Auswirkungen der astrozytären mitochondrialen Dysfunktion auf die Bildung reaktiver Astrozyten und den neuronalen Zelltod nach photothrombotischen ...
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Auswirkungen der astrozytären mitochondrialen Dysfunktion auf die
Bildung reaktiver Astrozyten und den neuronalen Zelltod nach
photothrombotischen Läsionen
Der Medizinischen Fakultät
der Friedrich-Alexander-Universität
Erlangen-Nürnberg
zur
Erlangung des Doktorgrades Dr. med.
vorgelegt von
Christian Matthias Fiebig
aus Biberach an der Riß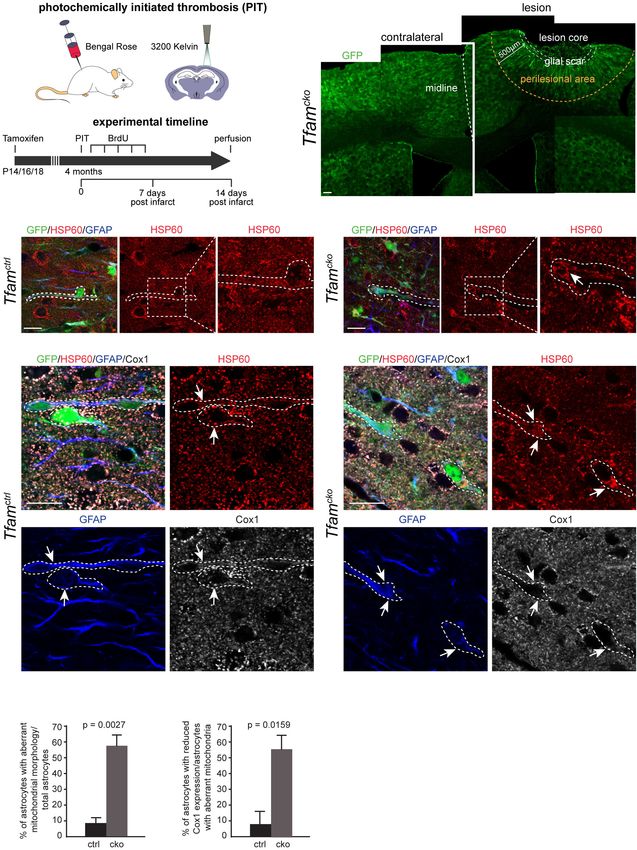
Als Dissertation genehmigt von der Medizinischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg Tag der mündlichen Prüfung: Vorsitzender des Promotionsorgans: Prof. Dr. med. Markus F. Neurath Gutachter: Prof. Dr. Dieter Chichung Lie Gutachter: Prof. Dr. Jürgen Winkler Gutachterin: Dr. Ruth Beckervordersandforth-Bonk Gutachterin: Prof. Dr. Leda Dimou Tag der mündlichen Prüfung: 11. Januar 2022
Inhaltsverzeichnis Zusammenfassung ............................................................................................ 1 Hintergrund ..................................................................................................... 1 Methoden ........................................................................................................ 1 Ergebnisse ...................................................................................................... 1 Schlussfolgerungen ....................................................................................... 2 Einleitung und Einordnung in den fachwissenschaftlichen Kontext ............ 3 Rolle und Funktion der Astrozyten im Zentralen Nervensystem .............. 3 Photochemisch-induzierte Thrombose als Modell für Schlaganfälle ....... 4 Tfam-Funktionsverlust als Modell für mitochondriale Dysfunktion .......... 7 Einordnung der Ergebnisse in den fachwissenschaftlichen Kontext ....... 9 Literaturverzeichnis ........................................................................................ 15 Originalpublikation .......................................................................................... 21 Verzeichnis der Veröffentlichungen............................................................... 34 Danksagung ..................................................................................................... 35
Zusammenfassung
Hintergrund
Mitochondrien sind Schlüsselorganellen zur Regulation des metabolischen
Haushaltes einer Zelle. Dabei ist die in den Mitochondrien stattfindende
oxidative Phosphorylierung im Gehirn der vorherrschende Mechanismus für
Neurone, um Adenosintriphosphat (ATP) zu generieren. Als gesichert gilt die
Annahme, dass die neuronale Funktion stark abhängig vom mitochondrialen
Metabolismus ist. Weitestgehend ungeklärt ist jedoch, inwiefern Astrozyten auf
die Funktion von Mitochondrien angewiesen sind.
Methoden
In der vorliegenden Publikation mit dem Titel „Astrozytäre mitochondriale
Dysfunktion beeinträchtigt die Bildung reaktiver Astrozyten und verstärkt den
neuronalen Zelltod im Kortex nach photothrombotischen Läsionen“ wurde die
Notwendigkeit einer funktionierenden Elektronentransportkette und oxidativen
Phophorylierung in Bezug auf die Funktion von Astrozyten unter
physiologischen und Verletzungsbedingungen untersucht. Hierfür dienten
immunhistochemische Färbungen, um das Vorhandensein einer
Elektronentransportkette und den an der oxidativen Phosphorylierung
beteiligten Komplexen „in vivo“ zu zeigen. Die genetische Unterdrückung der
mitochondrialen Transkription durch konditionelles Ausschalten des
mitochondrialen Transkriptionsfaktors A (Tfam) führte zu einer Dysfunktion der
Atmungskette und der oxidativen Phosphorylierung, welche sich morphologisch
in einer Schwellung der astrozytären Mitochondrien äußerte. Zur Untersuchung
des Einflusses mitochondrialer Dysfunktion unter Verletzungsbedingungen
diente die photochemisch-induzierte Photothrombose als Modell für
ischämische Hirninfarkte.
Ergebnisse
Unter physiologischen Bedingungen beeinträchtigt die astrozytäre
mitochondriale Dysfunktion nicht das Überleben von Astrozyten selbst,
verursacht jedoch eine reaktive Astrogliose im Kortex. Nach Induktion eines
ischämischen Schlaganfalls durch photochemische Thrombose ließen sich
1stark verzweigte mitochondriale Netzwerke in reaktiven Astrozyten periläsional
beobachten. Zudem verringerten dysfunktionelle Mitochondrien signifikant die
Anzahl an neu gebildeten Astrozyten und erhöhten den neuronaler Zelltod in
dieser Region.
Schlussfolgerungen
Die vorliegenden Ergebnisse bekräftigen die Notwendigkeit einer
funktionierenden Elektronentransportkette und oxidativen Phophorylierung in
Astrozyten zur Aufrechterhaltung der astrozytären Proliferation sowie der
neuroprotektiven Funktion unter Verletzungsbedingungen.
2Einleitung und Einordnung in den fachwissenschaftlichen
Kontext
Rolle und Funktion der Astrozyten im Zentralen Nervensystem
Astrozyten zählen zu den Gliazellen und nehmen mit etwa einem Drittel einen
großen Teil der Zellpopulation im zentralen Nervensystem (ZNS) ein (Sofroniew
and Vinters, 2010; Liddelow and Barres, 2017). Dort spielen sie eine wichtige
Rolle in der Aufrechterhaltung der Homöostase, beispielsweise in der
Regulation des Glutamat-, Ionen- und Wasserhaushaltes. Zu den
Kernaufgaben gehören zudem Bildung und Modulation von Synapsen,
Reparatur von Gewebeschäden, Energiespeicherung und Abwehr von
oxidativem Stress (Volterra and Meldolesi, 2005; Belanger and Magistretti,
2009). Erreichen können Astrozyten dies aufgrund ihrer speziellen Lage im
ZNS. Durch Verknüpfen der feinen Endverzweigungen mit den neuronalen
Synapsen einerseits und der Umhüllung der Mikrovaskulatur mit den Endfüßen
andererseits bilden sie eine Brücke zwischen Gefäßen des Gehirns und den
Neuronen (Iadecola and Nedergaard, 2007; Oberheim et al., 2009). Sie sind
damit an der Regulation des Gefäßtonus und der Aufrechterhaltung der Blut-
Hirn-Schranke beteiligt (Attwell et al., 2010). Mithilfe dieser speziellen
morphologischen Eigenschaften und ihrer räumlichen Verbindung zu den
umliegenden Zellen sind Astrozyten mittels Zytokinen, Wachstumsfaktoren,
Transportern und Ionenkanälen in der Lage, neuronale Aktivitäten an der
Synapse wahrzunehmen. Über ihre Endfüße an den Blutgefäßen können sie
zudem mithilfe von Glucose und Aquaporin 4 Transportern entsprechend auf
den Stoffwechsel reagieren (Attwell et al., 2010; Zhao et al., 2015).
Es gilt als allgemein gesichert, dass Neurone zur Aufrechterhaltung des
eigenen ATP-Haushaltes auf die mitochondriale oxidative Phosphorylierung
angewiesen sind. Astrozyten hingegen sichern sich den eigenen
Energiehaushalt über Glykolyse (Belanger et al., 2011). Interessanterweise
besitzen Astrozyten jedoch ähnlich viele Mitochondrien wie Neurone (Lovatt et
al., 2007). Durch Transkriptomanalysen verdichtete sich der Hinweis, dass
3Astrozyten zu einem oxidativen Metabolismus fähig sind. Dennoch ist bis heute
unklar, ob und in welchem Ausmaß Astrozyten in vivo von der oxidativen
Phosphorylierung abhängig sind (Lovatt et al., 2007; Cahoy et al., 2008).
Photochemisch-induzierte Thrombose als Modell für Schlaganfälle
Schlaganfälle zählen zu den weltweit häufigsten Todesursachen (Krishnamurthi
et al., 2013). Schwerwiegende Folgeerkrankungen wie Lähmungen führen
oftmals in die Pflegebedürftigkeit (Murray et al., 2012). Mehrere
Veröffentlichungen konnten zwar eine altersstandardisierte Abnahme der
weltweiten Schlaganfallmortalität in den letzten zwei Jahrzehnten zeigen,
gleichzeitig stieg jedoch im gleichen Zeitraum die Anzahl an Menschen, welche
einen Schlaganfall erlitten, signifikant an (Krishnamurthi et al., 2013; Feigin et
al., 2014). Der ischämische Hirninfarkt machte dabei mit ca. 80% den Großteil
der Schlaganfälle aus (Bejot et al., 2016). Damit steigt die weltweite Anzahl an
Menschen, welche nach einem Schlaganfall mit körperlichen Einschränkungen
zu kämpfen haben. Die tiefergehende Erforschung der Pathophysiologie des
ischämischen Schlaganfalles könnte daher in Zukunft neue
Behandlungsstrategien aufzeigen, um die Folgen der Erkrankung abzumildern
(Bejot et al., 2016).
Zur systematischen Untersuchung der pathophysiologischen Vorgänge
während und nach einem Schlaganfall bedienten wir uns eines etablierten
Mausmodells. Die sogenannte photochemisch-induzierte Thrombose (PIT)
bietet eine Möglichkeit, im Maushirn einen ischämischen Schlaganfall gezielt zu
generieren. Das Prinzip besteht dabei in der intraperitonealen Injektion von
Bengalrosa mit anschließender Beleuchtung der freigelegten Schädeldecke
mittels einer Kaltlichtquelle (3200 Kelvin). Dies erzeugt eine photochemische
Reaktion der zuvor injizierten Bengalrosa-Lösung im Bereich der bestrahlten
Hirnregion mit Bildung von Radikalen. Diese wiederum sind verantwortlich für
eine Schädigung des Gefäßendothels mit anschließender
4Thrombozytenaggregation, welche letztlich zur Bildung von Gefäßthromben im
beleuchteten Bereich führt (Keiner et al., 2008; Watson et al., 1985).
Die pathophysiologische Reaktion des Hirngewebes auf einen solchen
ischämischen Infarkt lässt sich in drei verschiedene Phasen einteilen (zitiert
nach Burda and Sofroniew, 2014):
Die erste Phase beschreibt die ersten Sekunden bis Stunden nach Stattfinden
des Schlaganfalls. Charakteristisch sind dabei die durch die toten
beziehungsweise apoptotischen Zellen freigesetzten Signalmoleküle, wie
beispielsweise Neurotransmitter (Glutamat/Stickstoffmonoxid), Zytokine,
Chemokine, danger-associated molecular patterns (DAMPs), Kalium,
Hitzeschockproteine, Desoxyribonukleinsäure (DNA) und Ribonukleinsäure
(RNA). Einige dieser Moleküle, wie beispielsweise Glutamat, sind in höheren
Konzentrationen toxisch für Neuronen. Andere, wie Kalium und
Hitzeschockproteine (HSP), wirken proinflammatorisch und versetzen
Astrozyten in einen reaktiven Zustand, die sogenannte reaktive Astrogliose
(Hamby et al., 2012; Zamanian et al., 2012). Durch mechanische Schädigung
der Blutgefäßendothelien sowie durch die Inflammation selbst verliert die
Bluthirnschranke (BBB) teilweise ihre Funktion und kann große und polare
Moleküle nicht mehr im Blutkreislauf halten. Es kommt folglich zur
Einwanderung von Albumin, Fibrin und Leukozyten in das Hirngewebe (Argaw
et al., 2009; Argaw et al., 2012; Seo et al., 2013). Makrophagen phagozytieren
toten Zellabfall im Hirngewebe (Mosser and Edwards, 2008), während aktivierte
T-Zellen unterschiedliche Reaktionen hervorrufen. Diese reichen von der
Induktion des Zelltodes bis hin zu protektiven Effekten bei der Reparatur des
Gewebes (Maciver et al., 2013; Mills, 2011; Walsh and Kipnis, 2011).
Die zweite Phase, welche sich etwa für ein bis zwei Wochen an die vorherige
anschließt, ist gekennzeichnet durch Proliferation und Migration von Zellen mit
dem Ziel, die Inflammation zu limitieren und den Gewebsschaden zu
begrenzen. Molekülsignale wie Thrombin, Endothelin und ATP aus
Serumproteinen und lokalen Zellen stimulieren die reaktiven Astrozyten zur
Proliferation (Gadea et al., 2008; Neary et al., 2003; Shirakawa et al., 2010;
Sirko et al., 2013). Die anschließend neu gebildeten und proliferierenden
Astrozyten migrieren in Richtung der Läsion und formen eine geschlossene
5Glianarbe bestehend aus dicht gedrängten Astrozyten. Diese neu gebildete
Barriere trennt das tote Zellmaterial aus dem Zentrum der Schlaganfallläsion
von dem peripheren und potentiell vitalen Hirngewebe (Faulkner et al., 2004;
Wanner et al., 2013).
Die dritte Phase beginnt ca. zwei Wochen nach dem Schlaganfallereignis und
beschreibt die Neuorganisation des Hirngewebes in den darauffolgenden
Monaten. Die volle Ausbildung der Glianarbe und die Reparatur der Blut-Hirn-
Schranke sind in diesem Zeitraum die vorherrschenden Mechanismen. In dieser
Phase lassen sich drei Regionen (zitiert nach Burda and Sofroniew, 2014)
unterscheiden:
1. Infarktkern: Besteht hauptsächlich aus Fibroblasten, Endothelzellen,
Entzündungszellen sowie unwiederbringlich abgestorbenen neuronalen
Zellen. Im weiteren Verlauf bildet sich der Bereich in eine Zyste um
(Tuszynski and Steward, 2012).
2. Glianarbe: Eine von neu proliferierenden Astrozyten gebildete Barriere
zwischen dem Infarktkern und der periläsionalen Region. Sie bildet eine
funktionelle und strukturelle Grenze zwischen dem toten, nicht
neuronalen Gewebe und dem vitalen peripheren Gewebe und wirkt
damit neuroprotektiv (Wanner et al., 2013; Sofroniew and Vinters, 2010).
Die Dichte der Astrozyten ist in dieser Region etwa doppelt so hoch
verglichen mit normalem Hirngewebe (Wanner et al., 2013).
3. Periläsionale Region: Enthält das an die Glianarbe angrenzende vitale
Hirngewebe. Die Stärke der reaktiven Astrogliose ist am
ausgeprägtesten in der Nähe des Infarktkerns und nimmt zur Peripherie
hin ab (Wanner et al., 2013). In dieser Region sind im Gegensatz zum
Infarktkern noch lebende Neurone vorhanden (Bush et al., 1999; Wanner
et al., 2013).
In der Vergangenheit konnte gezeigt werden, dass eine rehabilitative Therapie
bei Mäusen nach ischämischen Infarkten im Bereich der periläsionalen Region
das Überleben neu gebildeter Astrozyten steigerte und für eine verbesserte
funktionelle Genesung sorgte (Keiner et al., 2008). Diese Region erscheint
damit potentiell als geeignet für zukünftige Therapieansätze. Ebenfalls konnte
in aktuellen Veröffentlichungen gezeigt werden, dass die transgene Störung der
6Glianarbenbildung zu einer Vergrößerung der Schlaganfallläsion und
verstärktem neuronalen Zelltod führt (Bush et al., 1999; Faulkner et al., 2004;
Herrmann et al., 2008; Li et al., 2008; Wanner et al., 2013). Übereinstimmend
berichten alle Publikationen über eine reaktive Astrogliose in der periläsionalen
Region. Die genaue Rolle der Astrogliose ist bis heute nicht geklärt. Eine
kürzlich veröffentliche Studie zeigte jedoch, dass reaktive Astrozyten ihre für
Neuronen unterstützende Rolle verlieren und eher schädlich in Bezug auf den
Verlauf einer neurodegenerativen Erkrankung zu sein scheinen (Liddelow and
Barres, 2017). Über die zellulären und molekularen Mechanismen, die Einfluss
auf diese Prozesse haben, ist bisher wenig bekannt. In der vorliegenden Arbeit
liegt der Fokus daher auf Erforschung der Rolle astrozytärer Mitochondrien
innerhalb der periläsionalen Region mit dem Ziel, ein tiefgreifendes Verständnis
für deren Funktion im Rahmen der Neuroprotektion nach einem Schlaganfall zu
erlangen.
Tfam-Funktionsverlust als Modell für mitochondriale Dysfunktion
Eine fundamentale Aufgabe der Mitochondrien besteht in der Bereitstellung von
Energie in Form von ATP. Dies geschieht durch Aufbau eines
Protonengradienten im intermembranösen Raum entlang der vier Komplexe der
Atmungskette. Die dazu benötigten Reduktionsäquivalente werden über den
Zitratzyklus zu Verfügung gestellt. Im letzten Schritt, der oxidativen
Phosphorylierung, wird dieser Protonengradient von dem letzten Komplex V
(FoF1-ATP-Synthase) genutzt, um Adenosindiphosphat (ADP) zu ATP zu
phosphorylieren (Vernochet et al., 2012). Die vier Komplexe der Atmungskette
und die ATP-Synthase setzen sich insgesamt aus über 90 Proteinen
zusammen, die letztlich den Prozess der oxidativen Phosphorylierung
bestimmen (Gaspari et al., 2004). Die mitochondriale Desoxyribonukleinsäure
(mtDNA) kodiert dabei 13 dieser 90 Proteine (welche Untergruppen der
Komplexe I, III, IV und V bilden); die übrigen sind nukleär kodiert (Larsson et al.,
1998).
7Im Rahmen der Transkription kann die mitochondriale RNA Polymerase nicht
alleine an den Promotor der mtDNA binden, sondern ist auf die gleichzeitige
Bindung mit Tfam und einen der beiden Transkriptionsfaktoren B1 (TFB1M)
oder B2 (TFB2M) angewiesen. Der Humane Transkriptionsfaktor A (Tfam)
gehört zur Gruppe der High-Mobility-Group (HMG) Proteine. Er ist in der Lage,
an die Promotorregion zu binden, diese zu entwinden und damit die
Transkription zu initiieren (Gaspari et al., 2004).
Ohne Tfam ist eine Expression der mitochondrial kodierten Gene nicht möglich
(Gaspari et al., 2004). Die damit einhergehende, nur unvollständige Expression
der Proteine der Atmungskette führt beim Mausmodell im homozygoten
Funktionsverlust zum Erliegen der oxidativen Phosphorylierung und zum
embryonalen Tod der Maus (Larsson et al., 1998). Ein gezielter
Funktionsverlust von Tfam in verschiedenen Geweben konnte bereits
morphologische Veränderungen als Hinweis für dysfunktionale Mitochondrien
nachweisen, beispielweise in Skelettmuskelzellen (Sorensen et al., 2001),
Fettzellen (Vernochet et al., 2012) und in Neuronen (Beckervordersandforth et
al., 2017). Unklar ist allerdings bisher der Einfluss dysfunktionaler
Mitochondrien auf die Astrozyten selbst, sowie auf deren umliegenden Zellen.
Ziel der vorliegenden Arbeit war daher die Analyse dysfunktionaler
Mitochondrien, welche ausschließlich in Astrozyten lokalisiert sind. Dazu
wurden TfamloxP/loxP Mäuse (Larsson et al., 1998) mit der Astrozyten-
spezifischen Cre-Linie GLAST::CreERT2 (Mori et al., 2006) und CAG-CAT-
EGFP Reporter Mäusen (Nakamura et al., 2006) gekreuzt. Der L-Glutamat/L-
Aspartat Transporter (GLAST) wird spezifisch in Astrozyten und radialen
Gliazellen des erwachsenen Gehirns exprimiert und eignet sich daher für den
konditionellen Funktionsverlust in kortikalen Astrozyten (Chaudhry et al., 1995;
Lehre et al., 1995; Schmitt et al., 1997; Torp et al., 1994; Berger and Hediger,
1998; Robel et al., 2011). Der konditionelle Funktionsverlust von Tfam in den
Astrozyten (Tfamcko) wird durch Gabe von Tamoxifen induziert, das in den
Tagen 14, 16 und 18 nach der Geburt intraperitonial verabreicht wurde
(Ekstrand et al., 2007; Beckervordersandforth et al., 2017). Als Kontrolle
dienten GLAST:CreERT2; CAG-CAT-EGFP Mäuse, welche die wildtypischen
Allele für Tfam tragen (Tfamctrl).
8Einordnung der Ergebnisse in den fachwissenschaftlichen Kontext
Eine kürzlich veröffentlichte Publikation konnte zeigen, dass unter
physiologischen Bedingungen der konditionelle Funktionsverlust eines
einzelnen Komplexes der Elektronentransportkette (Cytochrom C Oxidase
(COX)) in Astrozyten keinen Einfluss auf das Langzeitüberleben der Astrozyten
selbst hat und auch sonst keine Pathologie im Gehirn hervorruft (Supplie et al.,
2017). Mithilfe einer GLASTCreERT2::Cox10flox/flox Mauslinie wurde konditionell die
Expression der Farnesyltransferase Cox10 in GLAST exprimierenden
Astrozyten inhibiert. Ohne Cox10 kann eine funktionelle Cytochrom C Oxidase
(COX) nicht exprimiert werden (Funfschilling et al., 2012; Fukui et al., 2007).
Daraus folgerten die Autoren, dass Astrozyten physiologisch nicht auf eine
oxidative Phosphorylierung angewiesen sind (Supplie et al., 2017).
Unbeantwortet bleibt dabei jedoch die Frage, ob Astrozyten nicht unter
pathologische Stressbedingungen, wie beispielsweise im Rahmen eines
Traumas oder Schlaganfalles, auf die Atmungskette als Energielieferant
angewiesen sind.
Um die Auswirkung mitochondrialer Dysfunktion in Astrozyten unter
pathologischen Bedingungen weiter zu untersuchen, wurden folgende
Fragestellungen adressiert:
- Exprimieren kortikale Astrozyten auf Proteinebene die Komponenten der
Atmungskette zur Durchführung der Oxidativen Phosphorylierung?
- Welchen Einfluss hat der Tfam-Funktionsverlust auf Astrozyten und
Mitochondrien unter sowohl physiologischen als auch pathologischen
Bedingungen im Schlaganfallmodell?
- Hat die mitochondriale Dysfunktion einen Einfluss auf die
Proliferationsfähigkeit der Astrozyten sowie das Überleben der
umliegenden Neurone im periläsionalen Schlaganfallgewebe?
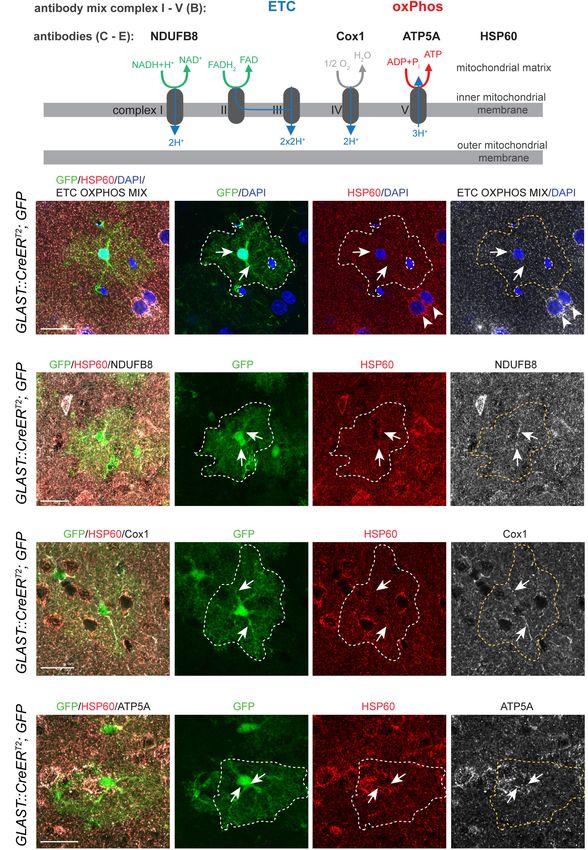
Um zunächst zu überprüfen, ob die Komponenten der Atmungskette und
oxidativen Phosphorylierung auch auf Proteinebene in den Astrozyten
exprimiert werden, wurden immunhistochemische Färbungen im erwachsenen
Maushirngewebe durchgeführt. Diese Charakterisierung wurde an
GLAST::CreERT2; CAG CAT GFP Mäusen durchgeführt (Tfamctrl), die ein
9starkes GFP Signal im Zytoplasma exprimieren. Die Mitochondrien wurden
durch Antikörper gegen das 60-kDa-Hitzeschockprotein (HSP60) dargestellt,
welches in Eukaryoten ausschließlich in Mitochondrien lokalisiert ist (Bukau and
Horwich, 1998). Zur Detektierung der einzelnen Komplexe wurde zunächst eine
Antikörpermischung verwendet, welche alle Komplexe der Atmungskette (I-V)
enthält. Hierbei zeigte sich bei den GFP positiven Astrozyten eine gleichmäßige
Verteilung der Atmungskettenkomplexe in den HSP60 positiven Mitochondrien.
Zur weiteren Differenzierung der einzelnen Komplexe wurden Färbungen mit
Antikörpern gegen Komplex I (Ubichinon Oxidoreduktase Untereinheit B8,
NDUFB8), Komplex IV (Cytochrom C Oxidase Untereinheit 1, COX1) und
Komplex V (ATP Synthase Untereinheit 5A, ATP5A) vorgenommen. Auch diese
ließen sich in allen beobachteten HSP60 positiven Mitochondrien feststellen.
Das Vorhandensein dieser Komplexe weist darauf hin, dass kortikale
Astrozyten die Proteine der ETC und OxPhos in vivo exprimieren und damit
potentiell die Voraussetzungen besitzen, eine oxidative Phophorylierung zu
betreiben.
Um herauszufinden, ob eine vorhandene Elektronentransportkette in Astrozyten
für das Überleben dieser überhaupt funktionstüchtig sein muss, kam ein
weiteres Mausmodell zur Anwendung, in welchem die Atmungskette durch
einen konditionellen Funktionsverlust von Tfam genetisch inhibiert wurde
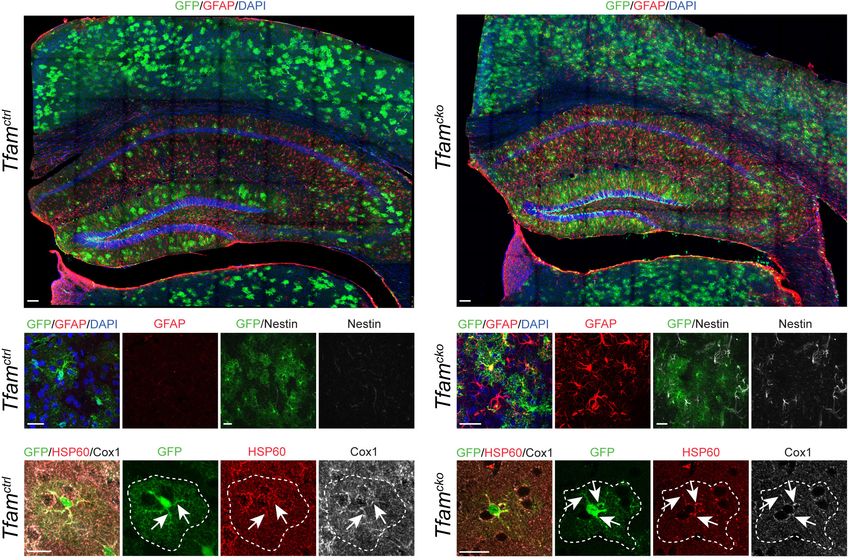
(Tfamcko). Vergleicht man vier Monate und ein Jahr nach Rekombination, zeigt
sich dabei keine signifikant unterschiedliche Anzahl an kortikalen Astrozyten in
den Tfamcko Tieren verglichen mit den Kontrolltieren (Tfamctrl). Dies weist darauf
hin, dass das Überleben der Astrozyten unter physiologischen Bedingungen
unabhängig von der Funktion der Atmungskette ist, was zuvor bereits für den
konditionellen Funktionsverlust vom Cox10 beschrieben wurde (Supplie et al.,
2017). Interessanterweise zeigte sich bei den Tfamcko Mäusen die Expression
des sauren Gliafaserproteins (GFAP) und des Nestins. Beide Proteine sind
Bestandteil der astrozytären Intermediärfilamente und werden nach Stand der
aktuellen Literatur unter physiologischen Bedingungen nicht im erwachsenen
Kortex exprimiert. Die Hochregulation dieser Intermediärfilamentproteine gilt in
der Literatur weitestgehend als markantes Merkmal für den Zustand der
sogenannte reaktiven Astrogliose. Dieser Zustand ist typisch für Astrozyten
nach Kontakt mit pathologischen Reizen, wie beispielsweise bei Entzündungen
10oder Gewebsverletzungen (Robel et al., 2011). Unklar bleibt damit noch die
Auswirkung des Tfam-Funktionsverlustes auf die Mitochondrien selbst. Um
diesen zu bestimmen, wurden Färbungen gegen HSP60 und die einzelnen
Komponenten der Atmungskette angefertigt. 55% der rekombinierten
cko
Astrozyten aus Tfam Mäusen enthielten Mitochondrien mit stark veränderter
geschwollener Morphologie und verstärktem HSP60 Signal. In der Literatur wird
dieser geschwollene Phänotyp nach Tfam-Funktionsverlust auch in anderen
Zelltypen beschrieben, wie beispielsweise in Neuronen (Beckervordersandforth
et al., 2017), in epidermalen Stammzellen (Baris et al., 2011), in braunem
Fettgewebe (Vernochet et al., 2012) und in Herzstammzellen (Chung et al.,
2007). Er gilt als Indikator für mitochondriale Dysfunktion. Überraschenderweise
exprimierten die Mitochondrien rekombinierter kortikaler Astrozyten nach dem
Tfam-Funktionsverlust weiterhin das Protein COX1. Dieses ist als mitochondrial
kodiertes Protein für dessen Expression auf das Vorhandensein von Tfam
angewiesen. Im Gegensatz zu Astrozyten war COX1 in neugeborenen
Neuronen des Gyrus Dentatus nach Tfam-Funktionsverlust in einer früheren
Publikation nicht mehr nachweisbar (Beckervordersandforth et al., 2017). Eine
Erklärung hierfür könnte der erhöhte Zellumsatz in einer sich stark
prolifierierenden Zellpopulation, wie den neugeborenen Neuronen, sein. Diese
sind auf eine verstärkte Proteinbiosynthese zur Bildung neuer Zellen im
Rahmen der Zellteilung angewiesen. Dazu gehört auch die Expression von
neuen Komplexen der Elektronentransportkette, um eine funktionierende
Atmungskette in den neu entstandenen Zellen zur Verfügung zu stellen. Im
Gegensatz dazu teilen sich jedoch postmitotische kortikale Astrozyten nicht
mehr. Sie können daher noch, im Gegensatz zu einer sich eben geteilten Zelle,
auf die noch vor dem Tfam-Funktionsverlust exprimierten Komplexe der
Atmungskette zurückgreifen. Somit könnte sich ein Funktionsverlust von Tfam
in neugeborenen Neuronen schneller bemerkbar machen als in kortikalen
Astrozyten.
Wie eingangs beschrieben, spielen Astrozyten eine wichtige Rolle in der
Neuorganisation des Hirngewebes nach Schlaganfällen. Um den Einfluss
geschädigter Mitochondrien auf die Funktion der Astrozyten im verletzten
Hirngewebe zu untersuchen, bedienten wir uns eines Schlaganfallmodells, der
sogenannten photochemisch-induzierten Thrombose (PIT). Der ischämische
11Schlaganfall wurde an vier Monate alten Tfamctrl und Tfamcko Mäusen induziert
und deren periläsionale Regionen zwei Wochen später immunhistochemisch
untersucht. Zunächst wurde analysiert, inwiefern die astrozytären Mitochondrien
morphologisch durch den induzierten Schlaganfall beeinflusst wurden. Unter
physiologischen Bedingungen konnte in den Tfamctrl Tieren kein einziger
Astrozyt beobachtet werden, der morphologisch abweichende Mitochondrien
aufgewiesen hätte. Im Schlaganfallmodell wiesen allerdings im Mittel 8,6% der
Astrozyten in den Tfamctrl Mäusen morphologisch abweichende Mitochondrien
auf. Bei den Tfamcko Tieren zeigten unter physiologischen Bedingungen im
Mittel 54,6% der Astrozyten abweichende Mitochondrien, während bei den
Tfamcko Tieren im Schlaganfallmodell im Mittel 57,2% auffällige Astrozyten zu
beobachten waren. Die Anzahl an Astrozyten mit morphologisch abweichenden
Mitochondrien ist damit in den Tfamcko Tieren sowohl unter physiologischen
Bedingungen als auch im Schlaganfallmodell nicht signifikant unterschiedlich
(t(7)=0,2158, p=0,834). Interessanterweise ließ sich unter physiologischen
Bedingungen dabei im Tfamcko lediglich ein abweichender Phänotyp
beobachten: Geschwollene Mitochondrien. Im Gegensatz dazu waren im
Schlaganfallmodell zwei unterschiedliche Arten von morphologischen
Abweichungen in den Mitochondrien zu beobachten: zum einen geschwollene
und zum anderen stark elongierte Mitochodrien, welche in manchen Zellen die
Form eines verzweigten Netzes bildeten.
Eine weitere spannende Beobachtung betrifft die Expression von Komponenten
der Atmungskette in den astrozytären Mitochondrien im Schlaganfallmodell. Bei
den Tfamctrl Tieren zeigte sich im Mittel bei 6,6% der astrozytären
Mitochondrien ein Verlust der COX1 Expression. Im Gegensatz dazu stieg der
Verlust der COX1 Expression in den Tfamcko Tieren signifikant auf im Mittel
55,6% (exakter Mann-Whitney-U-Test: U=0,5, p=0,0159) der astrozytären
Mitochondrien an. Eine Erklärung hierfür könnte der durch den Schlaganfall
ausgelöste reaktive Zustand der Astrozyten sein, welcher zu einer verstärkten
Proliferation der dieser führt. Die erhöhte Proliferation könnte für einen früher
sichtbaren Verlust von COX1 ursächlich sein, vergleichbar mit dem oben
beschriebenen Verlust der COX1 Expression in neugeborenen Neuronen (s.
Seite 11f). Diese Ergebnisse deuten darauf hin, dass unter schweren
12Krankheitsbedingungen wie dem Schlaganfall der abweichende Phänotyp der
mitochondrialen Dysfunktion noch weiter verstärkt wird.
Daran anschließend stellt sich die Frage, ob der verstärkte mitochondriale
Phänotyp im Rahmen des Schlaganfallmodells auch eine Auswirkung auf die
astrozytäre Funktion hat. Um dies weiter zu untersuchen, wurde zunächst die
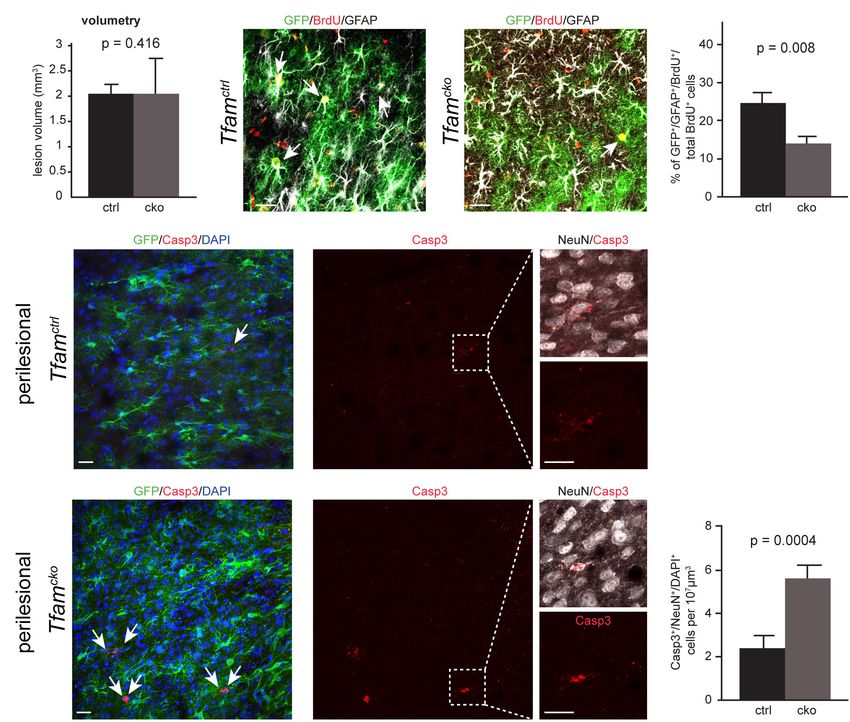
Anzahl an rekombinierten Astrozyten in der periläsionalen Region zwei Wochen
nach Induktion des Infarktes bestimmt. Dabei war kein signifikanter Unterschied
((t(8)=1,276, p=0,4241) zwischen Tfamctrl (221±12,45, n=5) und Tfamcko
(250,9±19,16, n=5) Mäusen zu erkennen, was darauf hinweist, dass auch unter
Verletzungsbedingungen das Gesamtüberleben der Astrozyten nicht von der
mitochondrialen Atmungskette abhängt. Die Proliferationsfähigkeit von
Astrozyten ist in verletztem Gehirngewebe nach Schlafanfällen ein
bedeutsamer Faktor für dessen Regenerationsfähigkeit. Proliferierende
Astrozyten bilden die für den Schutz des vitalen Hirngewebes notwendige
Glianarbe (Wanner et al., 2013; Sofroniew and Vinters, 2010). Zur Bestimmung
neu gebildeter Astrozyten in der periläsionalen Region wurde den Tieren in den
Tagen 2-6 nach Induktion der Photothrombose Bromdesoxyuridin (BrdU)
injiziert. BrdU ist ein Analogon des Nukleosids Thymidin. Nach Aufnahme in die
Zelle kann dieses anstelle des Nukleotids Desoxythymidintriphosphat in die
DNA eingebaut werden (Gratzner, 1982; Kee et al., 2002). Anschließend lassen
sich immunhistochemisch mittels eines Antikörpers gegen BrdU die Zellen
darstellen, welche zuvor BrdU inkorporiert haben. Diese Methode liefert somit
einen Hinweis auf proliferierende Zellen im untersuchten Gewebe (Gratzner,
1982; Kee et al., 2002). Die Zählung BrdU-positiver Zellen wurde 14 Tage nach
Induktion der Photothrombose vorgenommen. Dies entspricht dem oben
beschriebenen Zeitraum, in welchem Astrozyten in einen reaktiven Zustand
versetzt werden, proliferieren und die schützende Glianarbe ausbilden. Hierbei
konnte eine signifikante Reduktion (t(7,739)=3,539, p=0,008) der BrdU
inkorporierenden reaktiven Astrozyten im Verhältnis zur Gesamtanzahl der
BrdU-postiven Zellen (BrdU+ GFAP+ GFP+/gesamt BrdU+ Zellen) in den Tfamcko
(14,25±1,373, n=8) Tieren im Vergleich zu den Tfamctrl (24,67±2,603, n=6)
Tieren beobachtet werden. Ebenfalls erwähnenswert ist, dass im Rahmen der
ausgeführten Auszählungen nicht ein einziger rekombinierter Astrozyt
beobachtet werden konnte, welcher gleichzeitig Casp3 positiv war. Diese Daten
13unterstützen die These, dass zwar nicht das Gesamtüberleben, jedoch die
Neubildung von Astrozyten in der periläsionalen Region des induzierten
Schlaganfalls durch mitochondriale Dysfunktion vermindert ist.
Mit dem Wissen, dass die Proliferation von Astrozyten gestört ist, stellte sich
folglich die Frage, in welcher Form die neuroprotektive Funktion der Astrozyten
durch dysfunktionelle Mitochondrien beeinflusst wird. Konkret sollte untersucht
werden, ob die Sterblichkeit der Neurone von der Funktion der Mitochondrien in
den umliegenden Astrozyten abhängt. Um die entsprechende Zellpopulation zu
identifizieren, wurden immunhistochemische Doppelfärbungen gegen aktivierte
Caspase 3 (Casp3) und Neuronal Nuclei (NeuN) in der periläsionalen Region
des induzierten Schlaganfalls angefertigt. Caspase 3 ist als Protease am Abbau
von Schlüsselenzymen der Zelle beteiligt und damit mitverantwortlich für den
Ablauf der Apoptose (Porter and Janicke, 1999). NeuN eignet sich als
neuronenspezifischer Marker zur Detektion der kortikalen Neuronen (Mullen et
al., 1992). Interessanterweise zeigte sich im Bereich um die Läsion eine
Verdopplung der Anzahl an Casp3-positiven und NeuN-positiven Neuronen. Auf
der kontralateralen Hemisphäre konnte keine Veränderung in der Anzahl an
apoptotischen Neuronen festgestellt werden. Diese Beobachtungen legen
nahe, dass die astrozytäre mitochondriale Dysfunktion die Häufigkeit des
neuronalen Zelltodes im Schlaganfallmodell erhöht, und damit erheblich für die
Beeinträchtigung der neuroprotektiven Funktion der Astrozyten
mitverantwortlich ist.
Die vorliegende Arbeit bekräftigt damit die Vermutung, dass die mitochondriale
Dysfunktion das Überleben der kortikalen Astrozyten unter physiologischen
Bedingungen nicht beeinträchtigt. Im Rahmen eines Schlaganfallmodells
konnte gezeigt werden, dass die mitochondriale Dysfunktion in kortikalen
Astrozyten jedoch mit einer massiven mitochondrialen Änderung der
Morphologie einhergeht sowie eine erhöhte Häufigkeit des neuronalen
Zelltodes bewirkt.
14Literaturverzeichnis
Argaw, A. T., Asp, L., Zhang, J., Navrazhina, K., Pham, T., Mariani, J. N.,
Mahase, S., Dutta, D. J., Seto, J., Kramer, E. G., Ferrara, N., Sofroniew,
M. V. & John, G. R. 2012. Astrocyte-derived VEGF-A drives blood-brain
barrier disruption in CNS inflammatory disease. J Clin Invest, 122, 2454-
68.
Argaw, A. T., Gurfein, B. T., Zhang, Y., Zameer, A. & John, G. R. 2009. VEGF-
mediated disruption of endothelial CLN-5 promotes blood-brain barrier
breakdown. Proc Natl Acad Sci U S A, 106, 1977-82.
Attwell, D., Buchan, A. M., Charpak, S., Lauritzen, M., Macvicar, B. A. &
Newman, E. A. 2010. Glial and neuronal control of brain blood flow.
Nature, 468, 232-43.
Baris, O. R., Klose, A., Kloepper, J. E., Weiland, D., Neuhaus, J. F., Schauen,
M., Wille, A., Muller, A., Merkwirth, C., Langer, T., Larsson, N. G., Krieg,
T., Tobin, D. J., Paus, R. & Wiesner, R. J. 2011. The mitochondrial
electron transport chain is dispensable for proliferation and differentiation
of epidermal progenitor cells. Stem Cells, 29, 1459-68.
Beckervordersandforth, R., Ebert, B., Schaffner, I., Moss, J., Fiebig, C., Shin, J.,
Moore, D. L., Ghosh, L., Trinchero, M. F., Stockburger, C., Friedland, K.,
Steib, K., Von Wittgenstein, J., Keiner, S., Redecker, C., Holter, S. M.,
Xiang, W., Wurst, W., Jagasia, R., Schinder, A. F., Ming, G. L., Toni, N.,
Jessberger, S., Song, H. & Lie, D. C. 2017. Role of Mitochondrial
Metabolism in the Control of Early Lineage Progression and Aging
Phenotypes in Adult Hippocampal Neurogenesis. Neuron, 93, 560-573
e6.
Bejot, Y., Bailly, H., Durier, J. & Giroud, M. 2016. Epidemiology of stroke in
Europe and trends for the 21st century. Presse Med, 45, e391-e398.
Belanger, M., Allaman, I. & Magistretti, P. J. 2011. Brain energy metabolism:
focus on astrocyte-neuron metabolic cooperation. Cell Metab, 14, 724-
38.
Belanger, M. & Magistretti, P. J. 2009. The role of astroglia in neuroprotection.
Dialogues Clin Neurosci, 11, 281-95.
Berger, U. V. & Hediger, M. A. 1998. Comparative analysis of glutamate
transporter expression in rat brain using differential double in situ
hybridization. Anat Embryol (Berl), 198, 13-30.
Bukau, B. & Horwich, A. L. 1998. The Hsp70 and Hsp60 chaperone machines.
Cell, 92, 351-66.
Burda, J. E. & Sofroniew, M. V. 2014. Reactive gliosis and the multicellular
response to CNS damage and disease. Neuron, 81, 229-48.
Bush, T. G., Puvanachandra, N., Horner, C. H., Polito, A., Ostenfeld, T.,
Svendsen, C. N., Mucke, L., Johnson, M. H. & Sofroniew, M. V. 1999.
Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after
ablation of scar-forming, reactive astrocytes in adult transgenic mice.
Neuron, 23, 297-308.
15Cahoy, J. D., Emery, B., Kaushal, A., Foo, L. C., Zamanian, J. L.,
Christopherson, K. S., Xing, Y., Lubischer, J. L., Krieg, P. A., Krupenko,
S. A., Thompson, W. J. & Barres, B. A. 2008. A transcriptome database
for astrocytes, neurons, and oligodendrocytes: a new resource for
understanding brain development and function. J Neurosci, 28, 264-78.
Chaudhry, F. A., Lehre, K. P., Van Lookeren Campagne, M., Ottersen, O. P.,
Danbolt, N. C. & Storm-Mathisen, J. 1995. Glutamate transporters in glial
plasma membranes: highly differentiated localizations revealed by
quantitative ultrastructural immunocytochemistry. Neuron, 15, 711-20.
Chung, S., Dzeja, P. P., Faustino, R. S., Perez-Terzic, C., Behfar, A. & Terzic,
A. 2007. Mitochondrial oxidative metabolism is required for the cardiac
differentiation of stem cells. Nat Clin Pract Cardiovasc Med, 4 Suppl 1,
S60-7.
Ekstrand, M. I., Terzioglu, M., Galter, D., Zhu, S., Hofstetter, C., Lindqvist, E.,
Thams, S., Bergstrand, A., Hansson, F. S., Trifunovic, A., Hoffer, B.,
Cullheim, S., Mohammed, A. H., Olson, L. & Larsson, N. G. 2007.
Progressive parkinsonism in mice with respiratory-chain-deficient
dopamine neurons. Proc Natl Acad Sci U S A, 104, 1325-30.
Faulkner, J. R., Herrmann, J. E., Woo, M. J., Tansey, K. E., Doan, N. B. &
Sofroniew, M. V. 2004. Reactive astrocytes protect tissue and preserve
function after spinal cord injury. J Neurosci, 24, 2143-55.
Feigin, V. L., Forouzanfar, M. H., Krishnamurthi, R., Mensah, G. A., Connor, M.,
Bennett, D. A., Moran, A. E., Sacco, R. L., Anderson, L., Truelsen, T.,
O'donnell, M., Venketasubramanian, N., Barker-Collo, S., Lawes, C. M.,
Wang, W., Shinohara, Y., Witt, E., Ezzati, M., Naghavi, M., Murray, C.,
Global Burden of Diseases, I., Risk Factors, S. & The, G. B. D. S. E. G.
2014. Global and regional burden of stroke during 1990-2010: findings
from the Global Burden of Disease Study 2010. Lancet, 383, 245-54.
Fukui, H., Diaz, F., Garcia, S. & Moraes, C. T. 2007. Cytochrome c oxidase
deficiency in neurons decreases both oxidative stress and amyloid
formation in a mouse model of Alzheimer's disease. Proc Natl Acad Sci
U S A, 104, 14163-8.
Funfschilling, U., Supplie, L. M., Mahad, D., Boretius, S., Saab, A. S., Edgar, J.,
Brinkmann, B. G., Kassmann, C. M., Tzvetanova, I. D., Mobius, W., Diaz,
F., Meijer, D., Suter, U., Hamprecht, B., Sereda, M. W., Moraes, C. T.,
Frahm, J., Goebbels, S. & Nave, K. A. 2012. Glycolytic oligodendrocytes
maintain myelin and long-term axonal integrity. Nature, 485, 517-21.
Gadea, A., Schinelli, S. & Gallo, V. 2008. Endothelin-1 regulates astrocyte
proliferation and reactive gliosis via a JNK/c-Jun signaling pathway. J
Neurosci, 28, 2394-408.
Gaspari, M., Larsson, N. G. & Gustafsson, C. M. 2004. The transcription
machinery in mammalian mitochondria. Biochim Biophys Acta, 1659,
148-52.
Gratzner, H. G. 1982. Monoclonal antibody to 5-bromo- and 5-
iododeoxyuridine: A new reagent for detection of DNA replication.
Science, 218, 474-5.
16Hamby, M. E., Coppola, G., Ao, Y., Geschwind, D. H., Khakh, B. S. &
Sofroniew, M. V. 2012. Inflammatory mediators alter the astrocyte
transcriptome and calcium signaling elicited by multiple G-protein-
coupled receptors. J Neurosci, 32, 14489-510.
Herrmann, J. E., Imura, T., Song, B., Qi, J., Ao, Y., Nguyen, T. K., Korsak, R.
A., Takeda, K., Akira, S. & Sofroniew, M. V. 2008. STAT3 is a critical
regulator of astrogliosis and scar formation after spinal cord injury. J
Neurosci, 28, 7231-43.
Iadecola, C. & Nedergaard, M. 2007. Glial regulation of the cerebral
microvasculature. Nat Neurosci, 10, 1369-76.
Kee, N., Sivalingam, S., Boonstra, R. & Wojtowicz, J. M. 2002. The utility of Ki-
67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci
Methods, 115, 97-105.
Keiner, S., Wurm, F., Kunze, A., Witte, O. W. & Redecker, C. 2008.
Rehabilitative therapies differentially alter proliferation and survival of
glial cell populations in the perilesional zone of cortical infarcts. Glia, 56,
516-27.
Krishnamurthi, R. V., Feigin, V. L., Forouzanfar, M. H., Mensah, G. A., Connor,
M., Bennett, D. A., Moran, A. E., Sacco, R. L., Anderson, L. M., Truelsen,
T., O'donnell, M., Venketasubramanian, N., Barker-Collo, S., Lawes, C.
M., Wang, W., Shinohara, Y., Witt, E., Ezzati, M., Naghavi, M., Murray,
C., Global Burden of Diseases, I. R. F. S. & Group, G. B. D. S. E. 2013.
Global and regional burden of first-ever ischaemic and haemorrhagic
stroke during 1990-2010: findings from the Global Burden of Disease
Study 2010. Lancet Glob Health, 1, e259-81.
Larsson, N. G., Wang, J., Wilhelmsson, H., Oldfors, A., Rustin, P., Lewandoski,
M., Barsh, G. S. & Clayton, D. A. 1998. Mitochondrial transcription factor
A is necessary for mtDNA maintenance and embryogenesis in mice. Nat
Genet, 18, 231-6.
Lehre, K. P., Levy, L. M., Ottersen, O. P., Storm-Mathisen, J. & Danbolt, N. C.
1995. Differential expression of two glial glutamate transporters in the rat
brain: quantitative and immunocytochemical observations. J Neurosci,
15, 1835-53.
Li, L., Lundkvist, A., Andersson, D., Wilhelmsson, U., Nagai, N., Pardo, A. C.,
Nodin, C., Stahlberg, A., Aprico, K., Larsson, K., Yabe, T., Moons, L.,
Fotheringham, A., Davies, I., Carmeliet, P., Schwartz, J. P., Pekna, M.,
Kubista, M., Blomstrand, F., Maragakis, N., Nilsson, M. & Pekny, M.
2008. Protective role of reactive astrocytes in brain ischemia. J Cereb
Blood Flow Metab, 28, 468-81.
Liddelow, S. A. & Barres, B. A. 2017. Reactive Astrocytes: Production,
Function, and Therapeutic Potential. Immunity, 46, 957-967.
Lovatt, D., Sonnewald, U., Waagepetersen, H. S., Schousboe, A., He, W., Lin,
J. H., Han, X., Takano, T., Wang, S., Sim, F. J., Goldman, S. A. &
Nedergaard, M. 2007. The transcriptome and metabolic gene signature
of protoplasmic astrocytes in the adult murine cortex. J Neurosci, 27,
12255-66.
17Maciver, N. J., Michalek, R. D. & Rathmell, J. C. 2013. Metabolic regulation of T
lymphocytes. Annu Rev Immunol, 31, 259-83.
Mills, K. H. 2011. TLR-dependent T cell activation in autoimmunity. Nat Rev
Immunol, 11, 807-22.
Mori, T., Tanaka, K., Buffo, A., Wurst, W., Kuhn, R. & Gotz, M. 2006. Inducible
gene deletion in astroglia and radial glia--a valuable tool for functional
and lineage analysis. Glia, 54, 21-34.
Mosser, D. M. & Edwards, J. P. 2008. Exploring the full spectrum of
macrophage activation. Nat Rev Immunol, 8, 958-69.
Mullen, R. J., Buck, C. R. & Smith, A. M. 1992. NeuN, a neuronal specific
nuclear protein in vertebrates. Development, 116, 201-11.
Murray, C. J., Vos, T., Lozano, R., Naghavi, M., Flaxman, A. D., Michaud, C.,
Ezzati, M., Shibuya, K., Salomon, J. A., Abdalla, S., Aboyans, V.,
Abraham, J., Ackerman, I., Aggarwal, R., Ahn, S. Y., Ali, M. K., Alvarado,
M., Anderson, H. R., Anderson, L. M., Andrews, K. G., Atkinson, C.,
Baddour, L. M., Bahalim, A. N., Barker-Collo, S., Barrero, L. H., Bartels,
D. H., Basanez, M. G., Baxter, A., Bell, M. L., Benjamin, E. J., Bennett,
D., Bernabe, E., Bhalla, K., Bhandari, B., Bikbov, B., Bin Abdulhak, A.,
Birbeck, G., Black, J. A., Blencowe, H., Blore, J. D., Blyth, F., Bolliger, I.,
Bonaventure, A., Boufous, S., Bourne, R., Boussinesq, M., Braithwaite,
T., Brayne, C., Bridgett, L., Brooker, S., Brooks, P., Brugha, T. S., Bryan-
Hancock, C., Bucello, C., Buchbinder, R., Buckle, G., Budke, C. M.,
Burch, M., Burney, P., Burstein, R., Calabria, B., Campbell, B., Canter,
C. E., Carabin, H., Carapetis, J., Carmona, L., Cella, C., Charlson, F.,
Chen, H., Cheng, A. T., Chou, D., Chugh, S. S., Coffeng, L. E., Colan, S.
D., Colquhoun, S., Colson, K. E., Condon, J., Connor, M. D., Cooper, L.
T., Corriere, M., Cortinovis, M., De Vaccaro, K. C., Couser, W., Cowie, B.
C., Criqui, M. H., Cross, M., Dabhadkar, K. C., Dahiya, M., Dahodwala,
N., Damsere-Derry, J., Danaei, G., Davis, A., De Leo, D., Degenhardt, L.,
Dellavalle, R., Delossantos, A., Denenberg, J., Derrett, S., Des Jarlais,
D. C., Dharmaratne, S. D., et al. 2012. Disability-adjusted life years
(DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a
systematic analysis for the Global Burden of Disease Study 2010.
Lancet, 380, 2197-223.
Nakamura, T., Colbert, M. C. & Robbins, J. 2006. Neural crest cells retain
multipotential characteristics in the developing valves and label the
cardiac conduction system. Circ Res, 98, 1547-54.
Neary, J. T., Kang, Y., Willoughby, K. A. & Ellis, E. F. 2003. Activation of
extracellular signal-regulated kinase by stretch-induced injury in
astrocytes involves extracellular ATP and P2 purinergic receptors. J
Neurosci, 23, 2348-56.
Oberheim, N. A., Takano, T., Han, X., He, W., Lin, J. H., Wang, F., Xu, Q.,
Wyatt, J. D., Pilcher, W., Ojemann, J. G., Ransom, B. R., Goldman, S. A.
& Nedergaard, M. 2009. Uniquely hominid features of adult human
astrocytes. J Neurosci, 29, 3276-87.
Porter, A. G. & Janicke, R. U. 1999. Emerging roles of caspase-3 in apoptosis.
Cell Death Differ, 6, 99-104.
18Robel, S., Berninger, B. & Gotz, M. 2011. The stem cell potential of glia:
lessons from reactive gliosis. Nat Rev Neurosci, 12, 88-104.
Schmitt, A., Asan, E., Puschel, B. & Kugler, P. 1997. Cellular and regional
distribution of the glutamate transporter GLAST in the CNS of rats:
nonradioactive in situ hybridization and comparative
immunocytochemistry. J Neurosci, 17, 1-10.
Seo, J. H., Miyamoto, N., Hayakawa, K., Pham, L. D., Maki, T., Ayata, C., Kim,
K. W., Lo, E. H. & Arai, K. 2013. Oligodendrocyte precursors induce early
blood-brain barrier opening after white matter injury. J Clin Invest, 123,
782-6.
Shirakawa, H., Sakimoto, S., Nakao, K., Sugishita, A., Konno, M., Iida, S.,
Kusano, A., Hashimoto, E., Nakagawa, T. & Kaneko, S. 2010. Transient
receptor potential canonical 3 (TRPC3) mediates thrombin-induced
astrocyte activation and upregulates its own expression in cortical
astrocytes. J Neurosci, 30, 13116-29.
Sirko, S., Behrendt, G., Johansson, P. A., Tripathi, P., Costa, M., Bek, S.,
Heinrich, C., Tiedt, S., Colak, D., Dichgans, M., Fischer, I. R., Plesnila,
N., Staufenbiel, M., Haass, C., Snapyan, M., Saghatelyan, A., Tsai, L. H.,
Fischer, A., Grobe, K., Dimou, L. & Gotz, M. 2013. Reactive glia in the
injured brain acquire stem cell properties in response to sonic hedgehog.
[corrected]. Cell Stem Cell, 12, 426-39.
Sofroniew, M. V. & Vinters, H. V. 2010. Astrocytes: biology and pathology. Acta
Neuropathol, 119, 7-35.
Sorensen, L., Ekstrand, M., Silva, J. P., Lindqvist, E., Xu, B., Rustin, P., Olson,
L. & Larsson, N. G. 2001. Late-onset corticohippocampal neurodepletion
attributable to catastrophic failure of oxidative phosphorylation in MILON
mice. J Neurosci, 21, 8082-90.
Supplie, L. M., Duking, T., Campbell, G., Diaz, F., Moraes, C. T., Gotz, M.,
Hamprecht, B., Boretius, S., Mahad, D. & Nave, K. A. 2017. Respiration-
Deficient Astrocytes Survive As Glycolytic Cells In Vivo. J Neurosci, 37,
4231-4242.
Torp, R., Danbolt, N. C., Babaie, E., Bjoras, M., Seeberg, E., Storm-Mathisen,
J. & Ottersen, O. P. 1994. Differential expression of two glial glutamate
transporters in the rat brain: an in situ hybridization study. Eur J
Neurosci, 6, 936-42.
Tuszynski, M. H. & Steward, O. 2012. Concepts and methods for the study of
axonal regeneration in the CNS. Neuron, 74, 777-91.
Vernochet, C., Mourier, A., Bezy, O., Macotela, Y., Boucher, J., Rardin, M. J.,
An, D., Lee, K. Y., Ilkayeva, O. R., Zingaretti, C. M., Emanuelli, B.,
Smyth, G., Cinti, S., Newgard, C. B., Gibson, B. W., Larsson, N. G. &
Kahn, C. R. 2012. Adipose-specific deletion of TFAM increases
mitochondrial oxidation and protects mice against obesity and insulin
resistance. Cell Metab, 16, 765-76.
Volterra, A. & Meldolesi, J. 2005. Astrocytes, from brain glue to communication
elements: the revolution continues. Nat Rev Neurosci, 6, 626-40.
19Walsh, J. T. & Kipnis, J. 2011. Regulatory T cells in CNS injury: the simple, the
complex and the confused. Trends Mol Med, 17, 541-7.
Wanner, I. B., Anderson, M. A., Song, B., Levine, J., Fernandez, A., Gray-
Thompson, Z., Ao, Y. & Sofroniew, M. V. 2013. Glial scar borders are
formed by newly proliferated, elongated astrocytes that interact to corral
inflammatory and fibrotic cells via STAT3-dependent mechanisms after
spinal cord injury. J Neurosci, 33, 12870-86.
Watson, B. D., Dietrich, W. D., Busto, R., Wachtel, M. S. & Ginsberg, M. D.
1985. Induction of reproducible brain infarction by photochemically
initiated thrombosis. Ann Neurol, 17, 497-504.
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G. & Barres,
B. A. 2012. Genomic analysis of reactive astrogliosis. J Neurosci, 32,
6391-410.
Zhao, Z., Nelson, A. R., Betsholtz, C. & Zlokovic, B. V. 2015. Establishment and
Dysfunction of the Blood-Brain Barrier. Cell, 163, 1064-1078.
20ORIGINAL RESEARCH
published: 22 February 2019
doi: 10.3389/fnmol.2019.00040
Mitochondrial Dysfunction in
Astrocytes Impairs the Generation of
Reactive Astrocytes and Enhances
Neuronal Cell Death in the Cortex
Upon Photothrombotic Lesion
Christian Fiebig 1 , Silke Keiner 2 , Birgit Ebert 3 , Iris Schäffner 1,3 , Ravi Jagasia 3,4 ,
D. Chichung Lie 1,3 and Ruth Beckervordersandforth 1*
1
Institute of Biochemistry, Emil Fischer Center, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany,
2
Hans Berger Department of Neurology, Jena University Hospital, Jena, Germany, 3 Institute of Developmental Genetics,
Helmholtz Center Munich, German Research Center for Environmental Health, Munich, Germany, 4 F. Hoffmann-La Roche,
Ltd., CNS Discovery, Pharma Research and Early Development, Basel, Switzerland
Mitochondria are key organelles in regulating the metabolic state of a cell. In the
brain, mitochondrial oxidative metabolism is the prevailing mechanism for neurons
to generate ATP. While it is firmly established that neuronal function is highly
dependent on mitochondrial metabolism, it is less well-understood how astrocytes
function rely on mitochondria. In this study, we investigate if astrocytes require a
Edited by: functional mitochondrial electron transport chain (ETC) and oxidative phosphorylation
Christian Lange,
(oxPhos) under physiological and injury conditions. By immunohistochemistry we
Technische Universität Dresden,
Germany show that astrocytes expressed components of the ETC and oxPhos complexes
Reviewed by: in vivo. Genetic inhibition of mitochondrial transcription by conditional deletion of
Ilaria Decimo, mitochondrial transcription factor A (Tfam) led to dysfunctional ETC and oxPhos activity,
University of Verona, Italy
Federico Calegari, as indicated by aberrant mitochondrial swelling in astrocytes. Mitochondrial dysfunction
Technische Universität Dresden, did not impair survival of astrocytes, but caused a reactive gliosis in the cortex
Germany
under physiological conditions. Photochemically initiated thrombosis induced ischemic
*Correspondence:
stroke led to formation of hyperfused mitochondrial networks in reactive astrocytes of
Ruth Beckervordersandforth
ruth.beckervordersandforth@fau.de the perilesional area. Importantly, mitochondrial dysfunction significantly reduced the
generation of new astrocytes and increased neuronal cell death in the perilesional area.
Received: 28 August 2018
Accepted: 01 February 2019
These results indicate that astrocytes require a functional ETC and oxPhos machinery
Published: 22 February 2019 for proliferation and neuroprotection under injury conditions.
Citation:
Keywords: mitochondrial metabolism, astrocytes, stroke/photothrombotic lesion, electron transport chain,
Fiebig C, Keiner S, Ebert B, oxidative phosphorylation, reactive gliosis, Tfam
Schäffner I, Jagasia R, Lie DC and
Beckervordersandforth R (2019)
Mitochondrial Dysfunction
in Astrocytes Impairs the Generation
INTRODUCTION
of Reactive Astrocytes and Enhances
Neuronal Cell Death in the Cortex
Astrocytes are highly abundant in the brain (Nedergaard et al., 2003) and central to homeostasis
Upon Photothrombotic Lesion. of the nervous system by, e.g., regulating glutamate, ion and water homeostasis, synapse formation
Front. Mol. Neurosci. 12:40. and modulation, tissue repair, energy storage, and defense against oxidative stress (Volterra and
doi: 10.3389/fnmol.2019.00040 Meldolesi, 2005; Belanger and Magistretti, 2009). Astrocytes are also critically important for
Frontiers in Molecular Neuroscience | www.frontiersin.org 1 February 2019 | Volume 12 | Article 40Fiebig et al. Mitochondrial Dysfunction in Reactive Astrocytes
brain metabolism (Hertz et al., 2007; Belanger et al., 2011). the perilesional area, indicating that astrocytes require
Bridging between neurons and blood vessels, astrocytes functional mitochondrial machinery for proliferation after
are major components of neurovascular coupling. Fine injury and for protecting the neurons from the damage
astrocytic processes cover synaptic contacts on one side induced by stroke.
(Iadecola and Nedergaard, 2007; Oberheim et al., 2009),
while on the other side, astrocyte end-feet enwrap the brain
microvasculature and regulate the vascular tone as well MATERIALS AND METHODS
as blood–brain function (Attwell et al., 2010; Zhao et al.,
2015). These morphological characteristics and a special Experimental Model and Subject Details
regionalized molecular set-up allow astrocytes to sense neuronal All experiments were carried out in accordance with the
activity at the synapse (via receptors for neurotransmitters, European Communities Council Directive (86/609/EEC).
cytokines, growths factors, transporters, and ion channels), Animal experiments were approved by the Government
and react with the appropriate metabolic supply via their of Upper Bavaria. For all experiments, mice were group
end-feet on the blood vessels (via glucose transporters housed in standard cages under a 12 h light/dark cycle
and aquaporin 4), thereby coordinating synaptic needs and with ad libitum access to water and food. The astrocyte
metabolic supply. specific conditional Tfam knockout line and the control line
In contrast to neurons that sustain a high rate of oxidative (Tfamcko and Tfamctrl , respectively) were generated from
mitochondrial metabolism, astrocytes characteristically perform TfamloxP/loxP mice (Larsson et al., 1998), GLAST::CreERT2
glycolysis (Belanger et al., 2011). Still, astrocytes possess almost (Mori et al., 2006), CAG-CAT-EGFP reporter mice
as many mitochondria as neurons (Lovatt et al., 2007), and (Nakamura et al., 2006) and were described previously
transcriptome analysis indicated that astrocytes are equipped (Beckervordersandforth et al., 2017).
with the necessary molecular machinery to perform oxidative
metabolism (Lovatt et al., 2007; Cahoy et al., 2008). However,
it is an ongoing debate if and to which extent astrocytes
Tamoxifen Administration
Tamoxifen was dissolved at 10 mg/ml in corn oil (Sigma) and
in vivo perform and require oxPhos. A recent publication
animals were intraperitonially (i.p.) injected with 1 mg on
showed that conditional ablation of electron transport chain
postnatal days 14, 16, and 18 (Beckervordersandforth et al., 2017).
(ETC) neither affected long-term viability of astrocytes nor
caused any obvious brain pathology (Supplie et al., 2017). An
interesting question now is how astrocytes behave under Genotyping PCR
stress conditions. Astrocytes have a unique capacity to The following primers were used for genotyping: Tfam-
adapt to conditions of metabolic challenge and are able to A CTGCCTTCCTCTAGCCCGGG, Tfam-B GTAACAG
adjust their metabolic state to distinct injuries as assessed CAGACAACTTGTG, Tfam-C CTCTGAAGCACATGGTCAAT.
by transcriptome analysis (Hamby et al., 2012; Zamanian The expected size of PCR products for Tfamwt was 404 bp, for
et al., 2012). Furthermore, preservation of mitochondrial Tfamfloxed = 437 bp, and for Tfamcko = 329 bp.
respiratory function in astrocytes may be important for the
brain’s energy balance and for production of antioxidants Astrocyte Culture
that contribute to neuronal protection (Greenamyre et al., Primary astrocytes were isolated as previously described
2003; Dugan and Kim-Han, 2004). In many neurodegenerative (Heinrich et al., 2010). Briefly, postnatal day 5 (P5) CAG
disorders and under injury conditions, the astrocyte’s response CAT GFP; Tfamfl/fl mice were decapitated and cortices were
to injury and disease becomes increasingly recognized dissected in ice-cold dissection medium (HBSS with Hepes
because astrocytes bare the potential to enhance neuronal 10 mM) by carefully removing all meninges. Dissected slices
survival and regeneration (Sofroniew and Vinters, 2010; were minced into small tissue pieces, and further dissociated
Barreto et al., 2012). with a fire-polished Pasteur pipette into a single cell suspension.
Here, we investigated the impact of mitochondrial ETC After centrifugation (900 rcf, 5 min, RT), the supernatant
and oxPhos in astrocytes in vivo under pathological conditions was discarded, and the pellet resuspended in 10 ml astrocyte
in a stroke model of photochemically initiated thrombosis medium (DMEM/F12, 0,45% Glucose, 10%FBS, 5% horse serum,
(PIT). Toward this aim, we abolished ETC complexes I, B27, 10 ng/ml EFG and FGF). Cell suspension was transferred
III, and IV function as well as oxPhos complex V activity into in a medium sized flask (10 ml, 1T75) if two brains
in astrocytes by conditional deletion of the mitochondrial were pooled. Cells of one brain were transferred into a
transcription factor A (Tfam; Larsson et al., 1998). Deletion of small flask (5 ml, 1T25). Cells were incubated at 37 C with
Tfam did not impair survival of astrocytes but induced reactive 5% CO2 . Medium was changed every 4 days after shaking
gliosis in the cortex and led to morphological alteration of (200 rpm) at room tempertaure (RT) to remove unattached
mitochondria in reactive astrocytes. Upon photothrombotic tissue like microglia and oligodendrocytes. Cells were passaged
lesions, Tfam-deficiency worsened mitochondrial morphology by trypsination when cell density reached 70% confluence.
phenotypes and impaired the generation of new astrocytes in For immunostainings, cells were seeded onto PDL-coated glass
the perilesional area. Most notably, dysfunctional mitochondrial cover slips. Two days after passaging, cells were transduced
respiration in astrocytes increased neuronal cell death in with HTNCre protein (1, 2, or 4 µl). For immunochemistry,
Frontiers in Molecular Neuroscience | www.frontiersin.org 2 February 2019 | Volume 12 | Article 40Sie können auch lesen

















































