Katoh K, Wittamer V, Parmentier M, et al. Chemerin--a
new adipokine that modulates adipogenesis via its own receptor. Biochemical and biophysical
research communications. 2007;362(4):1013-8.
37. Takahashi M, Takahashi Y, Takahashi K, Zolotaryov FN, Hong KS, Kitazawa R, et al.
Chemerin enhances insulin signaling and potentiates insulin-stimulated glucose uptake in
3T3-L1 adipocytes. FEBS letters. 2008;582(5):573-8.
38. Yan Q, Zhang Y, Hong J, Gu W, Dai M, Shi J, et al. The association of serum
chemerin level with risk of coronary artery disease in Chinese adults. Endocrine.
2012;41(2):281-8.
39. Lehrke M, Becker A, Greif M, Stark R, Laubender RP, von Ziegler F, et al. Chemerin
is associated with markers of inflammation and components of the metabolic syndrome but
does not predict coronary atherosclerosis. European journal of endocrinology.
2009;161(2):339-44.
2040. Nakamura N, Naruse K, Kobayashi Y, Miyabe M, Saiki T, Enomoto A, et al.
Chemerin promotes angiogenesis in vivo. Physiol Rep. 2018;6(24):e13962.
41. Kaur J, Adya R, Tan BK, Chen J, Randeva HS. Identification of chemerin receptor
(ChemR23) in human endothelial cells: chemerin-induced endothelial angiogenesis.
Biochemical and biophysical research communications. 2010;391(4):1762-8.
42. Dimitriadis GK, Kaur J, Adya R, Miras AD, Mattu HS, Hattersley JG, et al. Chemerin
induces endothelial cell inflammation: activation of nuclear factor-kappa beta and monocyte-
endothelial adhesion. Oncotarget. 2018;9(24):16678-90.
43. Shin WJ, Pachynski RK. Chemerin modulation of tumor growth: potential clinical
applications in cancer. Discov Med. 2018;26(141):31-7.
44. Briana DD, Malamitsi-Puchner A. The role of adipocytokines in fetal growth. Annals
of the New York Academy of Sciences. 2010;1205:82-7.
45. Yoshimura T, Oppenheim JJ. Chemokine-like receptor 1 (CMKLR1) and chemokine
(C-C motif) receptor-like 2 (CCRL2); two multifunctional receptors with unusual properties.
Exp Cell Res. 2011;317(5):674-84.
46. Bonomini M, Pandolfi A. Chemerin in renal dysfunction and cardiovascular disease.
Vascul Pharmacol. 2016;77:28-34.
47. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO
2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney
Disease. Kidney inter., Suppl. 2013; 3: 1–150.
48. National Institute for Health and Care Excellence: Clinical Guidelines. Chronic
kidney disease in adults: assessment and management. London: National Institute for Health
and Care Excellence (UK)
Copyright © NICE 2020.; 2015.
49. Girndt M, Trocchi P, Scheidt-Nave C, Markau S, Stang A. Prävalenz der
eingeschränkten Nierenfunktion. Dtsch Arztebl International. 2016;113(6):85-91.
50. Mills KT, Xu Y, Zhang W, Bundy JD, Chen CS, Kelly TN, et al. A systematic
analysis of worldwide population-based data on the global burden of chronic kidney disease
in 2010. Kidney Int. 2015;88(5):950-7.
51. Hill NR, Fatoba ST, Oke JL, Hirst JA, O'Callaghan CA, Lasserson DS, et al. Global
Prevalence of Chronic Kidney Disease - A Systematic Review and Meta-Analysis. PloS one.
2016;11(7):e0158765.
52. Mills KT, Stefanescu A, He J. The global epidemiology of hypertension. Nature
Reviews Nephrology. 2020;16(4):223-37.
53. Lam DW, LeRoith D. The worldwide diabetes epidemic. Current Opinion in
Endocrinology, Diabetes and Obesity. 2012;19(2):93-6.
54. Tuot DS, Plantinga LC, Hsu C-Y, Jordan R, Burrows NR, Hedgeman E, et al. Chronic
kidney disease awareness among individuals with clinical markers of kidney dysfunction.
Clin J Am Soc Nephrol. 2011;6(8):1838-44.
55. Webster AC, Nagler EV, Morton RL, Masson P. Chronic Kidney Disease. Lancet.
2017;389(10075):1238-52.
56. Vanholder R, Davenport A, Hannedouche T, Kooman J, Kribben A, Lameire N, et al.
Reimbursement of Dialysis: A Comparison of Seven Countries. Journal of the American
Society of Nephrology. 2012;23(8):1291.
57. Gandjour A, Armsen W, Wehmeyer W, Multmeier J, Tschulena U. Costs of patients
with chronic kidney disease in Germany. PloS one. 2020;15(4):e0231375.
58. Chatfield G, Rickert K, Komm N. Jahresbericht 2018 zur Qualität in der Dialyse:
IQTIG – Institut für Qualitätssicherung und Transparenz im Gesundheitswesen; 2019
10.07.2019.
59. Breyer MD, Susztak K. Developing Treatments for Chronic Kidney Disease in the
21st Century. Seminars in nephrology. 2016;36(6):436-47.
21Sie können auch lesen

















































