Der DDP4- Inhibitor Linagliptin verbessert renalen Schaden und fördert die Auflösung einer halbmondbildenden Glomerulonephritis im Rattenmodell ...
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Der DDP4- Inhibitor Linagliptin verbessert
renalen Schaden und fördert die Auflösung
einer halbmondbildenden Glomerulonephritis
im Rattenmodell
Pathologisches Institut
Abteilung für Nephropathologie
Der Medizinischen Fakultät
der Friedrich-Alexander-Universität
Erlangen-Nürnberg
zur
Erlangung des Doktorgrades Dr. med.
vorgelegt von
Anna-Lena Sophie Mayer
Als Dissertation genehmigt von der Medizinischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg Vorsitzender des Promotionsorgans: Prof. Dr. Markus F. Neurath Gutachter: Prof. Dr. Christoph Daniel Gutachter: Prof. Dr. Mario Schiffer Gutachterin: Prof. Dr. Nicole Endlich Gutachterin: Prof. Dr. Catherine Meyer-Schwesinger Tag der mündlichen Prüfung: 22. März 2022
Meinen Eltern in Dankbarkeit

1. Zusammenfassung
Hintergrund und Ziele:
Dipeptidylpeptidase 4 (DPP4) - Inhibitoren sind eine Substanzklasse von blutzuckersenkenden
Medikamenten für die Behandlung von Diabetes mellitus Typ 2. In eigenen Vorstudien an
humanen Nierenbiopsien beobachteten wir eine hohe DPP4- Expression in frühen
glomerulären Halbmonden. Diese glomerulären Läsionen treten in verschiedenen
Nierenerkrankungen auf und sind ein Kennzeichen für einen schweren Nierenschaden.
Deshalb untersuchten wir, ob und auf welche Art und Weise die DPP4 bei der Pathogenese
einer anti-GBM-Glomerulonephritis im Rattenmodell beteiligt ist.
Methoden:
Wistar Kyoto Ratten mit induzierter anti-GBM-Glomerulonephritis wurden in einem zwei- oder
achtwöchigen Therapieregime (je n=44) oral mit dem DPP4- Inhibitor Linagliptin behandelt (3
mg/kg Körpergewicht): entweder präventiv mit Therapiebeginn am Tag der
Krankheitsinduktion oder therapeutisch 7 Tage nach Studienbeginn in der Kurzzeit- bzw. nach
4 Wochen in der Langzeit-Studie bis jeweils zum Endpunkt nach 14 Tagen bzw. nach 8
Wochen. In beiden Experimenten wurden eine gesunde Kontroll- und eine unbehandelte anti-
GBM-nephritische Gruppe eingeschlossen. Alle Gruppen bestanden jeweils aus 11 Tieren. Die
Nierenfunktion, morphologische Veränderungen, Entzündung und Fibrose in den Nieren,
besonders in den Glomeruli, wurden zu mehreren Zeitpunkten untersucht.
Ergebnisse:
Im Langzeit- Experiment reduzierte eine präventive Linagliptin- Behandlung signifikant die
Anzahl an glomerulären Halbmonden, die Glomerulosklerose, den tubulären Schaden und die
renale Fibrose in den anti-GBM-nephritischen Ratten im Vergleich zu unbehandelten
erkrankten Ratten. In beiden Linagliptin- Gruppen wurde eine signifikante Abnahme an Pax8-
positiven Zellen auf dem glomerulären Tuft erreicht. Dies zeigte eine verstärkte Auflösung der
zellulären Halbmonde auf. Die Therapie mit Linagliptin konnte die Anzahl an Podozyten nicht
erhöhen, aber den podozytären Stress in beiden behandelten Gruppen vermindern. Insgesamt
erbrachte die therapeutische Intervention eine geringere Verbesserung der Nierenerkrankung
nach 8 Wochen verglichen zur präventiven Therapievariante.
Schlussfolgerungen:
Die DPP4- Inhibition mit Linagliptin verbessert renalen Schaden in der anti-GBM-
Glomerulonephritis im Rattenmodell. So stellt Linagliptin nicht nur eine sichere Therapieoption
gegen Diabetes mellitus Typ 2 dar, sondern kann auch die Auflösung und Ausheilung von
glomerulären Schäden in anderen, diabetesunabhängigen Nierenerkrankungen verbessern.
1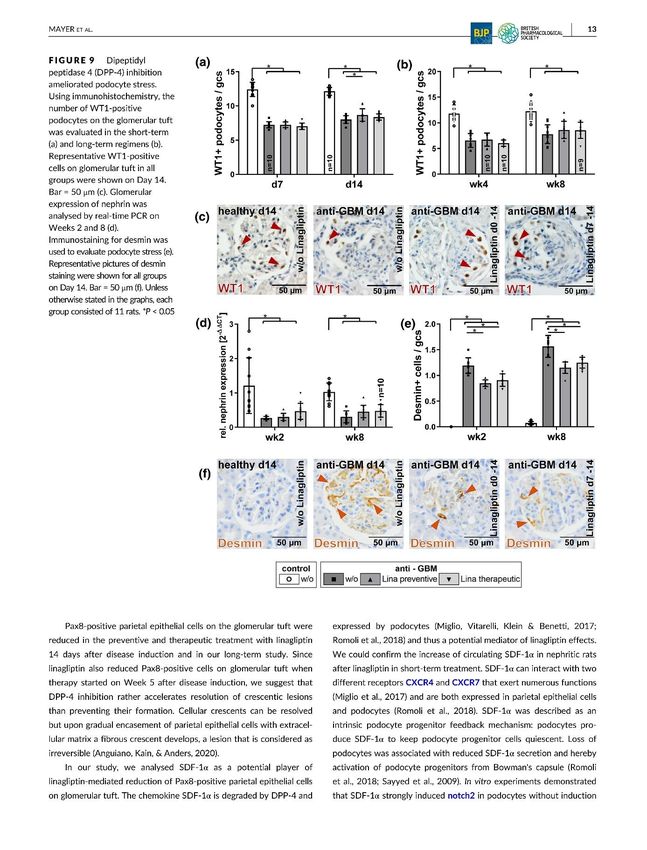
2. Einleitung
Eigenschaften und Therapie der rasch progedienten Glomerulonephritis (RPGN)
Eine Glomerulonephritis (GN) mit sogenannter Halbmondbildung (crescence formation oder
crescentic GN) ist ein typisches immunhistologisches Merkmal für die rasch progrediente
Glomerulonephritis (RPGN). Die RPGN zeichnet sich klinisch durch ein akutes nephritisches
Syndrom mit rascher Progression zu einem terminalen Nierenversagen aus (Moroni and
Ponticelli, 2014). Diese spezielle Glomerulonephritis (GN)- Form betrifft ca. 10- 15% aller
Patienten mit GN (Schena, 1997).
Charakteristischerweise sind mehr als 50% der Glomeruli einer renalen Biopsie von
extrakapillärer Proliferation betroffen (Moroni and Ponticelli, 2014). Histologisch zeichnen sich
diese Läsionen durch das Vorkommen von fibrinoiden Nekrosen des glomerulären
Kapillarkonvoluts und glomerulären Halbmonden (sog. crescents) aus, die Ausdruck eines
schweren glomerulären Schadens sind (Li and Chen, 2013, Smeets et al., 2009b). Dabei lässt
sich die halbmondbildende Glomerulonephritis klassischerweise immunologisch bzw.
immunhistologisch in vier Typen unterteilen: Typ I anti-glomeruläre Basalmembran (anti-
GBM)-Glomerulonephritis (ca. 10% aller RPGN- Fälle), Typ II Immunkomplex-
Glomerulonephritis (15- 20%), Typ III Pauci-Immune-Glomerulonephritis (60- 80%) und Typ IV
Doppel-Antikörper-Krankheit, die sehr selten ist und eine Mischform aus Typ I und Typ III
darstellt (Jennette, 2003, Couser, 1988, Moroni and Ponticelli, 2014, Erwig and Rees, 1999).
Die häufigste RPGN Form stellt die Typ III Pauci-Immune-GN dar (Schena, 1997). Sie
zeichnet sich durch erhöhte antineutrophile zytoplasmatische Antikörper (ANCAs, v.a.
Antiproteinase 3-ANCA oder Myeloperoxidase-ANCA) sowie eine systemische Vaskulitis aus,
die u.a. bei den ANCA- assoziierten Vaskulitiden wie der mikroskopischen Polyangiitis, der
Granulomatose mit Polyangiitis und der eosinophilen Granulomatose mit Polyangiitis
vorkommt (Sinico et al., 2006, Jennette and Falk, 1997, Jennette et al., 2013). Die Mehrheit
der Pauci-Immunen Patienten sind dabei pANCA- positiv (Moroni and Ponticelli, 2014).
Ca. 15- 20% der RPGN fallen auf die Typ II Immunkomplex- vermittelte GN zurück. Die
Immunkomplex-RPGN kann als Komplikation postinfektiös (poststreptokokken-GN, infektiöse
Endokarditis) (Moroni et al., 2002), bei einer Bindegewebsstörung (Lupus-Nephritis,
kryoglobulinämische GN) (Masani et al., 2005, Moroni et al., 2007, Borchers et al., 2012,
Tarantino et al., 1995) oder anderen Glomerulopathien (IgA-Nephropathie,
membranoproliferative GN, Purpura Schönlein-Henoch) auftreten (Kawasaki et al., 2004,
Coppo et al., 1997, Korzets et al., 1987).
Die Typ I anti-GBM-GN betrifft ca. 10% der RPGN und ist autoimmun vermittelt (Jennette,
2003). Dabei werden autoimmune Antikörper überwiegend der igG-Klasse (Bolton, 1996)
gegen Fragmente von Kollagen IV gebildet, das vorwiegend in der glomerulären
2
Basalmembran der Niere und in der Lunge vorkommt. Folglich kommt es zu einer
zellgebundenen Entzündungsreaktion. Ist nur die Niere betroffen, spricht man von einer anti-
GBM-Glomerulonephritis. Bei zusätzlicher Lungenbeteiligung mit alveolären Hämorrhagien
und konsekutiven Hämoptysen bezeichnet man die Krankheit als Goodpasture Syndrom, das
bei ca. 40- 60% der an Typ I erkrankten Patienten auftritt (McAdoo and Pusey, 2017). Das
Hauptziel dieser autoimmunen Antwort ist die non-collagenous 1 (NC1) Domäne der alpha-3
Kette von Typ IV Kollagen in der glomerulären Basalmembran und in der Membran der
pulmonalen Alveoli alpha-3(IV)NC1; auch das „Goodpasture Autoantigen” genannt (Saus et
al., 1988, Turner et al., 1992, Kalluri et al., 1995). Es konnten aber auch Antikörper gegen
alpha-5(IV)NC1 identifiziert werden (Pedchenko et al., 2010). Diese Autoimmunität scheint
stark mit HLA (human leukocyte antigen)- Genen assoziiert zu sein (Phelps and Rees, 1999,
Fisher et al., 1997). Jedoch sind diese Suszeptibilitätsallele weit verbreitet und auch mit vielen
anderen autoimmunen Krankheiten vergesellschaftet, sodass zusätzliche Faktoren zur
Genese der anti-GBM-GN notwendig sind. Hierbei könnten Zigarettenrauch, virale
respiratorische Infekte, sowie eine Inhalation von Hydrocarbon als Trigger eine Rolle spielen
(Donaghy and Rees, 1983, Kelly and Haponik, 1994). Obwohl die humorale Antwort auf den
direkten Glomerulusschaden der Hauptauslöser für die Aktivierung der parietalen Epithelzellen
ist, scheint auch eine T-Zell- Antwort und Regulation eine pathogene Rolle zu spielen (Tarzi
et al., 2011). Bisherige Studien konnten das Vorhandensein von autoreaktiven T-Lymphozyten
direkt gegen alpha-3NC1 aufzeigen (Pedchenko et al., 2010, Merkel et al., 1996).
Die Inzidenz der Typ I RPGN beträgt allgemein ca. 0,5- 1 pro 1 Millionen Population pro Jahr
in der europäischen Bevölkerung (McAdoo and Pusey, 2017, Fomegne et al., 2006), wobei
eine Studie aus Irland über 10 Jahre eine Krankheitsrate von 1,64 Fällen auf 1 Millionen
Einwohner aufzeigen konnte (Canney et al., 2016). Die Erkrankung weist eine bimodale
Altersverteilung auf: Ein Maximum in der dritten Dekade sowie ein zweiter Anstieg in der
sechsten/ siebten Dekade (Fischer and Lager, 2006, Moroni and Ponticelli, 2014).
Klinische Symptome treten meist langsam progredient auf. Dabei sind Schwäche, Müdigkeit,
Fieber, Übelkeit, Erbrechen, Anorexie, Arthralgie und Bauchschmerzen typisch (Moroni and
Ponticelli, 2014). Viele der Betroffenen klagen über akute influenzaähnliche Beschwerden, die
meist innerhalb von wenigen Wochen in einer raschen Verschlechterung der Nierenfunktion
bis im Nierenversagen mit einer schweren Oligurie enden (L'Imperio et al., 2017). Meistens
wird die RPGN durch das Vorliegen eines akuten Nierenversagens im Rahmen eines
nephritischen Syndroms mit Hämaturie, Proteinurie und dysmorphen Erythrozyten auffällig.
Zur Diagnose werden eine Hämaturie sowie Erythrozytenzylinder/-sediment in der
Urinanalyse, ein erhöhtes Serumkreatinin, eine Anämie und Leukozytose im Blutbild
herangezogen. Zudem können jeweils die spezifischen Antikörper (z.B. anti-GBM-AK, Anti-
DNA-AK, Antistreptolysin-O-AK) erhöht in der Serologie gemessen werden (Moroni and
3
Ponticelli, 2014). Die Detektion von zirkulierenden Autoantikörpern gelingt in 95% der Fälle
anhand von ELISA (Enzyme-linked Immunosorbent Assay) oder Multiplex Flow
Immunoassays (L'Imperio et al., 2017, McAdoo and Pusey, 2017). Die Nierenbiopsie mit den
typischen glomerulären Halbmonden stellt den Goldstandard der Diagnose dar. Im
Durchschnitt weisen in der anti-GBM-GN ca. 75% aller Glomeruli eine glomeruläre
Halbmondbildung auf (Jennette, 2003). Bei der anti-GBM-Glomerulonephritis können lineare
IgG- Ablagerungen an der glomerulären Basalmembran anhand von Immunfluoreszenz-
Mikroskopie dargestellt werden (Sethi and Fervenza, 2019).
Essentiell für eine erfolgreiche Behandlung ist ein rascher Therapiebeginn (Moroni and
Ponticelli, 2014), ansonsten entwickeln mehr als zwei Drittel der unbehandelten Patienten
innerhalb von 6 Monaten einen Progress bis zur terminalen Niereninsuffizienz (Hellmark and
Segelmark, 2014).
Zurzeit stehen klinisch nur wenige Präparate zur effektiven Behandlung von RPGN zur
Verfügung. Entsprechend der Immunpathogenese besteht die Therapie bei allen Typen der
RPGN aus einer hochdosierten, aggressiven Immunsuppression mit Corticosteroiden
gegebenenfalls in Kombination mit dem zytotoxischen Mittel Cyclophosphamid und eventuell
Plasmapherese (Li and Chen, 2013). Jedoch gibt es Behandlungsgrenzen beim Einsatz einer
hochaggressiven Immunsuppression: z.B. bei einem Serumkreatinin über 7 mg/dl zu Beginn.
Für diese Patienten könnte solch eine Therapie mehr Risiken als Vorteile bringen (L'Imperio
et al., 2017). Es gibt einzelne Berichte über den Nutzen des anti-CD20-Antikörpers Rituximab
als „add-on“ oder bei Intoleranz der Standardtherapie (Touzot et al., 2015, Davies et al., 2013).
Zudem gibt es Behandlungsversuche mit TNF-α (Tumornekrosefaktor)- Inhibitoren (Khan et
al., 2005, Karkar et al., 2001, Le Hir et al., 1998). Aber es gibt aktuell keine ausreichende
Evidenz für deren Gebrauch in der first-line-Therapie.
Speziell bei der anti-GBM-GN wird vorwiegend die Plasmapherese in Kombination mit
Prednison und Cyclophosphamid eingesetzt und für mindestens 3 Monate beibehalten, um die
vorliegenden Antikörper aus dem Körper zu eliminieren und einer neuerlichen
Antikörperbildung entgegenzuwirken (Lahmer and Heemann, 2012, Moroni and Ponticelli,
2014, Podracka and Matousovic, 2013, Hruskova and Tesar, 2018). Die Plasmapherese (ein
Austausch von täglich 2- 4 l Blut) ist durch die schnelle Eradikation von freien Antikörpern,
intakten Immunkomplexen und Entzündungsmediatoren (z.B. Fibrinogen, Komplement)
wirksam. Bei bereits eingesetzter, ausgeprägter Niereninsuffizienz ist häufig eine
Dialysebehandlung notwendig (Levy et al., 2001). Eine Studie mit einer
Kombinationsbehandlung von Plasmaaustausch und Hochdosisimmunsuppression zeigte bei
Patienten mit einer Serumkreatinin- Konzentration von unter 5,7 mg/dl eine Überlebensrate
von 100% bzw. 95% Nierenüberleben über ein Jahr. Bei einem höheren Serumkreatinin fielen
diese Werte auf 83% bzw. 82% innerhalb eines Jahres. Dagegen wiesen dialysepflichtige
4
Betroffene eine 1- Jahres Überlebensrate von 65% bzw. Nierenüberleben von 8% auf. Bei
ubiquitärem Glomerulibefall innerhalb einer Nierenbiopsie blieben diese erkrankten Personen
dialysepflichtig (Moroni and Ponticelli, 2014). Ein Rückfall nach erreichter Remission tritt in der
anti-GBM-GN selten auf. Die größte Langzeit- follow-up Kohortenstudie am Hammersmith
Hospital, London, mit 71 Patienten konnte eine Wiedererkrankung in unter 3% der Fälle
registrieren (Levy et al., 2001).
Eine Nierentransplantation ist für alle RPGN-Typen wirksam, wobei ein Wiederauftreten der
Krankheit im Transplantat möglich ist. Daher sollte mindestens ein Zeitraum von sechs
Monaten persistierender Seronegativität erreicht werden, bevor eine Nierentransplantation bei
Patienten mit terminalen Nierenversagen durch eine anti-GBM-GN durchgeführt wird. Unter
diesen Voraussetzungen und laufender Immunsuppression ist ein Relapse sehr selten, wie
auch die Registerstudie ANZDATA mit einer Rate von 2,7% aufzeigen konnte (Tang et al.,
2013).
Da jedoch eine Nierentransplantation nur selten umgesetzt werden kann und bisher keine
hocheffektiven Medikamente für eine sichere vollständige Remission vorliegen, ist die
Erforschung neuer Therapieoptionen bei dieser Erkrankung klinisch weiterhin von großer
Bedeutung. Zudem treten glomeruläre Halbmonde wie bereits genannt auch in anderen,
häufigeren Nierenerkrankungen wie der IgA-Nephritis, Lupus-Nephritis oder in postinfektiösen
Glomerulonephritiden auf (Jennette, 2003).
Dipeptidylpeptidase 4 (DPP4) als mögliche Therapie zur Behandlung der RPGN
Eigene Vorarbeiten legen den Schluss nahe, dass die Dipeptidylpeptidase 4 (DPP4) ein neues
Target bei der Behandlung von Glomerulonephritiden mit Halbmondbildung sein könnte, da
diese in besonderem Maße in den Halbmonden exprimiert wird und bereits in eigenen
Voruntersuchungen gezeigt werden konnte, dass ein Fehlen der DPP4 in einem 5/6-
Nephrektomie- Modell der chronischen Nierenerkrankung anti-inflammatorisch wirkt.
Das Enzym Dipeptidylpeptidase 4 ist vor allem als Target in der Therapie des Diabetes mellitus
Typ 2 bekannt. DPP4- Inhibitoren sind in den letzten Jahren zu einem festen Bestandteil in der
Therapie des Diabetes mellitus Typ 2 geworden (Mikhail, 2012, Mikhail, 2008). Unabhängig
von ihrer Rolle bei Diabetes mellitus wird die DPP4 in vielen Organen exprimiert.
Untersuchungen wie beispielsweise von Wang et al. (Wang et al., 2014) oder Fuchs et al.
(Fuchs et al., 2009) konnten zeigen, dass die DPP4 überwiegend in der Niere lokalisiert ist.
Dort wurde auch eine besonders hohe enzymatische Aktivität und Expression vor allem im
Bürstensaum proximaler Tubuluszellen und in geringerem Maße auch von Podozyten
nachgewiesen. Eine ausgeprägte Expression und Aktivität von DPP4 in glomerulären Zellen
wurde ebenfalls bereits durch Fukasawa et al. (Fukasawa et al., 1981) und durch weitere
Gruppen bestätigt (Mentzel et al., 1996, Kettmann et al., 1992). Außerdem lässt sich eine
5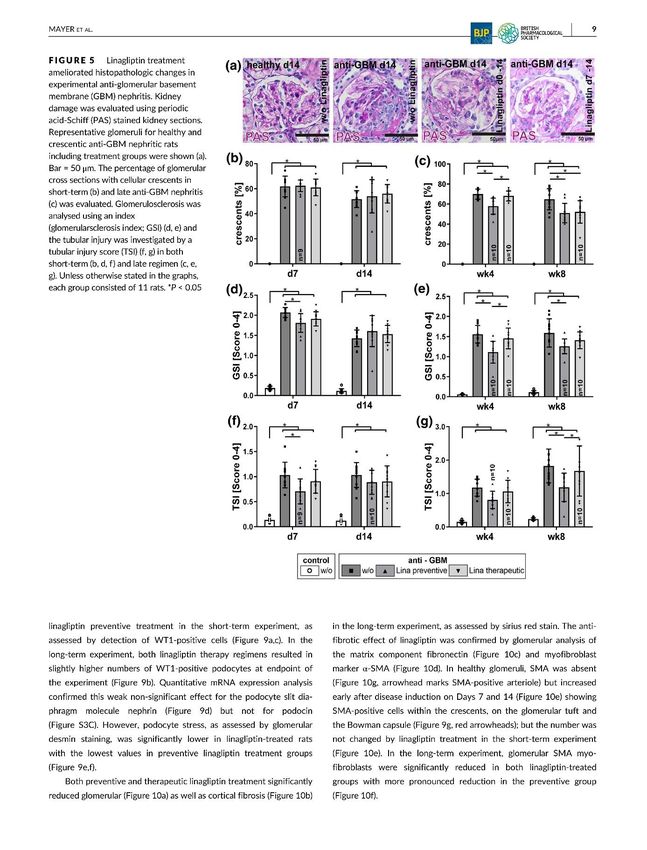
Dipeptidylpeptidase- Aktivität in verschiedenen Zelltypen des Immunsystems wie T-Zellen
(Mentzel et al., 1996), B-Zellen (Buhling et al., 1995) und natürlichen Killer- Zellen (Buhling et
al., 1994) nachweisen. Dabei scheint die DPP4 auch eine wichtige Funktion in der Aktivierung
und der Migration von Makrophagen zu spielen (Ta et al., 2010).
Eigene Untersuchungen der nephropathologischen Abteilung konnten in Nierenbiopsien von
Patienten mit unterschiedlichen Nierenerkrankungen zeigen, dass die DPP4 besonders stark
in glomerulären Halbmonden gebildet wird, die eine entscheidende Bedeutung in der
Progression von Nierenerkrankungen haben.
Welche genauere Rolle die DPP4 bei der Halbmondbildung spielt, ist bislang jedoch unklar.
Dies sollte in der vorliegenden Arbeit näher untersucht werden. Speziell wurde geprüft, ob eine
Inhibition der DPP4 eine Halbmondbildung in einer experimentellen anti-GBM-
Glomerulonephritis im Rattenmodell (Glomerulonephritis-Modell mit Halbmondbildung)
verhindert oder reduziert.
Dabei wäre ein indirekter Einfluss der DPP4 durch ihre enzymatische Aktivität und Degradation
von Substraten, aber auch ein direkter Mechanismus durch Interaktionen der DPP4 mit
anderen Molekülen und Zellen, z.B. als Rezeptor möglich. Durch die Gabe eines DPP4-
Inhibitors zu verschiedenen Zeitpunkten nach Induktion der Nierenerkrankung sollte weiter
geklärt werden, ob eine DPP4- Inhibition die Entstehung und/oder die Progression
entzündlicher Nierenerkrankungen mit Halbmondbildung günstig beeinflussen kann.
Eines der initialen Schlüsselereignisse bei der Entstehung einer Halbmondnephritis scheint
die Aktivierung von PECs (parietal epithelial cells) zu sein, welche die Bowman‘sche Kapsel
auskleiden. Diese bilden zunächst „Brücken“ zwischen der Bowman’schen Kapsel und dem
glomerulären Tuft (Smeets et al., 2009b). Vermutlich ist es ein spezieller Subtyp, der aus
weniger differenzierten PECs besteht, die einen unreifen progenitorähnlichen Phänotyp
aufweisen. Dieser besitzt eine starke Fähigkeit zur hyperplastischen Expansion (Smeets et al.,
2009a). Die Aktivierung dieses speziellen PECs- Subtypens wird durch eine Schädigung der
glomerulären Filtrationsbarriere verursacht (Ryu et al., 2012).
Grundsätzlich bestehen die zellulären Halbmonde im Glomerulus aus mehr als zwei Schichten
von proliferierenden Zellen im Bowman’schen Raum zwischen den Zellen des glomerulären
Tufts und der Bowman’schen Kapsel. Durch verschiedene Immunangriffe, unter anderem
durch zytotoxische Stoffe wie Immunkomplexe oder extrazelluläre Histone, kann es zu einer
Entzündungsreaktion und mikrovaskulären Schäden kommen. Zunächst entwickeln sich durch
diese endokapillare Inflammation mit einer Ablösung des Endothels von der Basalmembran
Lücken in der Kapillarwand, der GBM und der Bowman’schen Kapsel (Ryu et al., 2012, Mulay
et al., 2016). Diese Ruptur ermöglicht den Eintritt von Plasmaproteinen und
Koagulationsfaktoren aus den Gefäßen in den glomerulären Raum (Fogo et al., 2016). Diese
Verletzung der vaskulären Integrität im Glomerulus und die eintretenden Blutbestandteile
6
bewirken eine Hyperplasie von PECs (Ryu et al., 2012). Diese stellen damit die zelluläre
Hauptkomponente in den entstehenden Halbmonden dar (Smeets et al., 2009b). Zudem
kommt es zur Zellimmigration von beispielsweise Monozyten, Lymphozyten, Makrophagen
und Fibroblasten und zu einer Fibrosierung innerhalb des Raums, sodass letztlich fixe zelluläre
Halbmonde zwischen Endothel, Podozyten und dem äußeren Blatt der Bowman’schen Kapsel
entstehen (L'Imperio et al., 2017, Smeets et al., 2009b, Jennette, 2003). Schließlich kommt es
zu einem Zusammenbruch des glomerulären Tufts, sodass die GFR (glomeruläre
Filtrationsrate) eines einzelnen Nephrons sinkt und schließlich insgesamt die renale
Filtrationsleistung abnimmt (Anguiano et al., 2020).
Die genauen Mechanismen, die diese Brückenbildung der PECs hervorrufen, wie die PECs
aktiviert werden und über welche Moleküle sie verknüpft werden, sind noch unklar. In der
frühen Phase der Erkrankung der experimentellen anti-GBM-Nephritis ist das
inflammatorische Geschehen zunächst von einer Th17- später im Verlauf von einer Th1-
Reaktion geprägt (Paust et al., 2012). Bei humanen Glomerulonephritiden mit
Halbmondbildung ist das immunologische Geschehen zumindest zum Zeitpunkpunkt der
Diagnose vom Th1-Typ (Tipping and Kitching, 2005), vermutlich spielen hier ebenfalls Th17-
Zellen in der initialen Phase eine Rolle. Die Erkrankung ist jedoch bei den humanen Proben in
der Regel bereits weiter fortgeschritten, sodass eine exakte Aussage zum
Entstehungsmechanismus schwierig ist.
In zellulären Halbmonden eines Glomerulus konnten Podozyten, PECs, Makrophagen und T-
Zellen nachgewiesen werden (Moeller et al., 2004, Jennette and Hipp, 1986, Morita et al.,
1973, Min et al., 1974, Boucher et al., 1987). Linage tracing Experimente in Mausmodellen
konnten zeigen, dass der überwiegende Teil der Zellen, die den Halbmond bilden, ihren
Ursprung in proliferierenden PECs haben (Smeets et al., 2009b, Wheeler et al., 1993). Auch
laut Shankland et al. sind Pax8 (paired box Protein 8)- positive PECs durch Migration und
Proliferation die dominierenden Zellen in den zellulären Halbmonden (Shankland et al., 2014).
Nach Ruptur der parietalen Basalmembran finden sich auch Makrophagen in den zellulären
Halbmonden (Boucher et al., 1987, Lan et al., 1992). Podozyten werden bei der Pathogenese
durch die inflammatorischen Prozesse geschädigt, können aber selbst zusätzlich auch an der
Immunmodulation beteiligt sein. Es konnte bereits gezeigt werden, dass Podozyten im Verlauf
einer anti-GBM-Nephritis als Antigen-präsentierende Zellen fungieren und so wesentlich die
Halbmondbildung und Progression der Erkrankung bestimmen können (Goldwich et al., 2013).
Doch der konkretere Pathomechanismus mit den beteiligten Reaktionspartnern ist bisher noch
nicht geklärt. Unseren Vorarbeiten nach könnte die DPP4 möglicherweise involviert sein.
Die Dipeptidylpeptidase 4 (DPP4) ist ein Glykoprotein, das in einer löslichen im Blut
zirkulierenden sowie membrangebunden Form (dort als CD26 bezeichnet) vorliegt (De
Meester et al., 2009). Als Serin-Exopeptidase spaltet sie Dipeptide am N-Terminus von
7Polypeptiden ab, wenn ein spezifisches Peptid mit den Aminosäuren Alanin und Prolin in
Position 2 vorhanden ist. Die DPP4 hat einige Substrate. Der bekannteste Regelkreis spielt
eine wichtige Rolle im Glukosehaushalt der Zellen. Durch die DPP4 werden die Inkretin-
Hormone GLP-1 (glucagon-like peptide 1) und GIP (glucose-dependent insulinotropic peptide
bzw. gastric inhibitory polypeptide) gespalten und somit inaktiviert (Deacon, 2005). Zudem
inaktiviert sie einige Chemokine wie RANTES (Regulated And Normal T cell Expressed and
Secreted), MCP-1 (monocyte chemotactic protein 1) und SDF-1α (stromal-derived factor 1 α)
(De Meester et al., 1999). Weitere Substrate sind Glukagon, NPY (Neuropeptid Y), BNP (brain
natriuretic peptide) oder auch der Neurotransmitter Substanz P (De Meester et al., 2000).
Weiterhin fungiert die DPP4 als Protein-Bindungspartner für ADA (Adenosindeaminase) im
Rahmen der Immunmodulation und für Integrine, wie z.B. bei der TGF-ß (transforming growth
factor)- Aktivierung (Lambeir et al., 2003, Kameoka et al., 1993). In der membrangebundenen
Form (CD26) dient sie zudem als Oberflächenrezeptor und Co-stimulierendes Protein, v.a. bei
der Modulation von Immunreaktionen. So wirkt CD26/DPP4 als Co-stimulierendes Signal bei
der Aktivierung von T-Zellen (Kahne et al., 1999, Gorrell et al., 2001). Die Aktivierung wird
dabei über die Bindung an Caveolin-1, das auf Antigen-präsentierenden Zellen lokalisiert ist,
vermittelt (Ohnuma et al., 2009, Ohnuma et al., 2007). Interessanterweise konnte auch auf
parietalen Epithelzellen eine Expression von Caveolin-1 nachgewiesen werden (Ostalska-
Nowicka et al., 2007). Möglicherweise sind auch parietale Epithelzellen an der Aktivierung von
T-Zellen beteiligt.
Bisher stellen DDP4- Inhibitoren eine etablierte Wirkstoffklasse zur Behandlung von Diabetes
mellitus Typ 2 dar (Baetta and Corsini, 2011). Durch ihre hemmende Wirkung auf die DPP4
werden die Inkretinhormone GIP sowie GLP-1 nicht mehr durch die DPP4 degradiert und ihre
Konzentrationen steigen. Erhöhte GIP-/ GLP-1- Spiegel im Serum führen zu einer vermehrten
Insulinausschüttung durch stimulierte β-Zellen im Pankreas. Das sekretierte Insulin kann
letztlich den Blutglukosespiegel senken (Lankas et al., 2005, Mulvihill, 2018).
Zusätzlich zu den GIP-/ GLP-1- vermittelten Effekten wurde nach DPP4- Inhibition bei
Diabetes mellitus bereits eine moderate Senkung des systolischen Blutdrucks, verbesserte
Lipidparameter, reduzierte hsCRP (hochsensitives C-reaktives Protein)- Werte und eine
verbesserte endotheliale Dysfunktion, aber auch anti-inflammatorische Effekte festgestellt
(Fadini and Avogaro, 2011, Rizzo et al., 2009). Eine CKD (chronical kidney disease)- Studie
mit einem 5/6- Nephrektomie- Rattenmodell und urämischer Kardiomyopathie konnte bereits
zeigen, dass nach einer viertägigen oralen Therapie mit einem DPP4- Inhibitor die mRNA-
Expressionen profibrotischer Gene im Vergleich zur Placebo-behandelten Gruppe signifikant
reduziert waren (Chaykovska et al., 2011).
Eine regrediente Albuminurie durch eine DPP4- Inhibition mittels verschiedener Gliptine wurde
bereits sowohl in präklinischen (Sharkovska et al., 2014, Spencer et al., 2018) als auch in
8klinischen Studien beschrieben. Hattori et al. zeigte, dass eine 6-monatige Therapie mit dem
DPP4- Inhibitor Sitagliptin sowohl eine Mikro- als auch Makroalbuminurie in Patienten mit
Diabetes mellitus Typ 2 signifikant reduzierte (Hattori, 2011). Eine retrospektive Arbeit
analysierte vier klinische Phase III Studien zwischen Januar 2008 und Mai 2010 und konnte
eine signifikante Reduktion der Albuminurie in Patienten mit Diabetes mellitus Typ 2 und
renaler Dysfunktion durch eine additive Gabe von Linagliptin zu RAAS (Renin-Angiotensin-
Aldosteron-System)- Inhibitoren zeigen. Diese Ergebnisse waren unabhängig vom
Blutglukoselevel und systolischen Blutdruck (Groop et al., 2013). Die randomisierte MARLINA-
T2D Studie untersuchte die Effekte von Linagliptin in Bezug auf die glykämische Kontrolle und
Albuminurie in Diabetes mellitus Typ 2 Patienten. Dabei erhielten 360 Patienten entweder
Linagliptin oder Placebo für 24 Wochen zusätzlich zu ihrer RAAS-Blockade- Therapie (Groop
et al., 2017). Jedoch konnten nur signifikante Verbesserungen in der glykämischen Kontrolle,
aber nicht für die Albuminurie registriert werden. Dennoch lassen post hoc Analysen der
MARLINA-T2D Daten vermuten, dass Linagliptin in einigen Patienten doch eine bessere
Reduktion bewirkte. Besonders Patienten, denen eine Nierenschädigung im Endstadium
drohte, schienen besonders zu profitieren (signifikante Reduktion des relativen Risikos für ein
terminales Nierenversagen um 17%). Hierbei muss berücksichtigt werden, dass die
bestehende Albuminurie durch verschiedene pathophysiologische Mechanismen verursacht
werden kann, nicht nur durch die bekannte Diabeteserkrankung und endotheliale Schädigung
(Satchell and Tooke, 2008). So könnten einige Teilnehmer mit anderen zugrundeliegenden
Pathomechanismen wie Podozytenschaden oder mesangialer Expansion unabhängig von
Diabetes von der Linagliptin- Behandlung renal profitiert haben.
So zeigte auch die Sicherheitsstudie SAVOR-TIMI 53 für den DPP4- Inhibitor Saxagliptin eine
ausgeprägtere Albuminurie- Reduktion in Teilnehmern, die bereits eine niedrigere GFR zu
Beginn der Therapie hatten (Mosenzon et al., 2017). So könnte die MARLINA-T2D
Studienpopulation zu wenig Individuen mit fortgeschrittener CKD eingeschlossen haben,
sodass der positive Effekt auf die Albuminurie durch Linagliptin nicht offensichtlich und
signifikant wurde (Groop et al., 2017). Ähnliche Ergebnisse waren auch in einer retrospektiven
Kohorten- Studie zu verzeichnen. Besonders Patienten mit einer manifesten Makroalbuminurie
zeigten einen signifikanten Rückgang ihrer Albuminlevel nach einjähriger Behandlung mittels
eines DPP4- Inhibitors. Nach vier Jahren mit dieser Medikation konnte eine Abnahme der GFR
in Patienten mit Mikroalbuminurie verlangsamt und in Patienten mit Makroalbuminurie
rückgängig gemacht werden (Kim et al., 2016).
Wie sich eine langfristige DPP4- Inhibition bei einer chronischen Nierenerkrankung
unabhängig von Diabetes mellitus Typ 2 auf die Nieren auswirkt, wurde jedoch bisher wenig
intensiv untersucht.
9Anzeichen für pleiotrope, renoprotektive Wirkungen des DPP4- Inhibitor Linagliptin konnten
bereits sowohl in diabetischen als auch in nicht- diabetischen Nephropathien im Tiermodell
aufgezeigt werden. Bereits Kanasaki et al. wies auf anti- inflammatorische als auch anti-
fibrotische Effekte von DDP4- Inhibitoren in chronischen Nierenerkrankungen hin und
postulierte eine zusätzliche Anwendungsmöglichkeit der DPP4- Inhibitoren bei chronischen
Nierenerkrankungen. Er vermutete antifibrotische Effekte durch eine Interaktion mit microRNA
und Integrinen (Kanasaki, 2018). Zudem verwies er auf antioxidative Eigenschaften von
Linagliptin zur Renoprotektion. Weitere Effekte wie beispielsweise die Unterbrechung des AGE
(advanced glycation endproducts)- RAGE (receptor for AGEs)- Signalwegs, erhöhte
Konzentrationen von Peptiden wie GLP-1, SDF-1α, die möglicherweise eine endotheliale
Dysfunktion verbessern und eine Inflammation reduzieren könnten, wurden bereits von ihm
als mögliche Erklärungen genannt (Kanasaki, 2018).
Aber ein definitiver Mechanismus, wie die DPP4 zu diesen positiven Effekten führen könnte,
wurde dabei noch nicht gefunden.
Einige präklinische Studien, die einen akuten renalen Ischämie- Reperfusionsschaden im
Tiermodel untersuchten, konnten bereits die nierenprotektiven Effekte der DPP4- Inhibitoren
bestätigen (Reichetzeder et al., 2017, Glorie et al., 2012, Chen et al., 2017). Dabei vermutete
Reichetzeder et al. eine Protektion gegen tubulären Schaden. Glorie et al. führte die protektive
Wirkung eines DPP4- Inhibitors auf antiapoptotische und antioxidative Effekte zurück.
Auch bei chronischen Nierenschäden konnte u.a. Hasan et al. und Tsuprykov et al. in einem
5/6- Nephrektomie- Tiermodell die Progression einer CKD durch eine DPP4- Inhibition
reduzieren (Hasan et al., 2019, Tsuprykov et al., 2016). In einer Arbeit über Gentamycin
verursachte Nephrotoxizität im Rattenmodell konnten renoprotektive Effekte durch Linagliptin
evaluiert werden (Helmy and Mouneir, 2019). Dabei bekamen Ratten Linagliptin oral für 14
Tage mit einer täglichen Dosis von 3 mg pro kg Körpergewicht. Während der Versuchszeit
konnten durch die Gentamycin- Schädigung erhöhte Plasma und renale DPP4-
Konzentrationen in den Tieren gemessen werden. Die DPP4- Inhibitorgabe verbesserte den
renalen Schaden und reduzierte oxidative, inflammatorische, apoptotische und
histopathologische Veränderungen in den Nieren (Helmy and Mouneir, 2019).
Jedoch wurde die Rolle der DPP4 in der Pathogenese einer Glomerulonephritis bisher wenig
untersucht. In einem Anti-Thy1-Glomerulonephritis Tiermodell wurden bereits anti-
inflammatorische Effekte durch die DPP4- Inhibitoren Alogliptin und Anagliptin durch eine
reduzierte Makrophageninfiltration angenommen, die aber laut Higashijima et al. direkt via
eines GLP-1- abhängigen Signalweges herbeigeführt wurden (Higashijima et al., 2015).
Um den Einfluss der DPP4 in der Immunkomplex- vermittelten Glomerulonephritis zu
untersuchen, induzierte Shinosaki et al. eine experimentelle Nephritis durch anti-Thy-1.1
monoklonale Antikörper in Wistar Ratten und F344 Ratten mit einem spontanen Mangel an
10DPP4 (Shinosaki et al., 2002). Mittels einer Gabe von DPP4- Antikörpern konnte er die
auftretende Proteinurie in den Wistar Ratten reduzieren, jedoch nicht in den defizienten F344
Ratten. Allerdings kann in diesem Modell nur ein bedingter Einfluss der DPP4 auf die
Albuminurie postuliert werden, da auch DPP4- defiziente Mäuse zunächst eine Proteinurie
entwickelten.
Diese bisherigen Erkenntnisse lassen vermuten, dass sich DPP4- Inhibitoren zusätzlich zu
ihrem aktuellen Nutzen in der Diabetestherapie zur Behandlung von chronischen
Nierenerkrankungen anbieten. Dabei zeigen die als Antidiabetika genutzten DPP4- Inhibitoren
unterschiedliche Eigenschaften u.a. in Bezug auf Dosierung, Halbwertszeit und
Bindungsspezifität. Wir entschieden uns in unserem Studienmodell für den DPP4- Inhibitor
Linagliptin, der einige Vorteile gegenüber anderen Substanzen besitzt.
Linagliptin ist ein Xanthin-basierter DPP4- Inhibitor (BI 1356) mit einer molekularen Masse von
472.5 Da und wurde von Boehringer Ingelheim Pharmaceuticals (Ingelheim, Deutschland)
entwickelt. Das Medikament hemmt die DPP4 kompetitiv und reversibel, wobei es eine
niedrige Dissoziationsrate vom aktiven Zentrum des Enzyms zeigt, sodass es nur einmal
täglich verabreicht werden muss. Es weist die niedrigste mittlere inhibitorische Konzentration
(IC50) von ca. 1 nM und somit die höchste Potenz innerhalb seiner Substanzklasse auf. Die
Selektivität für DPP4 im Vergleich zu anderen Subformen der DPP ist u.a. 40.000-mal höher
für DPP4 als für DPP8 (Thomas et al., 2008). Eine Studie von Holger Fuchs et al. konnte
zeigen, dass Linagliptin spezifisch an sein Zielenzym DPP4 bindet und dass die Stoffverteilung
im Gewebe zur Anzahl der Bindungen passt (Fuchs et al., 2009).
Ein weiterer, sehr wichtiger Vorteil ist der Unterschied im Metabolismus. Linagliptin wird über
den biliären Weg ausgeschieden, sodass keine Dosisanpassung bei einer Nierendysfunktion
notwendig ist (Fuchs et al., 2009). Dies ist gerade für unser Studienmodell von Bedeutung, da
in unserer Arbeit der Nutzen von DPP4- Inhibitoren in Nierenerkrankungen mit eingeschränkter
glomerulären Filtrationsrate evaluiert wird.
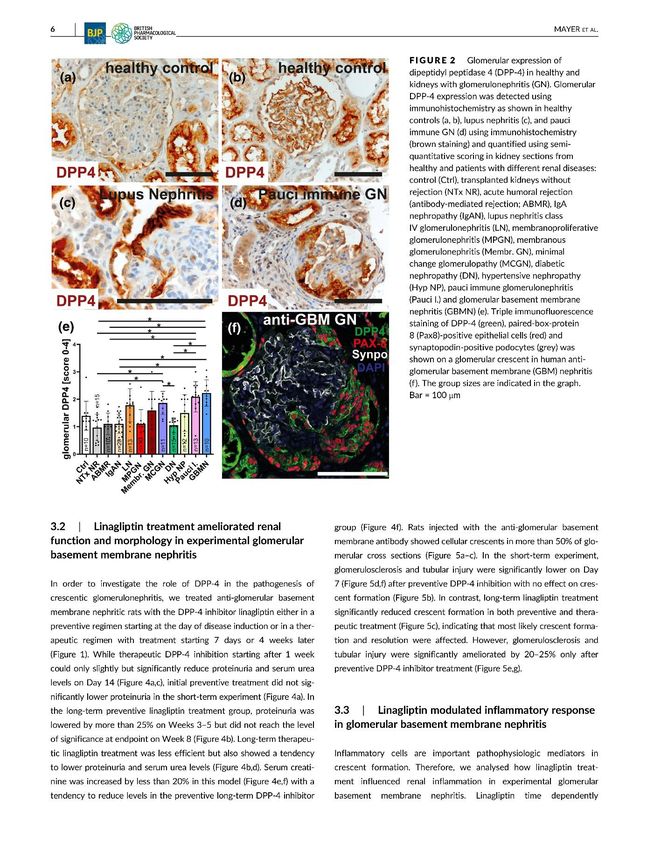
In unserer Pilotstudie wurden verschiedene renale Erkrankungen hinsichtlich ihrer
glomerulären DPP4- Expression untersucht. Dafür wurden archivierte Nierenbiopsien genutzt
und anhand von immunhistochemischen Färbungen das Ausmaß der DPP4- Expression
evaluiert. Diese war signifikant in humanen Biopsien von Pauci-Immune, Lupus-Nephritis,
MCGN (Minimal-Change-Glomerulonephritis) und anti-GBM-Nephritis Patienten im Vergleich
zu unauffälligen Transplantatnieren erhöht. Jedoch konnten keine erhöhten Werte in
Nierenbiopsien von Diabetes mellitus Patienten nachgewiesen werden. Am auffälligsten war
die DPP4 jedoch in den Halbmonden exprimiert.
Unser experimentelles Tiermodell wurde in einem Kurzzeit-Experiment über 14 Tage sowie in
einem Langzeit-Experiment über 8 Wochen mit jeweils 44 Ratten durchgeführt. Dafür wurden
jeweils diese 44 Tiere in 4 Gruppen aufgeteilt:
1111 Kontrolltiere, 11 an anti-GBM-GN erkrankte Tiere ohne Behandlung, 11 präventiv mit
Linagliptin behandelte Ratten vom Zeitpunkt der Krankheitsinduktion bis zum Endpunkt der
Studie (1. - 14. Tag bzw. 1. Tag bis Ende 8. Woche) sowie 11 therapeutisch behandelte Ratten
(7. - 14. Tag bzw. ab der 4. Woche bis Ende 8. Woche).
Als DPP4- Inhibitor wurde Linagliptin in einer Konzentration von 83 mg pro kg über das Futter
verabreicht, sodass eine Tagesdosis von ca. 5 mg Linagliptin pro kg Körpergewicht für die
Ratten erreicht wurde (Kroller-Schon et al., 2012). Das anti-GBM-Glomerulonephritis- Modell
wurde in 12 Wochen alten männlichen Wistar Kyoto Ratten induziert. Dieser spezielle
Rattenstamm zeigt eine hohe Empfänglichkeit für eine halbmondbildende Glomerulonephritis,
sodass sie sich sehr für unser Krankheitsmodell eignen (Smith et al., 2007).
Zur Krankheitsinduktion wurde jeweils den 33 Tieren 30 µg des monoklonalen anti-GBM-
Nephritis- Antikörpers systemisch in die Schwanzvene injiziert. 11 gesunde Kontrolltiere
erhielten PBS (phosphate-buffered saline/ buffer solution) als Placebo. Das direkte pathogene
Potential dieses Antikörpers wurde bereits 1967 von Lerner et al. aufgezeigt. Sie verabreichten
Primaten Antikörper, die sie aus Nieren von an anti-GBM-Nephritis Patienten isoliert hatten
(Lerner et al., 1999). Für die Untersuchungen zum Krankheitsverlauf führten wir
Überlebensbiopsien mit der Entnahme des unteren Nierenpols am 7. Tag (Kurzzeit-Regime)
bzw. nach 4 Wochen (Langzeit-Regime) durch. Um die Nierenfunktion anhand von
Urinanalysen zu untersuchen, wurden die Tiere über 24 Stunden in metabolischen Käfigen
gehalten und ihr Urin gesammelt. Zudem wurden Gewicht, Futter- und Trinkmenge registriert.
Weiterhin erfolgten regelmäßige Blutentnahmen an Tag 7 und 14 bzw. Woche 5 und 8, um
Serum- Kreatinin und Harnstoff als Surrogatmarker für die Nierenfunktion und um
Chemokinkonzentrationen im Blut bestimmen zu können.
Zunächst wurde das Kurzzeit-Experiment über 14 Tage durchgeführt, um unsere Hypothese
zur Rolle der DPP4 in der Pathogenese der halbmondbildenden anti-GBM-Glomerulonephritis
zu bestätigen. Eine erfolgreiche Induktion der anti-GBM-Glomerulonephritis konnte durch die
immunhistologische Auswertung der angefertigten PAS- Färbungen nachgewiesen werden. In
den an anti-GBM-Nephritis erkrankten Tieren zeigten über 50% der ausgewerteten Glomeruli
eine Halbmondbildung.
Anhand von in situ Aktivitätsmessungen, die an Kryoschnitten mit Gly-Pro 4-methoxy-β-
naphtylamid als Substrat der DPP4 durchgeführt wurden, konnte die DPP4- Aktivität in den
Nieren genau lokalisiert werden. In den pathologischen Halbmonden konnte eine signifikant
höhere Aktivität im Vergleich zu gesunden Glomeruli aufgezeigt werden. Wohingegen die
fehlende Anfärbung, d.h. nicht vorhandene Restaktivität in den mit Linagliptin behandelten
Tieren eine erfolgreiche und suffiziente Inhibierung des Enzyms DPP4 beweisen konnte.
Die erhöhte Aktivität zeigte sich vor allem in Arealen, in denen ein Glomerulus Kontakt zur
Bowman’schen Kapsel hatte. Dieser Kontakt ist als pathologisch zu werten und kann in diesem
12Zusammenhang als „tip lesion“ charakterisiert werden. Diese Bezeichnung wird vorwiegend
für eine histopathologische Läsion benutzt, die in Glomeruli von Patienten mit idiopathischem
nephrotischen Syndrom, v.a. bei MCGN und FSGS (fokal segmentale Glomerulosklerose) zu
finden ist (Stokes et al., 2004). Jedoch wird dieser Begriff mittlerweile ausgeweitet und nicht
mehr nur krankheitsspezifisch genutzt (Haas and Yousefzadeh, 2002). Diese Läsion stellt den
initialen Schaden dar, der sich weiter zu einem vollständigen krankhaften Halbmond im
Glomerulus entwickeln kann.
Zudem konnte die vollständige DPP4- Inhibition mittels Linagliptin durch Serum-
Aktivitätsmessungen bestätigt werden, die mit Hilfe eines fest im Labor etablierten
Aktivitätsassays durchgeführt wurden. Die Plasma DPP4- Aktivität war ursprünglich während
der anti-GBM-GN nicht erhöht. Dies bestärkt die Theorie, dass der Prozess der
Halbmondbildung lokal in der Niere reguliert wird. Wie bereits durch Fuchs et al. untersucht
(Fuchs et al., 2009) und nun auch durch unsere in situ Aktivitätsmessungen bestätigt, wird das
Enzym DPP4 überwiegend in der Niere exprimiert und zeigt dort eine besonders hohe
enzymatische Aktivität auf, die in entstehenden Halbmonden verglichen zu gesunden
Glomeruli nochmals stark erhöht ist. Dies lässt Rückschlüsse auf die Pathogenese der
Halbmondbildung ziehen.
Wie bereits in den humanen Biopsien von uns gezeigt, ist die glomeruläre DPP4- Expression
in der anti-GBM-GN erhöht, jedoch konnte in immunhistologischen Schnitten unserer
Rattennierenbiopsien keine Expressionsänderung durch eine DPP4- Inhibition vermerkt
werden. Eine erhöhte DPP4- Expression wurde auch bereits in einem Modell einer fettreichen
Diät mit Streptozotocingabe (Yang et al., 2007), in einem Mausmodell einer Diabetes mellitus
Typ 2 Nephropathie (Sharkovska et al., 2014) und in einem 5/6- Nephrektomie- Tiermodell
(Hasan et al., 2019) aufgezeigt. Auch Sakai et al. konnte in einer Studie mit salzsensitiven
hypertensiven Dahl- Ratten eine erhöhte DPP4- Aktivität in den geschädigten Nieren messen,
die durch die Gabe des DPP4- Inhibitors Saxagliptin signifikant gesenkt wurde. Zudem konnte
eine signifikante Reduktion der Albuminexkretion und eine leichte Verbesserung des
glomerulären Schadens durch Saxagliptin festgestellt werden (Sakai et al., 2015).
Da wir nicht nur die Beteiligung des Glykoproteins DPP4 in der Halbmondbildung, sondern
auch eine mögliche Therapieoption mit DPP4 als Target untersuchen wollten, entwickelten wir
zwei unterschiedliche Behandlungsregime. Eine sofortige, präventive Gabe des Inhibitors
Linagliptin könnte eine Bedeutung der DPP4 an der Entstehung der Halbmonde aufzeigen.
Für die Übertragbarkeit der positiven Wirkung einer DPP4- Inhibition auf Patienten und vor
allem für die Umsetzung als Therapeutikum müsste Linagliptin auch einen Effekt auf eine
bereits etablierte Erkrankung besitzen. Das bedeutet, dass der Inhibitor auch bei einer
therapeutischen Gabe während eines späteren Verlaufs der Krankheit eine positive Wirkung
zeigen müsste, um Linagliptin auch als Medikation bei einer bereits ausgebrochenen anti-
13GBM-Glomerulonephritis einsetzen zu können. So designten wir auch ein therapeutisches
Behandlungskonzept, das erst nach einer vollständig entwickelten anti-GBM-GN mit deutlicher
Halbmondbildung in das Krankheitsgeschehen eingreift. Die verzögerte Behandlung setzten
wir im Kurzzeit-Regime ab Tag 7, im Langzeit-Regime ab Beginn der 4. Woche ein. Anhand
des Langzeit-Experiments über 8 Wochen konnten die langfristigen Effekte einer DPP4-
Inhibition genauer evaluiert und unsere Ergebnisse aus dem Kurzzeit-Versuch bestätigt bzw.
weiterverfolgt und mögliche Mechanismen auf längere Dauer analysiert werden.
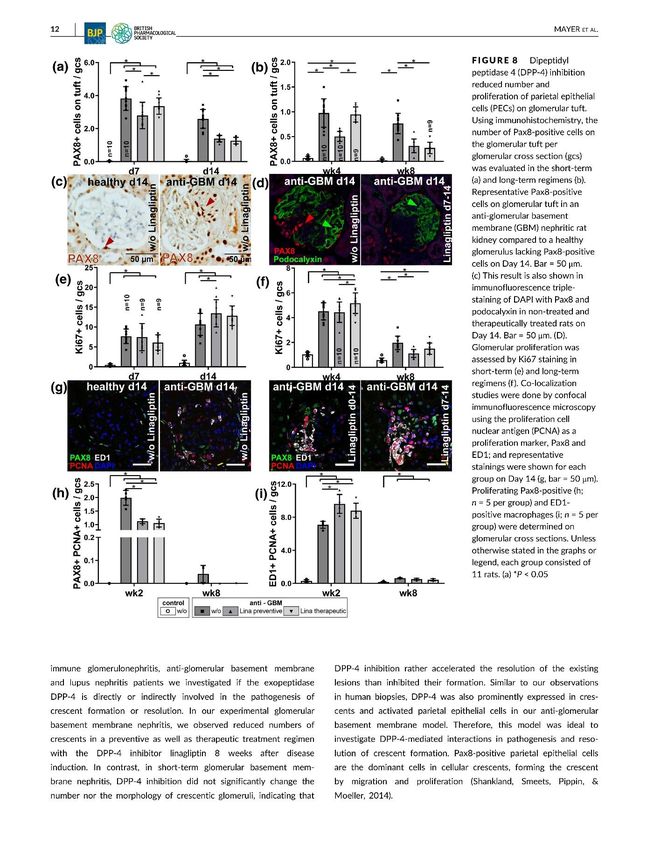
In unserem experimentellen anti-GBM-Nephritis- Modell konnte eine reduzierte Anzahl an
glomerulären Halbmonden sowohl im präventiven als auch therapeutischen
Behandlungsregime innerhalb von 8 Wochen nach Krankheitsinduktion verzeichnet werden.
Im Gegensatz dazu wurde die Anzahl der halbmondbildenden Glomeruli während des
Kurzzeit-Regimes über 14 Tage noch nicht signifikant verringert. Dies lässt annehmen, dass
eine DPP4- Inhibition eher die Auflösung von bestehenden Läsionen fördert, anstatt signifikant
die Entwicklung verhindert.
Der Pathomechanismus der glomerulären Halbmondbildung setzt sich aus drei Stadien
zusammen: Zunächst entwickelt sich ein zelluläres Konglomerat zwischen Bowman’scher
Kapsel und Tuft, im weiteren Verlauf nimmt die fibrozelluläre Komponente zu bis sich
schließlich ein fibröser, sklerosierter Halbmond bildet, bei dem die eingewanderten PECs mit
extrazellulärer Matrix umhüllt sind. In den Anfangsstadien können die glomerulären
Halbmonde wieder aufgelöst oder zumindest in ihrem Ausmaß reduziert werden. Jedoch ist
das fibrosierte Endstadium eines Glomerulus irreversibel (Nitta et al., 1999, Anguiano et al.,
2020). Diese Annahme konnten in unserer Arbeit bestätigt werden. Durch eine Linagliptin-
Therapie verringerte sich das Ausmaß des glomerulären Schadens signifikant um 21%, der
anhand eines GSI (glomerulärer Skleroseindex) ermittelt wurde und die Anzahl an
Halbmonden in den ausgewerteten Glomeruli reduzierte sich signifikant um 22%. Aber eine
komplette Auflösung von allen Halbmonden war nicht möglich, da bereits vollständig fibrosierte
Nierenkörperchen irreversibel verändert und geschädigt waren. Dies erklärt auch die
Ergebnisse bei der Nierenfunktion. Eine präventive Behandlung konnte nach 5 Wochen die
Proteinurie um über 25% reduzieren, erreichte aber zum Endzeitpunkt nach 8 Wochen kein
Signifikanzniveau mehr. Eine therapeutische Behandlung zeigte auch positive Effekte auf die
Proteinurie und die Ureakonzentration im Serum, die aber geringer ausfielen.
Durch eine Auflösung der Halbmonde kann die glomeruläre Filtrationsbarriere
wiederhergestellt werden. Dabei müssen die migrierten, pathologischen Zellen wieder den
glomerulären Tuft verlassen und dieser aus intakten Podozyten bestehen, die aus ihrem
verzweigten Aktin- Zytoskelett und mit ihren Fußfortsätzen die Filtermembran bilden (Reiser
and Sever, 2013). Ein Verlust dieser Fußfortsätze ist ansonsten eine Hauptursache für die
auftretende Proteinurie (Sicking et al., 2012). Für die Detektion von vitalen Podozyten wurden
14in unseren Immunhistologie- und Immunfluoreszenzfärbungen die Podozyten- Marker WT-1
(Wilms Tumor- 1) und Podocalyxin verwendet. Zudem bestätigten quantitative mRNA-
Expressionsanalysen für das Podozyten- Schlitzmembran- Molekül Nephrin und für Podocin
unsere Ergebnisse. Die Anzahl der vitalen Podozyten und mRNA- Expression waren in allen
anti-GBM-nephritischen Gruppen deutlich reduziert. Die längerfristige Linagliptin- Behandlung
konnte die Podozytenanzahl nur gering erhöhen. Jedoch konnte ein signifikant geringerer
Podozytenstress bei einer Reduktion von ca. 25% durch die präventive und um ca. 20% durch
die therapeutische Therapie anhand einer Immunhistologiefärbung für den Marker Desmin
registriert werden.
Eine besonders deutliche Verbesserung konnte bei der Evaluation von Pax8- positiven Zellen
auf dem glomerulären Tuft festgestellt werden. PECs und Podozyten besitzen einen
gemeinsamen Zellursprung bis zur S-förmigen Phase in der Glomerulogenese. Danach
beginnen sie spezifische Gene für ihre unterschiedlichen Zellfunktionen zu exprimieren.
Podozyten weisen den Transkriptionsfaktor WT-1 und das Aktin- Zytoskelett-bindende Protein
Synaptopodin auf. PECs exprimieren den Transkriptionsfaktor Pax8 und das Tightjunction-
Protein Claudin-1 (Narlis et al., 2007, Ohse et al., 2008). Die zellulären Halbmonde werden
durch Migration und Proliferation hauptsächlich aus Pax8- positiven PECs gebildet (Shankland
et al., 2014). Pax8- positive Zellen konnten lediglich auf den glomerulären Tufts von erkrankten
Tieren gefunden werden. Dagegen ließen sich keine Pax8- positiven Zellen auf dem Tuft von
gesunden Kontrolltieren nachweisen. Sowohl im Kurzzeit- als auch im Langzeit-Modell konnte
eine Behandlung mit Linagliptin in beiden Therapieoptionen die Zahl der Pax8- positiven Zellen
signifikant um ca. 50% an Tag 14 und um ca. 60% nach 8 Wochen reduzieren. Da Linagliptin
auch bei einer verspätet einsetzenden Behandlung erst 5 Wochen nach Krankheitsinduktion
die Anzahl der Pax8- positiven Zellen verringern konnte, gehen wir davon aus, dass eine
DPP4- Inhibition eher die Auflösung von bestehenden Läsionen fördert, anstatt die
Entwicklung verhindert.
Um den genaueren Mechanismus zu klären, wie Linagliptin die Veränderung in der
Differenzierung und Herunterregulation von Pax8- positiven Zellen sowie die konsekutive
verstärkte Auflösung von diesen Zellen auf dem glomerulären Tuft induzieren könnte, wurde
ein möglicher Mediator analysiert, der von der DPP4 reguliert wird. So untersuchten wir das
Chemokin SDF-1α, das von der Exopeptidase DPP4 gespalten und dadurch inaktiviert wird
(Lambeir et al., 2003). Zudem wird es von Podozyten sekretiert (Miglio et al., 2017) und ist
somit ein potentieller Mediator des DPP4- Effekts in der anti-GBM-Glomerulonephritis.
SDF-1α kann mit zwei verschiedenen Rezeptoren interagieren, CXCR4 und CXCR7 (Miglio et
al., 2017), die unter anderem eine Rolle bei der Mobilisierung und Chemotaxis von
Stammzellen sowie bei der Organogenese, der Angiogenese und der Wundheilung spielen
(Kawaguchi et al., 2019, Salcedo et al., 1999, Miglio et al., 2017). Auch bezüglich der Niere ist
15eine wichtige Rolle bei der Glomerulus- Entwicklung, der glomerulären Integrität und bei
regenerativen Prozessen bekannt (Mazzinghi et al., 2008, Stokman et al., 2010, Takabatake
et al., 2009). Beide Rezeptoren werden sowohl auf Podozyten als auch PECs exprimiert
(Romoli et al., 2018), sodass ein Mechanismus via SDF-1α möglich erscheint. Bereits frühere
Arbeiten postulierten, dass die SDF-1α /CXCR4- Achse bei der exzessiven epithelialen
Hyperplasie während der Halbmondbildung involviert ist (Lasagni and Romagnani, 2010,
Sicking et al., 2012).
Es konnten signifikant erhöhte SDF-1α Plasmalevel in allen anti-GBM-nephritischen Gruppen
verglichen zur gesunden Kontrollgruppe gemessen werden. Dies bestätigt die Ergebnisse
einer Studie von Balabanian et al., der eine besonders hohe Sekretion von SDF-1α in
geschädigten Podozyten messen konnte. Zudem wurde eine erhöhte Expression während
einer beginnenden Lupus-Nephritis registriert (Balabanian et al., 2003).
In unserer Arbeit zeigte bereits eine kurzzeitige DPP4- Inhibition und folglich gehemmte
Degradation von SDF-1α die Stabilisierung und einen signifikanten Anstieg von zirkulierendem
SDF-1α im Serum der nephritischen Ratten im Vergleich zu unbehandelten erkrankten Tieren,
sowohl bei der präventiven als auch therapeutischen Therapie. Auch die mRNA- Expression
von glomerulärem SDF-1α war in all unseren erkrankten Gruppen am Tag 14 im Vergleich zu
gesunden Kontrollen erhöht, wobei die präventive Behandlung keinen signifikanten
Unterschied zu Kontrolltieren an Tag 14 mehr zeigte. Immunfluoreszenz- Doppelfärbungen für
SDF-1α und den Podozyten- Marker Synaptopodin konnten die SDF-1α- Expression in
Podozyten bestätigen und einen Verlust an SDF-1α- Expression in Halbmonden, besonders
in sklerotischen Läsionen, korrespondierend zum Podozytenverlust aufzeigen. Die erhöhte
mRNA- Expression von SDF-1α in allen erkrankten Gruppen an Tag 14 kann als
Reparaturmechanismus, als Versuch der Kompensation des Schadens gesehen werden. Die
fehlende weitere Erhöhung in den behandelten Gruppen lässt sich durch die reduzierte
Degradation und die konsekutiv weitere Steigerung an freiem SDF-1α ohne notwendige
Steigerung der Expression erklären. Dies könnte auch ein Hinweis auf einen negativen
Feedback- Mechanismus sein. In Woche 8 zeigte die unbehandelte, erkrankte Gruppe noch
eine nicht-signifikante erhöhte SDF-1α- Expression, wohingegen sich die behandelten
Gruppen den Normwerten der gesunden Gruppe annäherten. Die Reduktion in den
unbehandelten, nephritischen Tieren lässt sich durch die reduzierte Anzahl und bereits
abgestorbenen Podozyten und somit geringeren SDF-1α- Sekretion erklären.
Während SDF-1α bereits protektive Eigenschaften in diabetischen Akita Mäusen zeigen
konnte (Takashima et al., 2016), wurde es schädlich in einem Diabetes mellitus Typ 2 Modell
und in einer Adriamycin- induzierten Nephritis angesehen (Romoli et al., 2018, Sayyed et al.,
2009). Dort wurde angenommen, dass eine Blockade von SDF-1α die Nierenfunktion
verbessert und die Regeneration von Podozyten durch Progenitorzellen erhöht. Wenn jedoch
16eine SDF-1α- Blockade die Nierenfunktion verbessern und die Podozytenreparatur verstärken
sollte, müssten hohe SDF-1α Werte einen Nierenschaden verschlechtern. Jedoch wurde in
unserem Modell keine progrediente Podozytenreduktion als Ergebnis einer SDF-1α-
Stabilisierung durch DPP4 beobachtet.
Auch im diabetischen Nephropathie- Modell von Takashima et al. verstärkte eine SDF-1α-
Inhibition eine Expansion des Mesangiums in Verbindung mit einem höheren glomerulären
Schaden und reduzierten Podozytenzahlen (Takashima et al., 2016).
Es ist bereits bekannt, dass SDF-1α Podozyten vor einer mitotischen Katastrophe bewahrt.
Die mitotische Katastrophe bezeichnet einen Vorgang, der bei einer glomerulären Schädigung
auftreten kann. Verletzte Podozyten treten häufig in den Zellzyklus ein und hypertrophieren
währenddessen. Aber dabei kommt es zum Versuch der Mitose, bei der das Aktin-Zytoskelett
mitotische Spindeln auszubilden versucht, wobei die podozytären Fußfortsätze nicht mehr
intakt gehalten werden, die Podozyten ihre Funktion verlieren und schließlich absterben
(Lasagni et al., 2013). In einigen Studien wurde SDF-1α als Teil eines intrinischen
Podozytenprogenitor- Feedback- Mechanismus beschrieben: Podozyten produzieren SDF-1α,
um die Podozyten- Progenitorzellen im Ruhezustand verweilen zu lassen (Romoli et al., 2018,
Sayyed et al., 2009). Bei einer Schädigung von Podozyten wird zunächst SDF-1α von den
noch verbleibenden intakten Podozyten vermehrt in die Bowman’sche Kapsel sekretiert.
Dadurch wird eine Aktivierung von Podozyten- Progenitorzellen und die Einleitung einer
mitotischen Katastrophe verhindert. Dabei wird auch die Mobilisation und Proliferation von
PECs auf den glomerulären Tuft gehemmt. Kommt es im längerfristigen Verlauf jedoch
trotzdem zu einem hohen Verlust an Podozyten, nimmt folglich auch die lokale SDF-1α-
Konzentration ab. Somit werden die Podozyten- Progenitorzellen aus der Bowman’schen
Kapsel aktiviert, doch auch die PECs- Proliferation steigt (Romoli et al., 2018, Sayyed et al.,
2009). Durch diesen Feedback- Mechanismus lassen sich auch unsere Ergebnisse bezüglich
der Zellarten auf dem glomerulären Tuft bzw. in den Halbmonden erklären. Die hohe SDF-1α-
Konzentration in den Nieren durch die DPP4- Inhibition wirkt als Podozyten- Überlebensfaktor,
indem es die Podozyten vor einer mitotischen Katastrophe bewahrt. Jedoch hemmt sie auch
die Heranreifung von Podozyten- Progenitorzellen, sodass man in unserem Modell keine
enorme Steigerung der Podozytenanzahl durch die Linagliptin- Gabe erwarten kann. Dafür
wird aber die Anzahl von PECs in den Glomeruli reduziert und die Auflösung von Halbmonden
gefördert. Dabei wäre eine durch SDF-1α induzierte Inhibition des Notch- Signalings eine
Methode, um die mitotische Katastrophe zu unterbinden. Das Notch- Signaling kann
normalerweise eine mitotische Katastrophe einleiten, da es ein wichtiger Regulator für
Podozyten- Progenitorzellen ist (Lasagni et al., 2010). Ein Adriamycin- Nephritis- Modell
zeigte, dass die Aktivierung des Notch- Signalwegs in Podozyten als Trigger für Zelltod wirkte,
zu einer gesteigerten PECs- Proliferation und zur Differenzierung zu Podozyten führte
17(Lasagni et al., 2010). Eine SDF-1α- Inhibition konnte Notch- bezogene Gene in Adriamycin-
stimulierten RPCs (renale Progenitorzellen) hochregulieren, zeigte jedoch keine
Veränderungen in differenzierten Podozyten. Dagegen kann intrinsisches SDF-1α das Notch-
Signaling supprimieren. Dabei wird bereits angenommen, dass sterbende Podozyten SDF-1α
abgeben und dadurch die Aktivität von RPCs und PECs reduzieren (Romoli et al., 2018).
Es wurde ein signifikanter Anstieg von Notch2 an Tag 14 erreicht, der aber nicht durch eine
Linagliptin- Therapie verändert wurde. In Woche 8 fiel die mRNA- Expression von Notch2 in
allen erkrankten Gruppen, ob behandelt oder unbehandelt signifikant unter die Werte der
gesunden Kontrollgruppe. Folglich lassen sich unsere Ergebnisse nicht eindeutig durch Effekte
mittels SDF-1α und Notch erklären.
Die Ergebnisse in der Studie von Romoli et al. (Romoli et al., 2018) und Sayyed et al. (Sayyed
et al., 2009) könnten sich durch den bereits beschriebenen Feedback- Mechanismus erklären
lassen: Die SDF-1α Blockade führt zunächst im Zeitraum der Schädigung zu einem Verlust an
Podozyten durch erhöhte mitotische Katastrophe, langfristig kann sie in der
Regenerationsphase einer glomerulären Erkrankung die Podozyten- Regeneration fördern.
Wir greifen mit unserer DPP4- Inhibition zum Zeitpunkt der Schädigung und Progression der
Erkrankung ein. Durch erhöhte SDF-1α- Werte wird ein weiterer Verlust an Podozyten durch
Blockade einer mitotischen Katastrophe verhindert und zusätzlich wird die Einwanderung von
Pax8- positiven PECs gestoppt, das Fortschreiten einer Fibrose gehemmt und folglich wird der
Funktionsverlust des Glomerulus reduziert. Auch begründen sich so unsere Ergebnisse in
Bezug auf die Podozyten- Anzahl. Durch höhere SDF-1α- Werte wird zwar die Podozyten-
Proliferation nicht gefördert, aber der Glomerulus wird vor der Proliferation von PECs bewahrt,
die ansonsten auf den glomerulären Tuft einwandern und Halbmonde bilden würden. Auf einen
bisher unbekannten Weg wird nicht nur die weitere Einwanderung von PECs verhindert,
sondern es wird auch die Auflösung von ihnen auf dem glomerulären Tuft gefördert, sodass
noch reparable Glomeruli vor einer fortschreitenden Halbmondbildung und Sklerose geschützt
werden. Nur können, wie bereits erläutert, bereits vollständig sklerosierte und destruierte
Glomeruli nicht mehr gerettet werden. Die SDF-1α- Expression fördert die Quieszenz von
lokalen Podozyten- Progenitorzellen. Dieser Mechanismus unterstützt die Homeostase,
limitiert aber auch die Kapazität an Regeneration im Fall des Verlusts von Podozyten.
Jedoch sehen wir als wichtigen positiven Faktor von SDF-1α, dass es die Migration von PECs
auf den glomerulären Tuft hemmt und in der frühen Phase einer Glomerulonephritis
möglicherweise durch eine Notch-Blockade weitere Podozyten vor einer mitotischen
Katastrophe bewahrt.
Auch eine Studie, die immortalisierte humane Podozyten und mesangiale Zellen nutzte, um
Effekte des DPP4- Inhibitors Linagliptin in der Niere zu analysieren, konnte korrespondierende
Ergebnisse verzeichnen (Miglio et al., 2017). Eine DPP4- Inhibition verlangsamte den
18Sie können auch lesen

















































