CRISPR/Cas9 Mediated Knock-Out Pancreatic Cancer Cell Lines - of KRASG12D Mutated - OPUS 4
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
CRISPR/Cas9 Mediated Knock-Out
of KRASG12D Mutated
Pancreatic Cancer Cell Lines
Der Medizinischen Fakultät
der Friedrich-Alexander-Universität
Erlangen-Nürnberg
zur
Erlangung des Doktorgrades Dr. med.
vorgelegt von
Eva Lentsch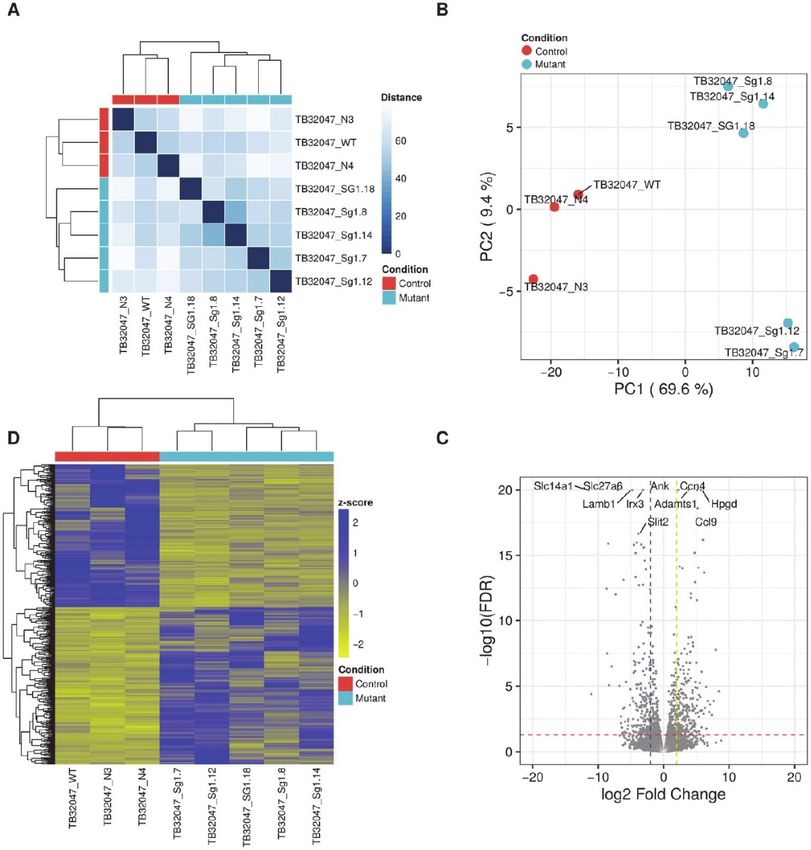
Als Dissertation genehmigt
von der Medizinischen Fakultät
der Friedrich-Alexander-Universität Erlangen-Nürnberg
Vorsitzender des Promotionsorgans: Prof. Dr. Markus F. Neurath
Gutachter: Prof. Dr. Robert Grützmann
Gutachter: PD Dr. Maximilian Brunner
Gutachter: Prof. Dr. Felix Rückert
Gutachter: PD Dr. Benjamin Frey
Tag der mündlichen Prüfung: 11. Januar 2022
Meinen Eltern gewidmet
Inhaltsverzeichnis
1. Zusammenfassung 1
1.1. Hintergrund und Ziele 1
1.2. Methoden 1
1.3. Ergebnisse und Beobachtungen 2
1.4. Schlussfolgerungen 2
2. Einordnung in den fachwissenschaftlichen Kontext 3
2.1. Das Pankreas – Aufbau und Funktion 3
2.2. Karzinogenese 4
2.2.1. Progressionsmodell 7
2.3. Das Pankreaskarzinom 8
2.3.1. Epidemiologie 8
2.3.2. Ätiologie und Risikofaktoren 8
2.3.3. Diagnostik 9
2.3.4. Tumorklassifikation 9
2.3.5. Therapie 11
2.4. Protoonkogene 13
2.4.1. KRAS 14
2.5. CRISPR/Cas-System 15
2.5.1. CRISPR/Cas9 16
2.6. Zielsetzung der Arbeit 17
2.7. Methoden 18
3. Publikation 19
4. Anhang 31
4.1. Literaturverzeichnis 31
4.2. Abkürzungsverzeichnis 36
4.3. Abbildungsverzeichnis 37
4.4. Tabellenverzeichnis 38
5. Danksagung 39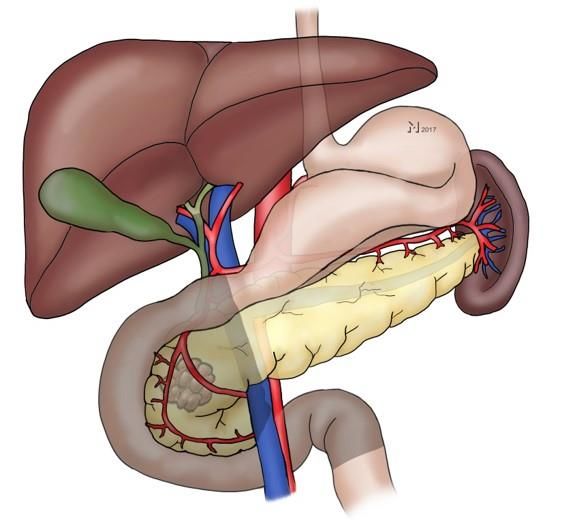
1. Zusammenfassung
Deutscher Titel: CRISPR/Cas9 vermittelter Knock-out von KRASG12D-Mutationen in
Pankreaskarzinomzellen
1.1. Hintergrund und Ziele
Das Pankreaskarzinom (PaCa) ist die vierthäufigste Krebstodesursache in
Deutschland. Die Überlebenschancen nach Diagnosestellung sind sehr gering, was
sich in der negativen 5-Jahres-Überlebensrate von nur circa 9% widerspiegelt. Es ist
bekannt, dass in rund 90% der PaCa-Fälle eine Mutation im Protoonkogen KRAS
nachweisbar ist. Als zentrales Element in verschiedenen Signalwegen ist KRAS in
der Regulation von Zellproliferation, -differenzierung und -überleben involviert. Eine
aktivierende Mutation kann zur kontinuierlichen Transmission eines Wachstums-
signals und damit zur Tumorentstehung führen. Ob KRAS jedoch zur
Aufrechterhaltung von Tumoren benötigt wird, ist fraglich. Mit dem CRISPR/Cas9-
System, welches die Modifikation von Genen ermöglicht, wurde analysiert, ob ein
Knock-out von mutiertem KRAS durchführbar ist. Weiterhin soll geklärt werden, wie
sich das Ausschalten des mutierten KRAS auf das Überleben der Zellen auswirkt.
1.2. Methoden
Zur Durchführung des CRISPR/Cas9 vermittelten Knock-outs und zur Überprüfung
der Rolle von mutiertem KRAS im Pankreaskarzinom wurden zwei humane Zelllinien
(Panc-1 und SUIT-2) und eine Zelllinie aus dem Mausmodell (TB32047) verwendet.
Alle drei Zelllinien enthalten die heterozygote KRASG12D-Mutation (c.35G>A). Nach
Transfektion, Puromycin Selektion und Einzelzell-Klonierung wurden Western Blots
durchgeführt, um den Knock-out zu bestätigen. Zur weiteren Überprüfung des Knock-
outs im Bereich von KRASG12D wurde die DNA aller Klone sequenziert. Auf Grund
der Expressionsveränderungen der Signaltransduktionsproteine der veränderten
TB32047 Zellen wurde die Genexpression dieser Klone mittels RNA-Sequenzierung
untersucht.
-1-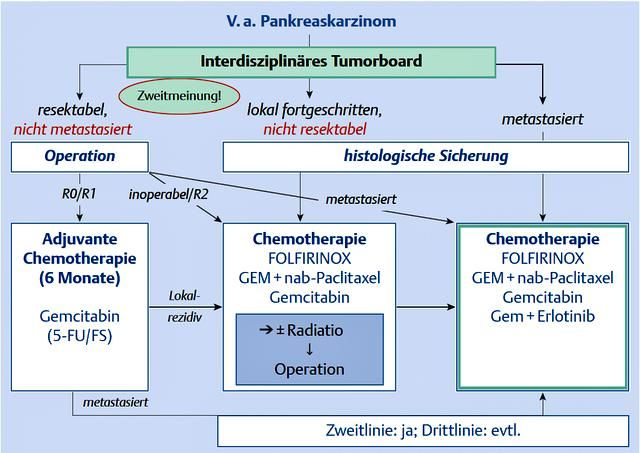
1.3. Ergebnisse und Beobachtungen
Mit Hilfe der DNA Sequenzierung konnten Insertions- und Deletionsmutationen
(Indel; variierend zwischen kleinen Indels unter 20 bp bis hin zu großen Indels über
20 bp) in der KRASG12D-Sequenz festgestellt werden. Mittels Western Blot war keine
Expression von KRASG12D nachweisbar. Daher ist von erfolgreichen Knock-outs in
allen drei getesteten Zelllinien auszugehen. Normales KRAS wurde dennoch von
allen Klonen exprimiert. Die Untersuchung der Signaltransduktionsproteine (Erk, Akt,
Stat3, AMPK, c-myc) zeigte eine heterogene Expression dieser Proteine. Panc-1
Klone zeigen eine stabile pErk Expression. Die Proteinexpression von pAkt ist bei
einigen Klonen reduziert. Bei SUIT-2 konnte ein Verlust der Akt-Phosphorylierung,
bei vorhandener pErk Expression, beobachtet werden. Konstante pErk- und pAkt-
Level waren bei den TB32047 Klonen sichtbar. Die RNA Sequenzierung der
TB32047 Klone offenbarte 417 unterschiedlich exprimierte Gene. Durch den Knock-
out des mutierten KRAS kann es zu einem langsameren Wachstum kommen.
Weiterhin zeigte die Zellkultur der Klone keinen Wachstumsstopp oder Apoptose.
1.4. Schlussfolgerungen
Diese Ergebnisse weisen darauf hin, dass KRAS wahrscheinlich nicht essentiell für
die Erhaltung vom Pankreaskarzinomzellen in vitro ist. Die Western Blots zeigen,
dass jeder Klon sich einzeln entwickelt hat und als individuell angesehen werden
sollte. Durch die RNA Sequenzierung konnten Einblicke in die veränderte
Genexpression der TB32047 Klone gewonnen werden. Zwei stark herunterregulierte
Gene waren Laminin B1 (Lamb1) und Solute Carrier Family 27, Member 6 (Slc27a6).
Diese Herunterregulierung kann die Angiogenese, Invasion und Energieversorgung
stark negativ beeinflussen und somit ein hilfreiches Werkzeug zur Bekämpfung vom
PaCa sein.
-2-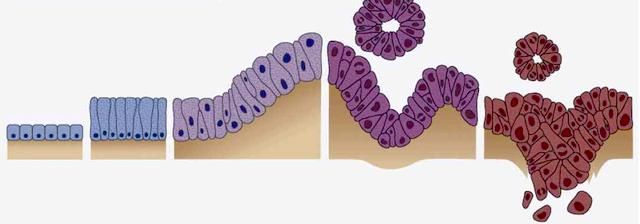
2. Einordnung in den fachwissenschaftlichen Kontext
2.1. Das Pankreas – Aufbau und Funktion
Die Bauchspeicheldrüse (Pankreas) ist eine Drüse mit endo- und exokriner Funktion,
welche sekundär retroperitoneal im Bauchraum liegt. Das Pankreas wiegt 70-80
Gramm, ist 13-18 cm lang und liegt auf Höhe des Lendenwirbelkörpers I-II (Aumüller
et al. 2010). Zwischen Bauchdecke und Wirbelsäule liegend, wird das keilförmige
Organ oben vom Magen, vorn vom Peritoneum, links von der Milz, rechts und unten
vom Duodenum, sowie auf der Rückseite von der Aorta abdominalis, der Vena cava
inferior, der Vena portae und der rechten Niere begrenzt (siehe Abbildung 1). Das
Pankreas begrenzt kaudal die Bursa omentalis, ein Nebenraum in der
Peritonealhöhle. Makroskopisch unterteilen kann man das Pankreas in Caput,
Corpus und Cauda (Aumüller et al. 2010). Die arterielle Gefäßversorgung erfolgt
durch Äste der Aorta. Der Truncus coeliacus gibt neben einem weiteren Ast, die
Arteria splenica und die A. hepatica communis ab, die sich wiederum in A. hepatica
propria und die A. gastroduodenalis aufteilt. Die A. pancreaticoduodenalis inferior,
ein Ast der A. mesenterica superior, die A. splenica und die A. gastroduodenalis
versorgen das Pankreas mit arteriellem Blut. Im venösen System fließt das Blut, je
nach Ursprung, aus den Venae pancreaticae in die V. splenica oder in die V.
mesenterica superior und von dort in die V. portae (Aumüller et al. 2010).
Abbildung 1. Makroskopischer Aufbau der Bauchspeicheldrüse. Die dunkle Markierung zeigt
die zu entfernenden Strukturen im Rahmen einer pyloruserhaltenden partiellen
Pankreatikoduodenektomie. (Quelle: Dr. med. Maak 2017)
-3-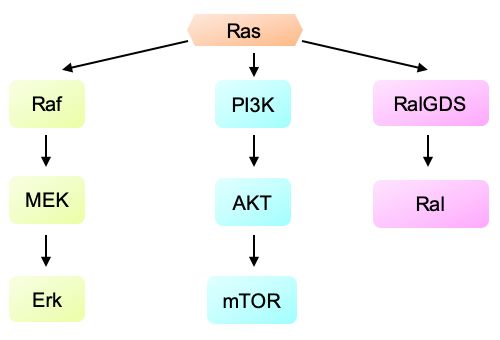
Die Innervation der Bauchspeicheldrüse erfolgt sympathisch durch das Ganglion
coeliacum und parasympathisch durch den Truncus vagalis posterior. Die
Nervenfasern treten gefäßbegleitend in das Organ ein. Die sympathische Aktivierung
durch Noradrenalin führt zu einer Hemmung der Insulinsekretion. Im Gegensatz dazu
wird durch den Parasympathikus (Acetylcholin) die Enzym- und die Insulinsekretion
gesteigert (Aumüller et al. 2010).
Das Pankreas erfüllt zwei wichtige Aufgaben: als seröse Drüse produziert sie bis zu
zwei Liter Verdauungssaft, in dem sich Vorstufen von Proteasen (z.B.: Trypsinogen),
Glykosidasen (α-Amylase), Nukleasen (Desoxy- und Ribonuklease) und Lipasen
befinden. Diese Enzyme werden über den Ductus pancreaticus, gemeinsam mit dem
Ductus choledochus, über die Papilla duodeni major in den Dünndarm sezerniert.
Dort werden die Vorstufen in ihre aktive Form gebracht, um wichtige
Nahrungsbestandteile spalten zu können. Damit der im Magen gebildete saure
Chymus neutralisiert werden kann, und um den Enzymen ein optimales Milieu zu
bieten, beinhaltet der Pankreassaft HCO3—-Ionen, die den pH-Wert bei 8 halten
(Lüllmann-Rauch und Paulsen 2012). Die verschiedenen endokrinen Zellen
übernehmen die Aufgabe der Hormonproduktion. Die A-, B-, D- und PP-Zellen lagern
sich als Langerhans-Inseln zusammen. Diese Inseln sind im ganzen Organ verteilt,
am häufigsten sind sie aber im Cauda pancreatis zu finden. Von den Zellen werden
Glukagon, Insulin, Somatostatin und das Pankreatische Polypeptid gebildet
(Aumüller et al. 2010, Lüllmann-Rauch und Paulsen 2012).
2.2. Karzinogenese
Um die Komplexität neoplastischer Erkrankungen, insb. bösartiger Neubildungen,
besser zu verstehen, sollte die Entstehung dieser genauer betrachtet werden.
Hanahan und Weinberg beschreiben sechs wesentliche Eigenschaften und neue
Besonderheiten einer entarteten Zelle (siehe Abbildung 2).
-4-Aufrechterhaltung der Umgehung von wachstums-
Wachstumssignale hemmenden Signalen
Umgestaltung des zellulären Abwenden immunogener
Energiehaushalts Destruktion
Ermöglichung der
Apoptoseresistenz replikativen
Unsterblichkeit
Genominstabilität& - tumorfördernde
mutation Inflammation
Induktion der Aktivierung der
Angiogenese Invasion und
Metastasierung
Abbildung 2. Grundlagen, fundamentale Eigenschaften und neu entstehende
Besonderheiten von Krebszellen. (Quelle: in Anlehnung an Hanahan und Weinberg 2011)
1. Aufrechterhaltung der proliferativen Signalübertragung
Normalerweise kontrolliert das Gewebe sorgfältig die Produktion und Freisetzung
von Wachstumssignalen. Dadurch entsteht eine Homöostase der Zellzahl, die die
Erhaltung der Gewebearchitektur ermöglicht. Durch Defekte in den negativen
Rückkopplungsmechanismen kann eine chronische Proliferation entstehen und
Krebszellen können durch diese Deregulierung unabhängig von den
Wachstumsfaktoren agieren.
2. Umgehung von wachstumshemmenden Signalen
Entartete Zellen müssen wachstumshemmende Faktoren umgehen können. Dazu
gehören beispielsweise Tumorsupressorgene, wie RB und TP53. Diese fungieren
ursprünglich als zentrale Elemente, die regeln, ob Zellen sich vermehren, Seneszenz
aktivieren oder in Apoptose gehen sollen.
3. Apoptoseresistenz
Um die Apoptose zu umgehen, entwickeln Tumorzellen eine Vielzahl von Strategien.
Häufig ist ein Verlust der TP53-Tumorsuppressorfunktion zu finden. Andererseits
können antiapoptotische Regulatoren (Bcl-2) oder Überlebenssignale (Igf1/2)
exprimiert oder erhöht und proapoptotische Faktoren (Bax, Bim) herunterreguliert
werden.
-5-4. Ermöglichung der replikativen Unsterblichkeit
Die Unsterblichkeit der entarteten Zellen kann auf verlängerte Telomere der DNA
zurückgeführt werden. Durch die stetige Verlängerung der Telomere kann die
Aktivierung der Seneszenz oder Apoptose vermieden werden. Oftmals wird dies
durch die gesteigerte Expression der Telomerase oder alternativ über eine
rekombinationsbasierte Aufrechterhaltung der Telomere erreicht.
5. Induktion der Angiogenese
Tumore benötigen, wie normales Gewebe, Nährstoffe und Sauerstoff, um zu
überleben. Darüber hinaus müssen Kohlendioxid und Stoffwechselabfälle
abtransportiert werden. Um dies zu ermöglichen, kann ein „angiogener Schalter“
aktiviert werden, welcher dazu führt, dass neue Gefäße wachsen. Diese können
dann das fortschreitende, neoplastische Wachstum unterstützen.
6. Aktivierung der Invasion und Metastasierung
Veränderungen in der Form von Krebszellen und in ihrer Bindung an andere Zellen
und der extrazellulären Matrix, z.B. durch den Verlust von E-Cadherin, führen zur
Invasion und Metastasierung dieser entarteten Zellen.
Grundlagen dieser sechs Eigenschaften sind Genominstabilität und Inflammation,
welche die Entstehung der o.g. Fähigkeiten einer Tumorzelle begünstigen. Zusätzlich
dazu sollten zwei weitere Merkmale, die Fähigkeit zur Neuprogrammierung des
Energiestoffwechsels und das Abwenden immunogener Destruktion, beachtet
werden. Durch die Möglichkeit der Umprogrammierung des Glukosestoffwechsels auf
die Glykolyse kann beispielsweise die Zellproliferation weiterhin stattfinden. Die
Zwischenprodukte der Glykolyse werden in verschiedene Biosynthesewege (z.B.:
Aminosäureproduktion) eingespeist. Dies wiederum führt zur Synthese von
Makromolekülen und Zellorganellen, welche für den Zusammenbau neuer Zellen
erforderlich sind (Hanahan und Weinberg 2011).
Besitzt eine Zelle einige oder alle der genannten Merkmale, besteht die Möglichkeit
der Entartung.
-6-2.2.1. Progressionsmodell
Die Zellen der Bauchspeicheldrüse durchlaufen eine Progression von normalem
Gangepithel über Vorläuferläsionen bis hin zum Karzinom. Intraepitheliale
Neoplasien (PanIN) sind ein Großteil der Vorläufer für infiltrierende Adenokarzinome
der Bauchspeicheldrüse. Eingeteilt werden die PanINs basierend auf dem
morphologischen Grad der Dysplasie – PanIN-1: geringgradig, PanIN-2: moderat,
PanIN-3: hochgradig (Wood und Hruban 2014). Pankreasgangläsionen mit diskreten
zytologischen Atypien, wie säulenförmiges Epithel, können eine aktivierende
Mutation im KRAS und eine erhöhte Expression von Her-2/neu aufweisen. Im
weiteren Verlauf erfolgt eine Inaktivierung des p16-Tumorsuppressorgens. Der
Zellaufbau wird zunehmend ungeordneter und es entstehen vermehrt nukläre
Atypien. Im letzten PanIN-Stadium tritt ein Verlust der Tumorsuppressorgene p53,
DPC4/SMAD und BRCA2 auf (Hruban et al. 2000, siehe Abbildung 3).
Normales PanIN-1: PanIN-2: PanIN-3: Duktales
Epithel Her-2/neu p16 p53, DPC4/SMAD, Adenokarzinom
KRAS BRCA2
Abbildung 3. Progressionsmodell für das Pankreaskarzinom. Die Überexpression von Her-
2/neu und eine aktivierende KRAS-Mutation treten im PanIN-1-Stadium auf, im Verlauf
erfolgt eine Inaktivierung der p16- (PanIN-2) und p53-, DPC4/SMAD- und BRCA2-
Tumorsuppressorgene (PanIN-3). (Quelle: in Anlehnung an Hezel et al. 2006 und Hruban et
al. 2000)
Ein geringer Anteil der duktalen Adenokarzinome des Pankreas entstehen hingegen
aus zystischen Vorläufern, wie der intraduktalen papillären muzinösen Neoplasie und
der muzinösen zystischen Neoplasien (Wood und Hruban 2014).
-7-2.3. Das Pankreaskarzinom
Durch maligne Entartung der Gangzellen des exokrinen Pankreas entstehen
Adenokarzinome, welche über 95% der Pankreaskarzinome ausmachen. Daneben
gibt es auch zystische, azinäre oder endokrine Tumore des Pankreas
(Leilinienprogramm 2013). Das duktale Adenokarzinom des Pankreas (PDAC) soll
Hauptgegenstand dieser Arbeit sein. Die Begrifflichkeit ‚Pankreaskarzinom‘ soll
hierbei insbesondere für PDAC stehen.
2.3.1. Epidemiologie
In Deutschland erkranken rund 18.400 Menschen jährlich an einem
Bauchspeicheldrüsenkrebs. Diese Erkrankung tritt im höheren Lebensalter auf, wie
man am mittleren Erkrankungsalter erkennen kann. Bei Männern wird das Karzinom
durchschnittlich mit 72 Jahren und bei Frauen mit 76 Jahren diagnostiziert. Bei dem
männlichen Geschlecht liegt die Inzidenz auf dem 10. Platz der Krebs-
neuerkrankungen, beim weiblichen Geschlecht auf dem 6. Platz, wobei der
Bauspeicheldrüsenkrebs die vierte Stelle aller Krebssterbefälle einnimmt (Zentrum
für Krebsregisterdaten 2016a). Die Mortalität entspricht annähernd der der Inzidenz.
Auch im Hinblick auf die 5-Jahres-Überlebensrate, welche beim Pankreastumor bei
9% liegt, wird deutlich, dass die Überlebenschance nach Feststellung der Krankheit
minimal ist (Zentrum für Krebsregisterdaten 2016b).
2.3.2. Ätiologie und Risikofaktoren
Die Entstehung des Bauchspeicheldrüsenkrebses ist noch nicht hinreichend geklärt
und bedarf weiterer Erforschung. Es gibt jedoch einige Risikofaktoren, die sich mit
der Zeit als wesentlich herausgestellt haben. Tabakkonsum, Adipositas und
exzessiver Alkoholismus stellen lebensartassoziierte Faktoren dar (Larsson et al.
2005, Talamini et al. 1999a). Daneben können auch andere Erkrankungen, wie
Diabetes Mellitus Typ II oder chronische Pankreatitis, die Entstehung eines solchen
Tumors beeinflussen (Huxley et al. 2005, Talamini et al. 1999b). Hereditäre
Syndrome, wie das Peutz-Jeghers-Syndrom, die hereditäre Pankreatitis, das
familiäre atypische multiple Muttermal- und Melanomsyndrom, das familiäre Mamma-
und Ovarialkarzinom aber auch das hereditäre nichtpolypöse kolorektale Karzinom
oder die familiäre adenomatöse Polyposis können eine Rolle in der Bildung eines
Pankreaskarzinoms spielen (Giardiello et al. 2000, Howes et al. 2004, Vasen et al.
2000, Friedenson 2005, Lilley und Gilchrist 2004, Giardiello et al. 1993).
-8-2.3.3. Diagonstik
Das Stellen der Diagnose ‚Pankreaskarzinom’ im Frühstadium ist schwierig, da
entsprechende Frühsymptome fehlen. Auch in späteren Stadien gibt es keine
eindeutigen Hinweise. Eine B-Symptomatik, also Fieber über 38°C, Nachtschweiß
und ungewollter Gewichtsverlust, kann Anhalt für ein Tumorgeschehen sein.
Symptome, wie neu aufgetretene gürtelförmige Rückenschmerzen, Appetitverlust,
ein schmerzloser Ikterus, sowie unspezifische Oberbauchschmerzen mit
Gewichtsverlust, können in Richtung des Vorhandenseins eines Tumors im Bereich
des Pankreas zeigen (Herold 2017). Verschließt der Tumor den Ductus choledochus,
kann das Courvoisier-Zeichen (tastbare prallelastische Gallenblase ohne
Schmerzreiz mit sichtbarem Ikterus) positiv ausfallen. Auch seltene Symptome, wie
eine pathologische Glukosetoleranz, ein neu aufgetretener Diabetes mellitus oder
eine erhöhte Thromboseneigung können auftreten (Herold 2017). Neben der
Anamnese spielt die körperliche Untersuchung eine wesentliche Rolle in der
Diagnostik. Gibt es dort Hinweise auf ein Pankreaskarzinom, sollten weitere
bildgebende Untersuchungen vorgenommen werden. Die Sonografie und die
Endosonografie sind Mittel der ersten Wahl (Leilinienprogramm 2013). Daneben
sollten auch laborchemische Parameter (Lipase, alkalische Phosphatase, ɣGT und
Bilirubin) und Tumormarker, wie das Carbohydrate-Antigen 19-9 oder das
Carcinoembryonale Antigen, präoperativ bestimmt werden (Bruch und Trentz 2008,
Nazli et al. 2000). Sie dienen auch im späteren Verlauf als Kontrolle (Distler et al.
2013, Herold 2017). Zur weiteren Abklärung oder bei einem starken Verdacht, stehen
neben CT und MRT auch MRCP oder ERCP zur Verfügung. Bei unklarem Status,
eingeschränkter Resektabilität oder einer palliativen Situation kann eine Biopsie
durchgeführt werden.
2.3.4. Tumorklassifikation
Um maligne Tumore, wie das Pankreaskarzinom, in der Medizin präzise einzuteilen,
wird die TNM-Klassifikation verwendet. Das TNM-System vereint die Ausdehnung
des Primärtumors (T), das Fehlen oder Vorhandensein von regionären
Lymphknotenmetastasen (N) und das Fehlen oder Vorhandensein von
Fernmetastasen (siehe Tabelle 1). Die TNM-Klassifikation kann zu unterschiedlichen
Zeiten während der Behandlung des Patienten ermittelt werden. Prätherapeutisch,
also nach klinischen Untersuchungen und bildgebenden Verfahren, wird die cTNM-
Klassifikation verwendet.
-9-Im Gegensatz dazu wird posttherapeutisch, nach Operation, die histopathologische
pTNM-Klassifikation verwendet (Bruch und Trentz 2008).
T Primärtumor
TX Primärtumor kann nicht beurteilt werden
T0 Kein Anhalt auf Primärtumor
Tis Carcinoma in situ
T1 Tumor begrenzt auf Pankreas, maximal 2 cm oder weniger im Durchmesser
T2 Tumor begrenzt auf Pankreas, 2 cm bis 4 cm im Durchmesser
T3 Tumor breitet sich jenseits des Pankreas aus, mindestens 4 cm im
Durchmesser
T4 Tumor infiltriert Truncus coeliacus, A. hepatica communis oder A. meseterica
superior
N Regionäre Lymphknoten
NX Regionäre Lymphknoten können nicht beurteilt werden
N0 Keine regionären Lymphknotenmetastasen
N1 Metastasen in 1 – 3 regionären Lymphknoten
N2 Metastasen in ≥ 4 regionären Lymphknoten
M Fernmetastasen
M0 Keine Fernmetastasen
M1 Fernmetastasen vorhanden
Tabelle 1. Aktuelle TNM-Klassifikation des Pankreaskarzinoms (modifiziert nach Cong et al.
2018).
Diese drei Aspekte werden zu vier Stadien gruppiert anhand derer man adäquate
therapeutische Entscheidungen aber auch prognostische Aussagen treffen kann
(siehe Tabelle 2). Am weitesten verbreitet ist die Einteilung nach der Union for
International Cancer Control zusammen mit dem American Joint Comittee on
Cancer.
- 10 -T N M
IA T1 N0 M0
IB T2 N0 M0
IIA T3 N0 M0
IIB T1-T3 N1 M0
III T4 jedes N M0
IV jedes T jedes N M1
Tabelle 2. Stadieneinteilung des Pankreaskarzinoms anhand der TNM-Klassifikation
(modifiziert nach Cong et al. 2018).
Mittels Grading kann ein Tumor nach seiner Malignität eingeteilt werden. Anhand
morphologischer Kriterien wird zwischen vier Stufen, von hochdifferenziert (G1) bis
undifferenziert (G4), unterschieden (Bruch und Trentz 2008).
Falls eine operative Entfernung des Tumors durchgeführt wird, beurteilt die
Residualtumorklassifikation (R-Klassifikation) den Resektionsrand nach Fehlen oder
Vorhandensein eines Resttumors (siehe Tabelle 3).
R-Status Residualtumor
RX Vorhandensein von Residualtumor kann nicht beurteilt werden
R0 Kein Residualtumor
R1 Mikroskopischer Residualtumor
R2 Makroskopischer Residualtumor
Tabelle 3. Residualtumorklassifikation.
2.3.5. Therapie
Um Therapieentscheidungen treffen zu können, wird das Pankreaskarzinom anhand
klinischer Untersuchung und bildgebenden Verfahren in drei – vier Kategorien
(resektabel, borderline, lokal- oder fernmetastasiert) eingeteilt (siehe Abbildung 4).
Da aktuell allein die Resektion des Pankreas potentiell kurativ ist, sollten resektable
Tumore operiert werden. Befindet sich das Karzinom im Kopf der
Bauchspeicheldrüse, besteht die Möglichkeit einer Kausch-Whipple-Operation oder
einer pyloruserhaltenden Pankreaskopfresektion (Distler und Grützmann 2012, Lin et
al. 2005). Basis beider operativer Verfahren ist die En-bloc-Entfernung des
tumortragenden Anteils des Pankreas. Die klassische Whipple-Operation beinhaltet
außerdem die Resektion der unteren 2/3 des Magens, des Duodenums inklusive der
- 11 -Gallenblase und der distalen Gallenwege und der proximalen Anteile des Jejunums
(Bruch und Trentz 2008, Henne-Bruns 2012). Im Gegensatz dazu bleiben bei der
pyloruserhaltenden Pankreaskopfresektion (PPPD) der Magen, der Pylorus und die
oberen 2 – 4 cm des Duodenums erhalten (siehe Abbildung 1). Die
Wiederherstellung des Abflusses der Pankreas- und Gallensäfte kann beispielsweise
durch eine Pankreatikojejunostomie und eine biliodigestive Anastomose
gewährleistet werden. Bei der Whipple-OP sollte zusätzlich z.B. eine Billroth-II-
Rekonstruktion erfolgen (Bruch und Trentz 2008, Henne-Bruns 2012). Eine subtotale
Pankreaslinksresektion oder eine totale Duodenopankreatektomie können beim
Pankreaskorpuskarzinom angewendet werden. Ist die Lokalisation im
Pankreasschwanz, wird eine Linksresektion, ggf. mit Splenektomie, durchgeführt
(Leitlinienprogramm 2013). Zusätzlich erfolgt bei allen Verfahren eine
Lymphknotendissektion. Ziel der Operationsverfahren sollte eine R0-Resektion sein,
da ein R1- oder R2-Status mit schlechterem Überleben assoziiert ist (Brunner et al.
2019). Nach der operativen Entfernung des Karzinoms wird leitliniengerecht eine
adjuvante Chemotherapie empfohlen. Je nach Zustand des Patienten kann entweder
eine Kombinationschemotherapie (z.B.: 5-Fluouracil mit Leucovorin, Irinotecan und
Oxaliplatin als FOLFIRINOX-Schema), Gemcitabin und/oder Capecitabin, eine
Vorstufe von 5-Fluouracil, für mindestens sechs Monate angewendet werden (Oettle
et al. 2007, Liao et al. 2013, Brunner et al. 2019).
Wird der Tumor als borderline eingestuft, sollte zuerst eine Biopsie durchgeführt
werden. Anschließend kann ggf. eine neoadjuvante (Radio-)Chemotherapie von
Vorteil sein, da durch ein down-staging des Tumors eine verbesserte Resektabilität
erzielt werden kann. Falls eine operative Exploration die Möglichkeit einer Resektion
zeigt, kann das Karzinom entfernt werden. Auch hier folgt eine adjuvante
Chemotherapie mit den oben genannten Substanzen (Brunner et al. 2019).
Ist der Primärtumor lokal metastasiert oder liegen Fernmetastasen vor, sollte
ebenfalls eine Biopsie durchgeführt werden. Anhand des Gesundheitszustands des
Patienten wird das weitere Therapieregime bestimmt. Die Primärbehandlung bei
Patienten mit ECOG 0 – 1 (Eastern Cooperative Oncology Group) wird, je nach
grundliegender Erkrankung, chemotherapeutisch mit FOLFIRINOX oder anderen
Kombinationen (z.B.: nab-Paclitaxel oder Capecitabin) mit Gemcitabin durchgeführt
(Klaiber et al. 2018, Sultana et al. 2007, Conroy et al. 2011). Sollte der ECOG-Status
höher sein, empfiehlt man Gemcitabin als Monotherapie. Nach dem ersten
Behandlungszyklus sollte ein Restaging erfolgen, um die Wirkung beurteilen zu
- 12 -können (Brunner et al. 2019). Schlägt diese Therapie nicht an oder kommt es zu
einem Progress der Grunderkrankung, sollte der Patient sekundär mit einem anderen
Chemotherapeutikum behandelt werden. Bei Patienten, die nicht mehr operativ oder
mittels Chemotherapie behandelt werden können, sollte eine palliative Versorgung
angestrebt werden. Dies umfasst auch die Behandlung von Komplikationen. Die
häufig auftretende Gallengangsobstruktion kann beispielsweise mittels Stent versorgt
werden.
Abbildung 4. Grobe Übersicht über den Behandlungsalgorithmus für Patienten mit
Adenokarzinom des Pankreas. (Quelle: Pelzer und Riess, 2015)
2.4. Protoonkogene
Protoonkogene kommen ubiquitär im menschlichen Organismus vor. Sie dienen der
Kontrolle und Steuerung von Wachstum, Teilung und Differenzierung der Zelle (Horn
et al. 2012). Beispiele für Protoonkogene sind Wachstumsfaktoren und deren
Rezeptoren, G-Proteine und Transkriptionsfaktoren. Solche Gene sind Teil eines
Signaltransduktionweges, bei dem durch Bindung eines Wachstumsfaktors an einen
extrazellulären Rezeptor ein Signal intrazellulär und spezifisch in den Zellkern
geleitet wird. Dort wird mittels Transkriptionsfaktoren die Transkription
zellteilungsspezifischer Gene aktiviert (Rassow et al. 2012). Findet nun eine
somatische Mutation (z.B.: Amplifikation, Deletions-, Punkt-, Insertionsmutation oder
Translokation) statt, kann das entweder zum Funktionsverlust dieses Gens und damit
zur Apoptose oder durch Aktivierung zum Onkogen führen. Durch diese Aktivierung
erhält die Zelle ein Funktionsgewinn, der mit einem Kontrollverlust einhergeht. Es
- 13 -findet eine übermäßige Expression statt. Die Zelle kann dadurch in der Lage sein,
sich unkontrolliert zu vermehren und zu teilen, was zur Tumorbildung beiträgt (Horn
et al. 2012, Rassow et al. 2012, Heinrich et al. 2014).
2.4.1. KRAS
Das KRAS ist ein Protoonkogen der RAS-Familie, welches zuerst im Kirsten rat
sarcoma virus entdeckt und identifiziert wurde. Es gehört zur Gruppe der G-Proteine.
Im Gegensatz zu den meisten heterotrimeren G-Proteinen liegt KRAS monomer vor.
Aktiviertes KRAS ist mit GTP beladen und kann Zielproteine und auch
Signaltransduktionsprozesse aktivieren. Gegenteilig sind mit GDP-beladene KRAS-
Proteine inaktiv. Durch Hilfsfaktoren, wie dem Guaninnukleotid-Austauschfaktor kann
GDP durch GTP ausgetauscht werden. Das Protoonkogen ist mittels Lipidanker an
dessen C-Terminus am inneren Blatt der Zellmembran befestigt (Rassow et al. 2012,
Hunter et al. 2015). Durch verschiedene nachgeschaltete Effektoren, wie z.B.
Raf/Mek/Erk, PI3K/PDPK1/Akt, RalGDS/p38MAPK, Rac, Rho und NF1, kann KRAS
eine Signalübertragung ermöglichen (Eser et al. 2014, siehe Abbildung 5).
Abbildung 5. Vereinfachte Darstellung des Ras Signalweges. (Quelle: in Anlehnung an Hezel
et al. 2006)
Die Funktion von KRAS liegt in der Kontrolle der Zellproliferation und –
differenzierung. Eine Punktmutation am Codon 12 (von GGT zu GAT; GTT oder
CGT) kann zu einer Inaktivierung der GTPase-Aktivität führen. Daneben kann es
auch zu Mutationen im Codon 13 oder 61 kommen. Durch die Mutation kommt es zur
Signalgebung und zur ständigen Weiterleitung nicht vorhandener Wachstumsreize
(Rassow et al. 2012, Hezel et al. 2006). Die aktivierende KRAS-Mutation greift zur
Übertragung von Signalen auf drei wesentliche Effektoren zurück: Raf, PI3K und
RalGDS. Die Raf-Kinase aktiviert durch eine Reihe von Phosphorylierungen die
MAPK/Erk-Kinase, welche zur Proliferation anregen kann. Durch den PI3K-
- 14 -Signalweg und dessen nachgeschaltete Effektoren (z.B.: Akt), kann das Überleben,
die Größe und die Proliferation von Zellen beeinflusst werden. Aktiviertes RAL fördert
die RAS-induzierte zelluläre Transformation (Hezel et al. 2006).
2.5. CRISPR/Cas-System
Schon im Jahr 1987 wurde von Wissenschaftlern der Osaka Universität eine
unbekannte Sequenz im Genom von Escherischia coli (E. coli) mit kurzen sich
wiederholenden Nukleotidsequenzen und angrenzenden einzigartigen Segmenten
beschrieben. Als in den letzten Jahren vermehrt das whole-genome sequencing
populär wurde, fand man CRISPR (clustered regularly interspaced short palindromic
repeat)-Sequenzen und CRISPR-assoziierte (Cas) Gene in verschiedensten
Bakterien und Archaeen (Charpentier und Doudna 2013). Die Entdeckung, dass
diese kurzen auffälligen Sequenzen der DNA von Viren oder Plasmiden ähneln, ließ
vermuten, dass es sich hierbei um ein adaptives Immunsystem der Bakterien
handelt, welches eine spezifische Abwehr gegen Angreifer bietet (Mojica et al. 2005).
CRISPR-Genloci umfassen ein CRISPR-Array mit identischen Wiederholungen und
eingelagerten Spacern, welche die CRISPR-RNA (crRNA) Komponenten kodieren.
Zudem gibt es ein Cas-Operon mit Genen, welches die Cas-Proteinkomponenten
verschlüsselt (Doudna und Charpentier 2014). Die crRNA bildet, je nach Typ, mit
einem oder mehreren Cas-Proteinen einen Komplex, um in die Virusproliferation
eingreifen zu können. Man vermutet, dass dieses adaptive Immunsystem dreiphasig
abläuft. In der ersten Phase findet die Insertion einer kurzen Angreifer-DNA-Sequenz
in den CRISPR-Komplex statt, diese dient dann als Spacer. Danach erfolgt die
Transkription einer Präkursor-crRNA (prä-crRNA), welche durch Reifung zu einer
individuellen crRNA wird. Die reife crRNA besteht dann aus einem Repeat- und
einem Spacer-Anteil. Der Spacer dient der Erkennung der fremden DNA. Zuletzt
kann durch Komplementarität der crRNA zur Angreifer-DNA eine Spaltung dieser
fremden DNA durch die Cas-Proteine stattfinden (Doudna und Charpentier 2014, van
der Oost et al. 2014). Die Tätigkeit vieler CRISPR-Systeme beruht auf dem
Vorhandensein von einem sequenzspezifischen protospacer adjacent motif (PAM),
welches in der Nähe der Ziel-crRNA Sequenz der eingedrungenen DNA liegt
(Doudna und Charpentier 2014). Basierend auf den unterschiedlichen Komponenten
und der Art der Funktion kann man das CRISPR/Cas-System in drei große Gruppen
aufteilen. Typ I und III des CRISPR/Cas-Systems enthalten mehrere Cas-Proteine,
die Komplexe mit der entsprechenden crRNA bilden (CASCADE-Komplex für Typ I
- 15 -und Cmr- oder Csm-RAMP-Komplex für Typ III), um die Erkennung und Zerstörung
der Ziel-DNA zu erleichtern. Bei den beiden Typen wird außerdem das prä-crRNA-
Transkript durch CRISPR-assoziierte Ribonukleasen gespalten, wodurch mehrere
kurze crRNA freigesetzt werden. Diese crRNA-Intermediate werden bei CRISPR Typ
III zusätzlich durch RNasen am 3‘-Ende weiter prozessiert, um das voll ausgereifte
Transkript zu erzeugen. Im Gegensatz dazu weist Typ II eine deutlich reduzierte
Anzahl an Cas-Proteinen auf. Es erfolgt eine Hybridisierung der transaktivierenden
CRISPR-RNA (tracrRNA) mit der crRNA zu einem tracrRNA:prä-crRNA-Duplex.
Dieser Duplex wird von RNase III und anderen Nukleasen zur reifen crRNA
prozessiert. Zusammen mit Cas9 wird ein Komplex gebildet, der die DNA abbauen
kann (Hsu et al. 2014).
2.5.1. CRISPR/Cas9
Das CRISPR-assoziierte Protein 9 (Cas9) wird am häufigsten unter den Cas-
Proteinen für genomische Veränderungen verwendet. Bei Cas9 handelt es sich um
eine RNA-gesteuerte Endonuklease. Diese ist Teil des CRISPR-Systems vom Typ II.
Es gibt eine große Vielfalt von Cas9 in verschiedenen bakteriellen CRISPR-
Systemen. Die Größe der Cas9-Nukleasen reicht von etwa 900 bis 1.600
Aminosäuren (Hsu et al. 2014). Die am häufigsten für das Genom-Engineering
verwendete Nuklease ist vom Streptococcus pyogenes (Wang et al. 2016). Cas9
setzt sich aus dem Nuklease(NUC)-Teil und dem α-helikalen Erkennungs(REC)-Teil
zusammen. Der NUC-Bereich umfasst die HNH-Nukleasedomäne, die den Ziel-DNA-
Strang spaltet, die RuvC-ähnliche Nukleasedomäne, die den Nicht-Ziel-Strang
spaltet, eine PAM-interagierende Domäne, die mit der PAM-Region der Angreifer-
DNA interagiert und zur Zielspezifität von Cas9 beiträgt, und eine Wedge-Domäne,
die wichtig für die orthogonale Erkennung der crRNA ist. Der REC-Bereich enthält
die Regionen, die zur Erkennung von crRNA:Angreifer-DNA-Duplexen benötigt
werden (Wang et al. 2016). Die Cas9-vermittelte Spaltung der Angreifer-DNA wird
durch eine Zusammenarbeit von zwei RNAs gesteuert. Es gibt einerseits die crRNA,
die die fremde DNA durch eine Watson-Crick-Basenpaarungsregion von etwa 20
Basenpaaren erkennt. Andererseits wird die tracrRNA gebraucht, die mit der crRNA
hybridisiert und entscheidend für die crRNA-Prozessierung und die Cas9-Bindung ist
(Hsu et al. 2014). Um dieses System zu vereinfachen, kann die crRNA:tracrRNA-
Verbindung in eine modifizierbare, chimäre Single-Guide-RNA (sgRNA) vereinigt
werden (Jinek et al. 2012). Der Cas9-sgRNA-Komplex ist in der Lage an jede Stelle
- 16 -der DNA zu binden, die eine Basenpaarung mit der sgRNA eingehen kann und an
eine PAM-Sequenz angrenzt. Dadurch entsteht ein leicht programmierbares System
zur Bearbeitung von Genen. Die Bindung der Einheit an der Ziel-DNA induziert
Doppelstrangbrüche, die die DNA-Reparatur beispielsweise durch nichthomologes
Endjoining (NHEJ) oder Homologie-gesteuerte Reparatur auslöst. NHEJ verursacht
Insertions- und/oder Deletionsmutationen an der Doppelstrangbruchstelle und kann
somit zum Kock-out von Genen führen. Durch Anpassen der sgRNA, damit diese
sich mit der DNA-Sequenz von Interesse paart, kann das CRISPR-System die Basis
eines Werkzeugs zur Veränderung des Genoms bilden (Wang et al. 2016).
2.6. Zielsetzung der Arbeit
Das Pankreaskarzinom ist aktuell Objekt der Forschung. Durch die fehlende
Frühsymptomatik, die fast unbekannte Ätiologie, die komplexe Therapie und die
schlechte 5-Jahres-Überlebensrate, muss man sich dieser Thematik widmen. Wie wir
nach derzeitigem Forschungsstand wissen, findet in circa 90% der
Pankreaskarzinome eine Aktivierung des Protoonkogens KRAS statt. Dies führt zur
Proliferationssteigerung und damit zum unkontrollierten Wachstum des Gewebes.
Mithilfe der neuen CRISPR/Cas-Methode, die es uns ermöglicht, in das vorhandene
Genom einzugreifen, wollten wir versuchen, ob dies prinzipiell in Zelllinien des
Pankreaskarzinoms möglich ist.
Dadurch stellen sich folgende Fragen:
- Funktioniert ein CRISPR/Cas9-vermittelter Knock-out von KRAS-Mutationen in
Pankreaszelllinien?
- Wie verhalten sich die behandelten Zelllinien (Klone) im Vergleich zum Wildtyp?
- In wie weit differenzieren sich Proteine der Klone von dem des Wildtyps?
- 17 -2.7. Methoden
Um die Zielsetzung der Arbeit praktisch umsetzen zu können, wurden in KRASG12D
heterozygote Zelllinien verwendet. Durch Nutzung von heterozygoten Zelllinien ist es
möglich, dass das mutierte KRAS mittels der CRISPR/Cas9 Methode ausgeschaltet
wird und sich durch einen loss of function/ expression auszeichnet. Das Wildtyp (WT)
KRAS bleibt hier erhalten und kann das Protein exprimieren.
Neben den im Paper veröffentlichten Methoden kommt es auf einen genauen Ablauf
der Transfektion an (siehe Tabelle 4). Die zu verwendenden Zelllinien werden am
ersten Tag mit einer Konzentration von 2,5x105 Zellen/Well ausplattiert. Die
Inkubationszeit beträgt 24 Stunden unter Kulturbedingungen. Am Tag danach findet
die Transfektion mittels Lipofectamine 3000 statt. Hierbei entstehen durch das
kationische Lipid, das die anionische Nukleinsäure komplexiert, Liposomen, die
durch ihre Lipophilie und ihre Ladung durch die Phospholipid-Doppelschicht der
Zellmembran nach intrazellulär gelangen. Das Lipofectamine 3000 wird nach
Herstelleranleitung vorbereitet und die Plasmid-DNA hinzugefügt. Nach einer
Inkubationszeit von 10 – 15 Minuten bei Raumtemperatur gibt man den DNA-Lipid-
Komplex zu den ausplattierten Zellen. Am Tag nach der Transfektion erfolgt ein
Mediumwechsel. Da das Plasmid durch den Vektor eine Puromycin-Resistenz
besitzt, wird Puromycin (Konzentration: 5ng/l) als Selektionsdruck verwendet. Um
die Überlebenschance der Zellen mit Plasmid zu erhöhen, wird außerdem Maus
Epidermaler Wachstumsfaktor (mEGF) hinzugefügt. Nach zwei Tagen Ruhe steht ein
erneuter Mediumwechsel, inkl. Gabe von mEGF, an. An Tag 8 werden die erfolgreich
transfizierten Zellen vereinzelt. Die Einzelzellklone werden über einen längeren
Zeitraum hochgezogen und dann mittels Protein- und DNA-Analyse verifiziert.
Zeitpunkt Aufgabe
Tag 1 Ausplattieren der Zelllinien
Tag 2 Transfektion mittels Lipofectamine 3000
Tag 3 Selektion mittels Puromycin, Gabe von mEGF
Tag 4 Ruhe
Tag 5 Ruhe
Tag 6 Mediumwechsel mit Gabe von mEGF, Weglassen von Puromycin
Tag 7 Ruhe
Tag 8 Ausplattieren der Klone (1 Zelle/Well)
Tabelle 4. Zeitlicher Ablauf der Transfektion.
- 18 -3. Publikation
Titel: CRISPR/Cas9 mediated knock-out of KRASG12D
mutated pancreatic cancer cell lines
Authors: Eva Lentsch, Lifei Li, Susanne Pfeffer, Arif B. Ekici,
Leila Taher, Christian Pilarsky, Robert Grützmann
Published: 14. November 2019
Journal: International Journal of Molecular Sciences
DOI: 10.3390/ijms20225706
- 19 -International Journal of
Molecular Sciences
Article
CRISPR/Cas9-Mediated Knock-Out of KrasG12D
Mutated Pancreatic Cancer Cell Lines
Eva Lentsch 1 , Lifei Li 2 , Susanne Pfeffer 1 , Arif B. Ekici 3 , Leila Taher 2 ,
Christian Pilarsky 1, * and Robert Grützmann 1, *
1 Department of Surgery, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) and
Universitätsklinikum Erlangen, 91054 Erlangen, Germany; eva.pilarsky@googlemail.com (E.L.);
susanne.pfeffer@uk-erlangen.de (S.P.)
2 Division of Bioinformatics, Department of Biology, Friedrich-Alexander Universität
Erlangen-Nürnberg (FAU), 91054 Erlangen, Germany; lilifeifiona@hotmail.com (L.L.);
leila.taher@fau.de (L.T.)
3 Institute of Human Genetics, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) and
Universitätsklinikum Erlangen, 91054 Erlangen, Germany; arif.ekici@uk-erlangen.de
* Correspondence: christian.pilarsky@uk-erlangen.de (C.P.); robert.gruetzmann@uk-erlangen.de (R.G.)
Received: 5 September 2019; Accepted: 9 November 2019; Published: 14 November 2019
Abstract: In 90% of pancreatic ductal adenocarcinoma cases, genetic alteration of the proto-oncogene
Kras has occurred, leading to uncontrolled proliferation of cancerous cells. Targeting Kras has
proven to be difficult and the battle against pancreatic cancer is ongoing. A promising approach to
combat cancer was the discovery of the clustered regularly interspaced short palindromic repeat
(CRISPR)/CRISPR-associated (Cas) system, which can be used to genetically modify cells. To assess
the potential of a CRISPR/CRISPR-associated protein 9 (Cas9) method to eliminate Kras mutations in
cells, we aimed to knock-out the c.35G>A (p.G12D) Kras mutation. Therefore, three cell lines with
a heterozygous Kras mutation (the human cell lines SUIT-2 and Panc-1 and the cell line TB32047
from a KPC mouse model) were used. After transfection, puromycin selection and single-cell
cloning, proteins from two negative controls and five to seven clones were isolated to verify the
knock-out and to analyze changes in key signal transduction proteins. Western blots showed a
specific knock-out in the KrasG12D protein, but wildtype Kras was expressed by all of the cells. Signal
transduction analysis (for Erk, Akt, Stat3, AMPKα, and c-myc) revealed expression levels similar
to the wildtype. The results described herein indicate that knocking-out the KrasG12D mutation
by CRISPR/Cas9 is possible. Additionally, under regular growth conditions, the knock-out clones
resembled wildtype cells.
Keywords: CRISPR/Cas9 system; G12D; Kras; Suit-2; Panc-1; TB32047
1. Introduction
Pancreatic cancer (PaCa) is the fourth leading cause of all cancer death in the United States
of America and Germany. Smoking, chronic pancreatitis, heavy alcohol consumption or genetic
predispositions, such as Lynch syndrome, are only a small variety of risk factors. Common symptoms,
including weight loss, abdominal or back pain, mean that identification during the early stages can be
difficult. With its disastrous five-year survival rate of about 8%, the chances of survival after diagnosis
are remote. Besides chemotherapy or radiation therapy as therapeutic options to alleviate pain or
extend survival, the only potentially curative prospect remains resection [1–4]. Therefore, a better
understanding of the functions of the tumor is fundamental to develop new treatment strategies and
identify new therapeutic targets.
Int. J. Mol. Sci. 2019, 20, 5706; doi:10.3390/ijms20225706 www.mdpi.com/journal/ijmsInt. J. Mol. Sci. 2019, 20, 5706 2 of 11
In about 90% of PaCa cases, a Kras mutation is detectable [5]. Kras is a proto-oncogene first
Int. J. Mol. Sci. 2019, 20, x 2 of 11
discovered and identified in Kirsten rat sarcoma virus. It is part of the G-protein family. In contrast
to most heterotrimeric G proteins, Kras is monomeric. As a central element in multiple signaling
to most heterotrimeric G proteins, Kras is monomeric. As a central element in multiple signaling
pathways, it is involved in the regulation of cell proliferation, differentiation, and survival. A point
pathways, it is involved in the regulation of cell proliferation, differentiation, and survival. A point
mutation
mutationininthe theprotein
proteinleads
leadstotoimpairment
impairment of of guanosine triphosphate(GTP)ase
guanosine triphosphate (GTP)aseactivity
activityand andthus
thus
continuous
continuous transmission
transmission of non-existent growthgrowth
of non-existent signals [6]. Activated
signals [6]. mutations
Activated inmutations
the proto-oncogene
in the
Kras are a hallmark of PaCa. Its activity leads to a major role in
proto-oncogene Kras are a hallmark of PaCa. Its activity leads to a major role in PaCa PaCa initiation but itsinitiation
importance butin
maintaining
its importance the in tumor is uncertain
maintaining [7–9]. is uncertain [7–9].
the tumor
InInrecent years, whole-genome
recent years, whole-genome sequencing led toled
sequencing the discovery of clustered
to the discovery regularly interspaced
of clustered regularly
short palindromic repeat (CRISPR) sequences and CRISPR-associated
interspaced short palindromic repeat (CRISPR) sequences and CRISPR-associated (Cas) genes in (Cas)
various bacteria
genes in
and archaea
various [10]. The
bacteria andidentification
archaea [10]. of Thethese sequences,ofresembling
identification DNA from
these sequences, viruses DNA
resembling or plasmids,
from
suggests
viruses that it is a bacterial
or plasmids, suggestsadaptive
that it isimmune system
a bacterial that immune
adaptive providessystem
particular
that defense
providesagainst viral
particular
intruders [11–13]. CRISPR-associated protein 9 (Cas9) is the most commonly
defense against viral intruders [11–13]. CRISPR-associated protein 9 (Cas9) is the most commonly used among the Cas
used among
proteins. the Cas proteins.
Cas9-mediated cleavage of Cas9-mediated
DNA operatescleavagewith twoofRNAs:DNA (i) operates
CRISPR-RNAwith two RNAs:which
(crRNA), (i)
CRISPR-RNA
recognizes foreign (crRNA), which recognizes
DNA through foreign DNA
a complementary regionthrough
20-basea complementary regioncalled
pairs (bps) in length, 20-base the
pairs (bps) in length, called the proto-spacer adjacent motif (PAM),
proto-spacer adjacent motif (PAM), and (ii) trans-acting CRISPR-RNA (tracrRNA), which hybridizes and (ii) trans-acting
CRISPR-RNA
with the crRNA (tracrRNA), which hybridizes
[14,15]. To facilitate this system,with thethe crRNA [14,15]. To
crRNA–tracrRNA facilitate
fusion thiscombined
can be system, the into
crRNA–tracrRNA
a chimeric single-guide fusion
RNA can(sgRNA)
be combined [16]. into a chimeric
By creating single-guide region
a 20-nucleotide RNA (sgRNA)
complementary[16]. Byto
creating
a DNA a 20-nucleotide
sequence of interestregion complementary
in the sgRNA, Cas9 to can a DNA
be aimedsequence
at anyofgenomic
interest in the with
locus sgRNA, Cas9
a suitable
can be aimed at any genomic locus with a suitable PAM sequence.
PAM sequence. After DNA tracking and cleavage, repair mechanisms can provoke insertion and After DNA tracking and cleavage,
repair mutations
deletion mechanisms can provoke
(indels), which mayinsertion
causeand deletion mutations
a knock-out (indels), whichproviding
[17]. With CRISPR/Cas9 may cause a newa
knock-out [17]. With CRISPR/Cas9 providing a new opportunity for modifying genes in general and
opportunity for modifying genes in general and the armed with the knowledge of overexpression of
the armed with the knowledge of overexpression of Kras in PaCa, the possible knock-out of mutated
Kras in PaCa, the possible knock-out of mutated Kras was investigated.
Kras was investigated.
2. Results
2. Results
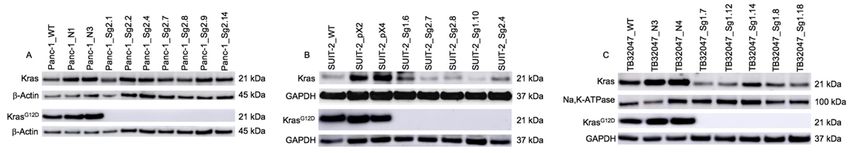
2.1. Expression of KrasG12D and Total Ras after CRISPR/Cas9-Mediated Knock-Out in PaCa Cell Lines
2.1. Expression of KrasG12D and Total Ras after CRISPR/Cas9-Mediated Knock-Out in PaCa Cell Lines
To verify the role of mutant Kras in pancreatic cancer cell lines, two human (Panc-1 and SUIT-2)
To verify the role of mutant Kras in pancreatic cancer cell lines, two human (Panc-1 and SUIT-2)
and a murine cell line (TB32047) were used to perform a CRISPR/Cas9-based knock-out of the mutated
and a murine cell line (TB32047) were used to perform a CRISPR/Cas9-based knock-out of the
allele. All cell lines contained a heterozygous KRASG12D (c.35G>A) mutation. After transfection,
mutated allele. All cell lines contained a heterozygous KRASG12D (c.35G>A) mutation. After
puromycin selection and single-cell cloning, we produced about five to seven clones (Kras clones).
transfection, puromycin selection and single-cell cloning, we produced about five to seven clones
Western blotting was performed to detect the knock-out effects introduced with the CRISPR/Cas9 gene
(Kras clones). Western blotting was performed to detect the knock-out effects introduced with the
editing system.
CRISPR/Cas9 gene In allediting
tested cell lines,Inthe
system. all results showed
tested cell lines,athe
knock-out with noaprotein
results showed expression
knock-out with noof
mutant G12D
proteinprotein Krasof mutant
expression . However,
protein aKras
normal total Krasaprotein
G12D. However, expression
normal total level was
Kras protein observed
expression levelfor
Panc-1 wildtype (WT) cell lines, negative controls (transfected with nonsense sgRNA
was observed for Panc-1 wildtype (WT) cell lines, negative controls (transfected with nonsense molecules; NCs)
and Kras clones. In the SUIT-2 cell line, WT and Kras clones resembled each other, whereas
sgRNA molecules; NCs) and Kras clones. In the SUIT-2 cell line, WT and Kras clones resembled each the protein
expression in the the
other, whereas negative
protein controls was higher.
expression Total Kras
in the negative expression
controls in the TB32047
was higher. clones
Total Kras was lower
expression in
than
thein the WTclones
TB32047 and negative
was lower controls
than in(Figure
the WT1). and negative controls (Figure 1).
Figure
Figure Kras
1. 1. expression
Kras by wildtype
expression cells,cells,
by wildtype negative controls
negative and clones
controls in Panc-1,
and clones SUIT-2SUIT-2
in Panc-1, and TB32047
and
cell lines. (A)
TB32047 cell Western
lines. (A)blots (WBs)
Western showed
blots (WBs) similar
showedbands
similarinbands
Panc-1. (B) SUIT-2
in Panc-1. negative
(B) SUIT-2 controls
negative
(N;controls
pX) expressed
(N; pX)more Kras protein
expressed than protein
more Kras wildtypethan(WT) cells and(WT)
wildtype clones. (C)and
cells TB32047 clones
clones. produced
(C) TB32047
slightly
clonesless Kras protein
produced than
slightly lesstheir
KrasWT and NC.
protein thanNo protein
their WT and expression, indicating
NC. No protein KrasG12Dindicating
expression, knock-out,
could
Krasbe
G12Dobserved
knock-out,in Panc-1,
could beSUIT-2 and TB32047
observed cell lines.
in Panc-1, SUIT-2β-actin, glyceraldehyde
and TB32047 3-phosphate
cell lines. β-actin,
dehydrogenase
glyceraldehyde(GAPDH) served
3-phosphate as loading controls.
dehydrogenase (GAPDH) served as loading controls.
2.2. DNA Sequencing from Knocked-Out ClonesInt. J. Mol. Sci. 2019, 20, 5706 3 of 11
2.2. DNA Sequencing from Knocked-Out Clones
DNA sequencing was performed on all knock-out clones to confirm the presence of the target
mutation in the sequence of KrasG12D . All knock-out clones had indel mutations in the sequence of
KrasG12D . Most clones (Panc-1 2.1, 2.2, 2.8 and 2.9; SUIT-2 1.6, 2.7, 2.8, and 1.10; TB32047 1.7, 1.12, 1.14,
and 1.18) had small indels (< 20 bp), while Panc-1 clones 2.7 and 2.14 had single-nucleotide insertions.
Only three clones, one from each cell line (Panc-1 2.4, SUIT-2 2.4 and TB32047 1.8), had large indels
(>20 bp) (see Table 1).
Table 1. Insertion and deletion mutation (indel) size of Panc-1, SUIT-2 and TB32047 clones. Indel size
varies between single nucleotide indel, small: < 20 base pair (bp) and large > 20 bp inserts
and/or deletions.
Panc-1 Clones Indel Size SUIT-2 Clones Indel Size TB32047 Clones Indel Size
2.1 small 1.6 small 1.7 small
2.2 small 2.7 small 1.12 small
2.4 large 2.8 small 1.14 small
2.7 single nucleotide 1.10 small 1.8 large
2.8 small 2.4 large 1.18 small
2.9 small
2.14 single nucleotide
2.3. Expression of Key Signal Transduction Proteins in CRISPR/Cas9-Edited PaCa Cell Lines
After confirming the KrasG12D protein knock-out, the effect on key signal transduction pathways
was analyzed. Specifically, the (non-)phosphorylated proteins Erk, Akt, Stat3, AMPKα and c-myc were
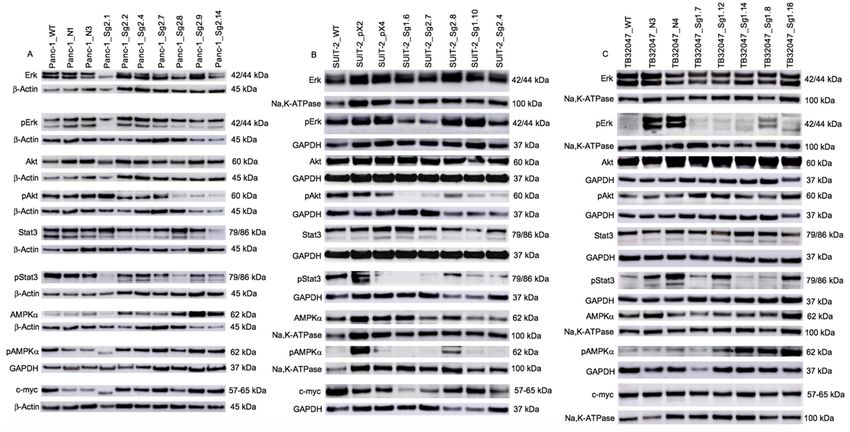
evaluated (Figure 2). We observed a heterogeneous expression pattern. Within Panc-1 cell clones pErk
levels remained comparatively stable, however pAkt, pStat3, pAMPKα and c-myc levels were reduced
in some Kras clones. This is contrary to the findings from the other cell lines. In the human cell line
SUIT-2, we observed a loss of Akt phosphorylation while Erk phosphorylation was retained. pStat3
and pAMPKα proteins were only produced by clone 2.6 and c-myc levels were reduced in some Kras
clones. However, within the murine KPC cell line TB32047, we found that the pErk levels and the
phosphorylation of Akt and c-myc remained constant, the pStat3 protein was only produced by two
clones, and most of the clones expressed pAMPKα.were reduced in some Kras clones. This is contrary to the findings from the other cell lines. In the
human cell line SUIT-2, we observed a loss of Akt phosphorylation while Erk phosphorylation was
retained. pStat3 and pAMPKα proteins were only produced by clone 2.6 and c-myc levels were
reduced in some Kras clones. However, within the murine KPC cell line TB32047, we found that the
pErk
Int. levels
J. Mol. and 20,
Sci. 2019, the5706
phosphorylation of Akt and c-myc remained constant, the pStat3 protein4 was
of 11
only produced by two clones, and most of the clones expressed pAMPKα.
Figure
Figure2.2.Expression
ExpressionofofErk,
Erk,Akt,
Akt,Stat3,
Stat3,AMPKα
AMPKα andand c-myc proteinsby
c-myc proteins bywildtype
wildtypecells,
cells,negative
negative
controls
controls and Kras clones in Panc-1, SUIT-2 and TB32047 cell lines. Here, the prefix “p” indicates the the
and Kras clones in Panc-1, SUIT-2 and TB32047 cell lines. Here, the prefix “p” indicates
phosphorylated
phosphorylated version
versionofofthe
the protein. (A)Panc-1
protein. (A) Panc-1 2.1,
2.1, 2.82.8
andand
2.142.14 expressed
expressed slightly
slightly less
less Erk Erk
than thethan
the others.
others. Clone
Clone 2.1 also
2.1 also produced
produced less pErk.
less pErk. Akt expression
Akt expression in showed
in Panc-1 Panc-1 showed no difference
no difference between
WT, NC and clones. pAkt was reduced in Panc-1 clones 2.8, 2.9 and 2.14. Stat3 was expressed by all
Panc-1 clones, except 2.14. Panc-1 showed reduced pStat3 expression in clone 2.1, 2.8 and 2.14. Lower
AMPKα expression could be observed in Panc-1 WT, NC and clone 2.1. The pAMPKα expression level
was lower in clone 2.1 than the other samples. Panc-1 NC and 2.1 produced less c-myc compared to
the others. (B) SUIT-2 clones expressed Erk similar to the wildtype cells. SUIT-2 clones 1.6 and 2.7
produced less pErk. Akt expression in SUIT-2 cells showed no difference in expression between WT,
NC and clones. Only WT cells, NC and clone 2.8 expressed pAkt. Clone 1.10 produced decreased
Stat3 levels. pStat3 was only produced by SUIT-2 WT, pX2 and clone 2.6. Lower AMPKα expression
could be observed in clones 1.10 and 2.4 compared to the WT cells. Just SUIT-2 pX2 and clone 2.6
produced pAMPKα. SUIT-2 clones 1.6, 2.7 and 2.4 produced less c-myc compared to the others. (C) Erk
expression in TB32047 clones was similar to the wildtype. pErk was only expressed by TB32047 NC.
Akt, pAkt, Stat3, AMPKα and c-myc protein levels were similar throughout all TB32047 samples.
pStat3 was only produced by TB32047 NC, clones 1.12 and 1.18. TB32047 clones 1.12, 1.14, 1.8 and 1.18
expressed higher pAMPKα than the remaining samples. β-Actin, GAPDH and Na,K-ATPase served as
loading controls.
2.4. TB32047 RNA Sequencing Results
Since the edited TB32047 cells demonstrated some changes in the expression levels of key signal
transduction proteins, we further investigated the differential gene expression of the CRISPR/Cas9
knock-out in this model system. RNA sequencing (RNA-seq) data from eight TB32047 samples (WT,
N3, N4, sg1.7, sg1.8, sg1.12, sg1,14, and sg1.18) were analyzed to determine RNA expression changes
in the KrasG12D knock-outs. Based on their whole transcriptomes, the WT and NC samples grouped
together and differed from the KrasG12D knock-out samples (see Figure 3A). A total of 417 genes were
differentially expressed between the two sample groups (see Supplementary data). Principal component
analysis (PCA) based on the expression of the differentially expressed genes (DEGs) confirmed that
most of the variance in the data (69.9%, PC1) was associated with expression changes between the
KrasG12D knock-out samples and the WT and NC samples (PC1, see Figure 3B). In addition, KrasG12D
knock-out samples were separated into two groups, although these differences were comparatively
smaller (9.4% of the variance, PC2). Approximately half of the DEGs (218) were up-regulated, while
the other half (199) were down-regulated (see Figure 3C). Among the down-regulated DEGs wereSie können auch lesen

















































