MARCO Institute for Clinical ...
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

22. Jahrgang • März • 1/2020
PM QM
Fachzeitschrift für
pharmazeutische Medizin
und Qualitätsmanagement 1
ZUR SACHE
Umgang mit Gesundheitsdaten
nach Inkrafttreten der DSGVO
BERICHTE + ANALYSEN + MEINUNGEN
ICH Guideline E19 – cui bono?
ARZNEIMITTELPRÜFUNG
Prozessmanagement zur Gewährleistung
der Datenqualität in klinischen Prüfungen
ARZNEIMITTELPRÜFUNG
Datenmanagement und Statistik stellen die Weichen für eine erfolgreiche Studiendurchführung
Prozessmanagement zur Ge-
währleistung der Datenqualität
in klinischen Prüfungen
Jede klinische Prüfung steht oder fällt mit der Qualität der erhobenen Daten. Hierbei sind insbesondere Daten-
Management (DM) und Statistik wichtige Partner, mit denen im Dialog frühzeitig die Weichen für eine erfolg-
reiche Studiendurchführung gestellt werden müssen. Eine konsequente Studienplanung berücksichtigt die
vielseitigen Möglichkeiten und Werkzeuge, die modernes DM und Statistik zum Studien-Monitoring bieten.
Einzelheiten dazu werden nachfolgend vorgestellt. In diesem Zusammenhang wird auch die Funktion des
Studien- bzw. Projektmanagers beleuchtet, der möglichst frühzeitig sowohl die Verantwortlichkeiten der
beteiligten Organisationen und Projektteams als auch die Interaktionen der Projektbeteiligten untereinander
klärt und definiert, um einen reibungslosen Informationsfluss und Datenaustausch zu gewährleisten.
| Dr. Manfred Wargenau, M.A.R.C.O. GmbH & Co. KG, Düsseldorf
Datenintegrität DM-CRO zum Sponsor nach vorab bandendaten (Case Report Form –
definierten Kriterien stattfindet. CRF) (Abbildung 1). Dabei ist insbe-
Von unterschiedlichen Seiten Die Qualität der Daten in einer sondere für den Einsatz eines elek-
(GCP- und ICH-Richtlinien, regula- Datenbank bemisst sich neben den tronischen CRF (eCRF) zu beachten,
torische Anforderungen) werden korrekten und vollständigen Ein- dass das eCRF-System vor Einschluss
hohe Ansprüche an die Datenqua- trägen auch nach der Reliabilität des ersten Patienten entwickelt, im-
lität/Datenintegrität in klinischen (Zuverlässigkeit) der zugrunde lie- plementiert, getestet und validiert
Prüfungen gestellt (siehe Tabelle 1). genden Messungen. Die Reliabilität ist. Dies erfordert in der Regel eine
Selbstverständlich liegt es auch im kann zum Beispiel eingeschränkt längere Vorbereitungsphase ver-
vitalen Interesse des Sponsors, eine sein, wenn ein analytisches Assay glichen mit dem Einsatz eines Pa-
hohe Datenqualität zu erreichen eine hohe Variabilität der Mess- pier-CRF. Dieser vermeintliche Zeit-
und die Daten in einer gut organi- ergebnisse liefert oder nicht hin- verlust, der bis zum Start der klini-
sierten und dokumentierten fina- reichend spezifisch ist. schen Studie in Kauf zu nehmen ist,
len klinischen Datenbank nach Ab- Das Kriterium der Nachvollzieh- wird allerdings mehr als aufgewo-
schluss der klinischen Studie zur Ver- barkeit bezieht sich auf die Doku- gen durch eine stark verkürzte Zeit
fügung zu haben. mentation von Datenänderungen zwischen dem Ende der klinischen
in der Datenbank (Audit Trail), die Phase (Last Patient Last Visit – LPLV)
neben der Änderung selbst auch und der Schließung der Datenbank,
Der Wert und Nutzen einer kli-
den Autor und den Zeitpunkt be- da die Daten bereits während der
nischen Studie, deren Evidenz und
inhaltet. Dateneingabe unmittelbar auf Feh-
Aussagekraft begründen sich maß-
lerfreiheit und Konsistenz geprüft
geblich auf die Qualität und Integri-
werden.
tät der erhobenen Daten.
Daten-Management-Schnittstellen
Im Fall, dass das Daten-Manage- Eine wichtige Voraussetzung zur Datenintegrität
ment vom Sponsor an eine DM-CRO Erreichung einer hohen Datenquali-
(Contract Research Organization) tät in der finalen klinischen Daten-
beauftragt wird, ist mithin rechtzei- bank ist eine gründliche und umfas- • Korrektheit
tig zu regeln, dass nach Abschluss sende Planung der Studie unter Ein- • Vollständigkeit
• Reliabilität
der Studie – spätestens nach Fina- beziehung aller Studienbeteiligten.
• Nachvollziehbarkeit
lisierung des klinischen Studien- Dies bezieht sich auf die Erstellung
berichts – ein zeitnaher Transfer des Prüfplans und des Erhebungs-
der finalen Studiendatenbank vom bogens für die Patienten- bzw. Pro- Tabelle 1: Kriterien zur Datenintegrität.
22 | PM QM 2020 | Jahrgang 22 | Heft 1 | März
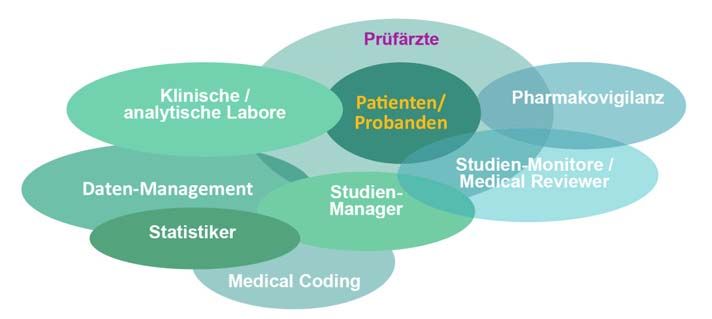
Das Zusammenspiel aller Projekt- Danach prüft der Datenmanager, Datenbank-Aspekte
beteiligten auch während der Durch- ob alle Prozesse korrekt durchge-
führung der klinischen Studie bildet führt wurden, z.B. ob es keine of- Es ist hervorzuheben – und in
eine Grundlage zur Gewährleistung fenen Queries mehr gibt, und führt Abbildung 3 bereits angedeu-
der Datenqualität (Abbildung 2). ggf. finale organisatorische und tet –, dass es im Verlauf des Da-
Hierbei spielen die Prüfzentren strukturelle Änderungen an der ten-Management-Prozesses nicht
eine zentrale Rolle, die für eine zeit- Datenbank durch. nur eine Datenbank gibt.
nahe, vollständige und korrekte Da- Nach Durchführung dieses Pro-
tenerfassung verantwortlich sind: zessschrittes wird die (e)CRF-Da- iDB – intermediate DataBase
tenbank geschlossen (soft lock). Zunächst wird eine Datenbank
Es folgt – unter Federführung generiert, die auf die Datenerfas-
ICH E6 (R2) – Abschnitt 4.9.1: “The
des Projektstatistikers – der finale sung ausgerichtet ist und in der
investigator should ensure the ac-
(ggf. verblindete) Daten-Review. üblicherweiser die von den klini-
curacy, completeness, legibility, and
Der Schwerpunkt liegt dabei auf schen Prüfzentren generierten Da-
timeliness of the data reported to
der Bewertung der Auswertbarkeit ten aufgenommen werden. Das ist
the sponsor in the CRFs and in all
der Daten. Speziell werden eventu- die intermediate DataBase (iDB).
required reports.”
elle Verletzungen des Prüfplans be- Darin werden z. B. Messungen aus
trachtet und deren Auswirkungen bioanalytischen Labors (z. B. Plas-
auf die Aussagekraft der Studie. makonzentrationen) nicht erfasst.
Ablauf der Klinischen Studie aus Die Patienten werden den im Prüf- Darüber hinaus unterscheidet
Daten-Management-Sicht plan definierten Analysepopulatio- sich die Struktur eines eCRF von
nen zugeordnet, üblicherweise zu der späteren Zieldatenbank meist
Abbildung 3 stellt den Daten-Ma- “Full-Analysis Set” und “Per-Proto- dadurch, dass Datensätze im eCRF
nagement-Prozess schematisch dar. col Set”. visitenweise organisiert sind und
Aus Sicht des klinischen Projektma- Die klinische Datenbank wird fi - die fi nale Struktur domainorien-
nagements ist dabei insbesondere nal geschlossen (hard lock), wenn tiert ist. Das heißt, Parameter, die
zu beachten, dass nach der Durch- alle daten- und analysebezogenen im Studienverlauf erhoben werden
führung der letzten Visite des letz- Entscheidungen getroffen und um- (z. B. Blutdruck als primärer Para-
ten Patienten (LPLV) für die Prüf- gesetzt wurden. meter) werden in nur einem Da-
zentren noch folgende Aufgaben
anstehen:
– Die dabei erhobenen Daten müs-
sen noch in das (e)CRF eingetra-
gen werden.
– Es müssen noch offene Rückfra-
gen (Queries) beantwortet wer-
den.
– Der Prozess der “SAE Reconcilia-
tion” muss noch abgeschlossen
werden, das heißt, der Abgleich
der Informationen zu schwerwie-
genden unerwünschten Ereignis-
sen (SAE – Abbildung 1: Schnittstellen zur Entwicklung eines eCRF.
Serious Adverse Events) zwischen
der Pharmakovigilanz-Datenbank
und der klinischen Datenbank.
Die “Data Cleaning Phase” gilt als
beendet, wenn:
– die Monitore die “Source Data
Verification (SDV)” beendet ha-
ben,
– es keine offenen Queries mehr
gibt,
– die klinischen Prüfärzte mit
ihrer Unterschrift abschließend
bestätigt haben, dass alle Daten
korrekt und vollständig eingege-
ben wurden. Abbildung 2: Schnittstellen bei der Durchführung der klinischen Studie.
März | Heft 1 | Jahrgang 22 | PM QM 2020 | 23
ARZNEIMITTELPRÜFUNG
LPLV
LPLV: Last Patient Last Visit, (e)CRF: electronical Case Report Form, iDB: intermediate DataBase, cDB: clinical DataBase, tDB: target DataBase,
DRM: Data Review Meeting, SAE: Serious Adverse Events
Abbildung 3: Schematischer Ablauf der klinischen Studie unter Daten-Management-Aspekten.
tensatz mit allen Visiten abgelegt Beantwortung der Studienfra- von pharmakokinetischen Da-
(Abbildung 4). gestellungen notwendig sind. ten zum Datenmanagement,
b) WELCHE Daten sollen im (ggf. iterativer) Prozess des
cDB: clinical DataBase eCRF-System erfasst bzw. SAE-Abgleichs.
Diese Datenbank enthält alle hochgeladen werden und wel-
in der Studie generierten Daten che Daten werden der cDB
und Informationen in einer wohl- unabhängig vom eCRF zuge- Spezielle Anforderungen an den
definierten Zielstruktur (siehe führt? Einsatz eines eCRF-Systems
auch Abbildung 5: Klinische Da- c) Alle elektronischen Daten-
tenbank). Transferprozesse, die das Beim Einsatz eines eCRF-Systems
Hochladen ins eCRF-System ist zu berücksichtigen:
tDB: target DataBase betreffen, sind vorab zu defi - – Visitenweise Unterschrift durch
Diese Datenbank basiert im We- nieren und zu validieren. den Prüfarzt.
sentlichen auf der klinischen Daten- d) Interaktive Prozesse zwischen – Die Applikation eines eCRF-
bank. Sie kann im Spezialfall auch Projektbeteiligten sind ad- Systems muss für jedes einzelne
mit dieser identisch sein. Sie ist hier äquat zu definieren und zu Studienprojekt explizit validiert
der Vollständigkeit halber erwähnt, implementieren, z. B. Transfer werden (Eingabe-Forms, pro-
da möglicherweise der Sponsor
oder die Behörden eine spezielle
Export-Struktur verlangen.
Eine typische Konstruktion zum
Management des Daten-Flows aus
unterschiedlichen Quellen illus-
triert Abbildung 5.
Daten- und prozessorientierte
Aspekte bei der eCRF-
Entwicklung
Folgende Kernfragen sind bei der
Planung zu adressieren – die Punkte
a) und d) gelten selbstverständlich
auch für ein papierbasiertes CRF: Domains: dm: Demographie, vs: Vital Signs, lb: Labor, ae: Adverse Events
a) Es sollten nur solche Daten
gesammelt werden, die zur Abbildung 4: Strukturvergleich (e)CRF versus klinische Datenbank.
24 | PM QM 2020 | Jahrgang 22 | Heft 1 | März
grammierte Datenprüfungen
etc.).
– Es ist ein “User-Acceptance Test“
(UAT) gemäß eines vorspezifi-
zierten Planes von einer quali-
fizierten Person durchzuführen
(z. B. Daten-Manager).
Die Datenverifizierung der in die
eCRFs eingetragenen Daten durch
einen Prüfer oder durch die von ihm
beauftragten ärztlichen Mitglieder
der Prüfgruppe muss regelmäßig
zeitnah zu den Visiten erfolgen, um
die an den Prüfer gerichtete Forde-
rung gemäß ICH-GCP 4.9.1 zu erfül-
len. Die fehlende Implementierung SAS ® : Software für Datenmanagement und Statistik (SAS Institute Inc.)
elektronischer Signaturen zur re-
gelmäßigen Datenverifizierung im Abbildung 5: Typisches Beispiel zum Management des Daten-Flows aus unterschiedlichen Quellen.
eCRF stellt daher einen schwerwie-
genden Mangel dar, der vom Spon- • Daten-Integritäts-Prüfung Interime Daten-Reviews
sor zu verantworten ist (ICH-GCP (z. B. mittels Check-Sum-
5.1.1 und 5.1.3). men generiert durch die Zur kontinuierlichen Prüfung der
MS-Windows Utility Datenqualität bietet es sich an,
Certutil-hashfile) während der klinischen Studie Da-
– Spezifikation und Beschrei- ten-Reviews seitens Daten-Manage-
Projekt-Management-Plan bung des Transfers der finalen ment und Statistik durchzuführen,
“comprehensive” Datenbank, die eine sinnvolle Ergänzung und
Möglichst vor Beginn einer kli- die alle Daten aus allen Daten- Unterstützung des klassischen Stu-
nischen Studie sollte seitens des quellen beinhaltet und die aus dien-Monitoring darstellen.
Projekt-Managements ein Plan strukturierten Datensätzen im Die GCP-Richtlinie ICH-E6(R2)
erstellt werden, der detailliert einheitlichen Format besteht empfiehlt ein solches Vorgehen
die beteiligten Projekt-Teams mit logisch verbundenen und unter dem Begriff “Centralized
und deren Verantwortlichkeiten organisierten Domains. Monitoring”.
aufführt (Tabelle 2).
Darüber hinaus sind Daten-
Transferprozesse konkret zu de- Organisation Funktion
finieren (Tabelle 3).
Diese Spezifikationen soll- NewDrug AG Sponsor
ten vorzugsweise in einem sog.
“Data Flow Document” darge-
legt werden, das folgende As- NewDrug AG Projekt-Management & Monitoring
pekte enthält:
– Spezifikation der Datenquel- ABC Center Phase I Unit
len und Ziele von Datentrans-
fers
– Beschreibung der Interaktio- EasyData Ltd eCRF Provider
nen und Datentransfers zwi-
schen Projektbeteiligten M.A.R.C.O. Institut Daten-Management und Statistik
– Zuordnung von Verantwort-
lichkeiten zu jedem Trans-
fer-Schritt QuickLab Billy Rubin Klinisches Zentral-Labor
– Detaillierte Beschreibung der
Transfer-Schritte mit folgen- Assay & More Center Pharmakokinetik
den Informationen:
• Transfer-Medium (z. B. CD-
ROM, E-Mail) Safety & Tolerance Group Pharmakovigilanz
• Datenschutz-Maßnahmen
(z. B. Passwortschutz) Tabelle 2: Beispiel für eine Übersicht über beteiligte Organisationen und Verantwortlichkeiten.
März | Heft 1 | Jahrgang 22 | PM QM 2020 | 25
ARZNEIMITTELPRÜFUNG
Format /
Daten-Quelle Transfer von … Transfer nach …
Transfer-Medium
Papier-CRF Monitoring (CRO_1) DM (CRO_2) Kurier
Klinische Labor Printouts Monitoring (CRO_1) DM (CRO_2) Kurier
Bioanalytical Lab
Bioanalytische Ergebnisse DM (CRO_2) csv-File / E-Mail
(CRO_3)
PK-File (Plasma-Konzen-
csv-File /
trationen mit aktuellen DM (CRO_2) PK (CRO_2)
via CRO_2 server
Abnahmezeiten)
csv-File /
PK-Parameter PK (CRO_2) DM (CRO_2)
via CRO_2 server
Finale klinische Datenbank DM (CRO_2) Stats (CRO_2) SAS® / via CRO_2 server
Export der finalen “com- Projekt-Management CD_ROM, passwortgeschützt,
DM (CRO_2)
prehensive” Datenbank (Sponsor) Kurier
Tabelle 3: Beispiel zur Beschreibung von Transferprozessen.
Diese interimen Daten-Reviews ha- – Ermöglichung eines gezielten kanzniveaus) und in keiner Weise
ben folgende Zielsetzung: Monitorings vor Ort. die Aussagekraft und Validität der
– Identifizierung von Prüfplan- Studienergebnisse beeinträchti-
Abweichungen, Ein relevanter Vorteil und Nutzen gen (Tabelle 4). Im Gegenteil, es
– Identifizierung von fehlenden der interimen Daten-Reviews liegt erhöht sich die Validität durch Op-
Werten, inkonsistenten Daten, Da- in der frühzeitigen Erkennung von timierung der Prozess- und Daten-
ten-Ausreißern, unerwartet gerin- Problemen, entsprechend in der qualität.
ger (oder auch hoher) Variabilität, Chance, Probleme frühzeitig zu lö-
– Untersuchung von Trends, Variabi- sen, und in einer Verkürzung der
lität innerhalb und zwischen Prüf- Zeitspanne zwischen dem Ende Fazit
zentren, der klinischen Prüfung (LPLV) und
– Evaluierung von Daten-Integri- der Schließung der Datenbank. Die Sicherstellung der Datenqua-
täts-Problemen, Es ist hervorzuheben, dass die lität und Datenintegrität beginnt
– Evaluierung von Performance interimen Daten-Reviews keine bereits bei der Planung der klini-
der klinischen Zentren (z. B. zeit- statistischen Implikationen ha- schen Studie, insbesondere bei der
nahe Datenerfassung), ben (z. B. Adjustierung des Signifi- Entwicklung und Implementierung
eines wohldefinierten Prozesses
zur Datenerhebung und Datenprü-
Interime Daten-Reviews fung. Dazu zählen nicht nur ein aus-
& gereifter und zielgerichteter Prüf-
Statistische Interim-Analyse plan sowie ein optimierter (e)CRF,
sondern auch eine frühzeitige Pro-
Interime Daten-Reviews sind charakterisiert durch: jektplanung, die alle Datenquellen
• Eine Entblindung der Daten findet NICHT statt. und Daten-Transferprozesse mit
• Es werden KEINE Behandlungsdifferenzen evaluiert. Nennung der Projektbeteiligten
• Sie führen zu KEINER Adaption des Studiendesigns. und deren Verantwortlichkeiten
berücksichtigt. Insbesondere sollte
Tabelle 4: Charakterisierung von interimen Daten-Reviews. gewährleistet sein, dass im Fall der
26 | PM QM 2020 | Jahrgang 22 | Heft 1 | März
Beauftragung eines externen DM- Quellen
CRO ein zeitnaher Transfer der fi - Good Clinical Practice Guide (compiled by AUTOR
nalen Studiendatenbank vom DM- MHRA) 2012.
CRO zum Sponsor nach Studienab- E6(R2) Good Clinical Practice: Integrated Dr. rer. nat. Man-
schluss gemäß vorab definierter Addendum to ICH E6(R1) Guidance for Industry, fred Wargenau ist
Kriterien geregelt ist. March 2018. seit Mai 1997 Inhaber
Darüber hinaus stellt die Durch- FDA Guidance for Industry: Computerized und Geschäftsführer
führung von interimen Daten-Re- systems used in clinical investigations, 2007. der M.A.R.C.O. GmbH
views während der klinischen Stu- FDA 21 CFR Part 11, 1997. & Co. KG (Institut für
die eine effiziente Maßnahme EMA/INS/GCP/454280/2010: Reflection paper Klinische Forschung
zur Optimierung der Datenqua- on expectations for electronic source data and und Statistik). Er hat
lität und Studien-Performance data transcribed to electronic data collection mehr als 25 Jahre Erfahrung in der Me-
dar. Diese werden seitens Daten- tools in clinical trials. dizinischen Statistik und im Klinischen
Management und Statistik geleis- Zentralstelle der Länder für Gesundheitsschutz Daten-Management mit Stationen u. a. in
tet und bieten eine sinnvolle Ergän- bei Arzneimitteln und Medizinprodukten (ZLG): der Universitätsklinik Hamburg-Eppen-
zung und Unterstützung des klas- V0500202, Datenverifizierung, elektronisches dorf und in internationalen pharmazeuti-
sischen Studien-Monitorings. Auch CRF, 26.02.2018. schen Unternehmen. Er leistete zahlreiche
empfiehlt die GCP-Richtlinie ICH- wissenschaftliche Beiträge hinsichtlich
E6(R2) ein solches Vorgehen unter statistischer Methoden und Strategien,
dem Begriff “Centralized Monito- Pharmakokinetik, klinisch-pharmakolo-
ring”. | gischer und klinischer Studien und führte
zudem Fortbildungskurse (u. a. Design
und Analyse von Biomarkerstudien)
durch.
Kontakt:
manfred.wargenau@marco-institut.de
ANZEIGE
Comfortable – Capture – Compliant
eCRF CTMS
Lab Data Management ePRO / eCOA
Device integration Randomization
AMEDON solutions
QoL Questionnaires SAE Reporting
INTEGRATED DATA SOLUTIONS FOR CLINICAL STUDIES
AMEDON GmbH
d data so a t e o wn e
Willy-Brandt-Allee 31c te riv
ra
P
lu
d
Integ
23554 Lübeck - Germany
t ion
Tel.: + 49 (0) 451 38 45 0 0
s
INTEGRATED DATA SOLUTIONS
Fax: + 49 (0) 451 38 45 0 11 Data security & more than 13 years
FOR CLINICAL STUDIES
info@amedon.de Data privacy QUALITY
Comfortable – Capture - Compliant www.amedon.de
März | Heft 1 | Jahrgang 22 | PM QM 2020 | 27

Sie können auch lesen

















































