Bedeutung des nicht-sekretorischen Renins als Modulator mitochondrialer und metabolischer Funktionen in kardialen Zellen
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Aus dem Institut für Physiologie
(Leiter Prof. Dr. med. Jörg Peters)
der Universitätsmedizin der Universität Greifswald
Bedeutung des nicht-sekretorischen Renins als Modulator
mitochondrialer und metabolischer Funktionen
in kardialen Zellen
Inaugural - Dissertation
zur
Erlangung des akademischen
Grades
Doktor der Medizin
(Dr. med.)
der
Universitätsmedizin
der
Universität Greifswald
2022
Vorgelegt von:
Alexander Albers
Geboren am 31.05.1993
in HamburgDekan: Prof. Dr. med. Karlhans Endlich
1. Gutachter: Prof. Dr. med. Jörg Peters
2. Gutachter: Prof. Dr. med. Heimo Ehmke
Ort, Raum: Greifswald, Seminarraum der Klinik für Innere Medizin B (Raum 6.0.8)
Tag der Disputation: 31.01.2023
2Inhalt
1 Einleitung ............................................................................................................. 5
1.1 Zirkulierendes (konventionelles) Renin-Angiotensin-System ....................... 5
1.2 Das lokale kardiale Renin-Angiotensin-System ............................................ 8
1.3 Ein alternatives Renin-Transkript ............................................................... 10
1.4 Mitochondrien als zentrale Zellorganellen .................................................. 13
1.5 Fragestellungen.......................................................................................... 18
2 Material und Methoden ...................................................................................... 19
2.1 Material und Geräte.................................................................................... 19
2.1.1 Chemikalien und Reagenzien ................................................................. 19
2.1.2 Weitere Materialien ................................................................................. 20
2.1.3 Puffer ...................................................................................................... 21
2.1.4 Zelllinie ................................................................................................... 21
2.1.5 Antikörper ............................................................................................... 21
2.1.6 Geräte und Software ............................................................................... 22
2.2 Methoden ................................................................................................... 23
2.2.1 Zellkultur ................................................................................................. 23
2.2.2 Transfektion der Zelllinie ......................................................................... 23
2.2.3 Ischämie-relevante Bedingungen ........................................................... 24
2.2.4 RNA-Quantifizierung ............................................................................... 25
2.2.5 Vitalitätsbestimmung ............................................................................... 26
2.2.6 Durchflusszytometrie .............................................................................. 27
2.2.7 Photometrie ............................................................................................ 29
2.2.8 Luminometrie .......................................................................................... 30
2.2.9 Messung der Sauerstoffverbrauchsrate .................................................. 31
2.2.10 Statistische Auswertung ...................................................................... 34
3 Ergebnisse ......................................................................................................... 35
33.1 Expression von Reninvarianten in H9c2-Zellen .......................................... 35
3.2 Einfluss der 1A-9Renin-Überexpression .................................................... 37
3.3 Zellüberleben unter OGD ........................................................................... 37
3.3.1 Vitalität .................................................................................................... 37
3.3.2 Apoptose ................................................................................................ 39
3.3.3 Nekrose .................................................................................................. 43
3.4 Mitochondriale Parameter unter OGD ........................................................ 45
3.4.1 MitoTracker ............................................................................................. 45
3.4.2 Mitochondriales Membranpotenzial ........................................................ 46
3.4.3 ATP-Gehalt (Aktivität von Komplex V der Atmungskette) ....................... 47
3.4.4 XTT-Färbung (Aktivität von Komplex I der Atmungskette) ...................... 48
3.5 Sauerstoffverbrauchsraten unter OGD ....................................................... 49
3.5.1 Zelluläre Atmung..................................................................................... 50
3.5.2 ATP-gekoppelte Atmung und Protonenleck ............................................ 52
3.5.3 Maximale mitochondriale Atmung und Reservekapazität ....................... 54
4 Diskussion .......................................................................................................... 56
4.1 OGD-induzierte Hochregulation beider Renintranskripte ........................... 56
4.2 Einfluss nicht-sekretorischen Renins auf die zelluläre Überlebensrate ...... 59
4.3 Einfluss nicht-sekretorischen Renins auf den zellulären O2-Verbrauch ...... 63
5 Zusammenfassung............................................................................................. 66
6 Abkürzungsverzeichnis ...................................................................................... 67
7 Abbildungsverzeichnis ....................................................................................... 70
8 Tabellenverzeichnis ........................................................................................... 71
9 Literaturverzeichnis ............................................................................................ 72
10 Danksagung ....................................................................................................... 82
41 Einleitung
Das Renin-Angiotensin-System (RAS) ist seit langem bekannt als wichtiger
Regulationsmechanismus von Blutdruck, Wasser- und Salzhaushalt. Auch
Entzündungs- und Fibrosierungsprozesse werden von diesem System beeinflusst.
Von vielen Bestandteilen des RAS sind mittlerweile weitere Funktionen auf Gewebe-
und Zellebene bekannt und Ziel aktueller Forschung.
Die Peptidase Renin ist das erste und geschwindigkeitsbestimmende Enzym der
konventionellen RAS-Kaskade und in seinen grundlegenden Zügen bereits seit 1898
durch Robert Tigerstedt und Per Bergmann beschrieben [Tigerstedt und Bergman
1898]. Das RAS zählt heute zu den bekanntesten Mechanismen der
Blutdruckregulation. Tigerstedt und Bergmann haben, in einem einfach und klar
strukturierten Experiment, Kaninchen kleinste Mengen eines Extrakts aus deren
eigenem Nierengewebe in den Blutstrom injiziert. Die Folge war ein deutlicher
Blutdruckanstieg. Durch diese Beobachtung konnten submikroskopische
Bestandteile der Niere direkt mit der Blutdruckregulation in Verbindung gebracht
werden. Diese Beobachtungen lieferten Ansatzpunkte für die weitere Erforschung
des Zusammenhangs von Bluthochdruck und Nierenerkrankungen, welcher bis dahin
lediglich empirisch beobachtet werden konnte. Die für die Wirkung dieses Extrakts
hauptverantwortliche und hier erstmals beschriebene Substanz erhielt, seinem
Ursprungsorgan entsprechend, den Namen „Renin“ (von lateinisch Ren für Niere).
Die seit über einem Jahrhundert kontinuierlich andauernde Forschung am RAS, zeigt
vor allem, dass das Zusammenspiel seiner verschiedenen Akteure deutlich
komplexer, als zunächst angenommen und weit entfernt von einer kompletten
Aufklärung ist. Die Entdeckung eines RAS auf Gewebe- und Zellebene, sowie neuen
Zielstrukturen des zirkulierenden RAS, geben eine Idee davon wie vielfältig dieses
System in verschiedenste Prozesse eingebunden ist und weit über die
Blutdruckregulation hinaus seine Wirkung entfaltet [Paul et al. 2006].
1.1 Zirkulierendes (konventionelles) Renin-Angiotensin-System
Die am längsten bekannte Aufgabe des RAS ist die Regulation des Blutdrucks. Bei
renaler Minderdurchblutung, gesteigerter Aktivität sympathischer Nierennerven sowie
bei Natriummangel wird die Freisetzung von Renin aus dem juxtaglomerulären
5Apparat der Nephrone der Niere erhöht. Die somit in die Blutzirkulation gelangte
Peptidase Renin spaltet nun von dem in der Leber kontinuierlich produzierten Protein
Angiotensinogen das Dekapeptid Angiotensin I (ANG I) ab. Für die Bildung des
hauptsächlichen Effektorpeptids ist eine weitere Abspaltung von zwei Aminosäuren
vom ANG I, durch das Angiotensin-Converting-Enzyme (ACE) notwendig. In diesem
Schritt entsteht das biologisch aktive Oktapeptid Angiotensin II (ANG II) [Romero und
Hoobler 1970].
In ihrer Gesamtheit führen die in Abbildung 1 schematisch dargestellten Effekte von
ANG II zu einer direkten Erhöhung des Blutdrucks einerseits sowie zu einer
vermehrten Aufnahme und Rückresorption von Wasser und Salz andererseits. Über
die damit verbundene intravasale Volumenerhöhung kommt es zu einer weiteren,
gewissermaßen indirekten, Blutdruckerhöhung. Die im Folgenden beschriebenen
Effekte von ANG II werden alle durch den Angiotensin II-Rezeptor Typ 1 (AT1R)
vermittelt. Die Erhöhung des Blutdrucks kommt durch Bindung von ANG II an den
AT1R sowohl direkt auf Gefäßmuskelzellen als auch auf adrenergen
Nervenendigungen zustande, wodurch die Ausschüttung von Noradrenalin steigt und
die glatten Gefäßmuskelzellen ihren Tonus erhöhen. Zentral führt ANG II zu einer
vermehrten Freisetzung des Antidiuretischen Hormons (ADH) aus dem
Hypophysenhinterlappen. ADH übt in ausreichend hoher Dosis über die
V1-Rezeptor-vermittelten Signalkaskaden, mit Ausnahme von Gehirn- und
Herzgefäßen, starke vasokonstriktorische Effekte aus. Die daraus resultierende
Erhöhung des Gefäßwiderstandes führt zur Erhöhung des Blutdrucks.
V2-Rezeptoren, welche auf Sammelrohrzellen der Niere exprimiert werden,
veranlassen nach Aktivierung durch ADH den Einbau von Aquaporin 2 in die apikale
Zellmembran, wodurch es zu einer verstärkten Wasserrückresorption kommt. ANG II
selbst verursacht eine verstärkte Natriumrückresorption im proximalen Tubulus.
Weiterhin bedingt es eine vermehrte Produktion des Mineralcorticoids Aldosteron in
der Nebennierenrinde. Dieses verursacht eine erhöhte Natriumrückresorption, durch
Einbau epithelialer Natriumkanäle (ENaC) in die Epithelzellen der Verbindungstubuli
und des Sammelrohres. Die Folge dieser Natriumretention ist die passive Resorption
von Wasser. Auch die orale Aufnahme von Wasser und Salz wird von ANG II durch
zentrale Effekte im Bereich des Hypothalamus gesteigert [Schmidt 2005].
6Andere weniger lang bekannte Effekte des ANG II werden durch den
Angiotensin II-Rezeptor Typ 2 (AT2R) vermittelt. Zu diesen zählen die Steigerung und
Modulation der Zellproliferation, Anti-Inflammation, Regeneration, Differenzierung
und Apoptose [Paul et al. 2006]. Knock-out-Modelle suggerieren sogar eine
antagonistische Beziehung zwischen den Effekten der beiden AT-Rezeptoren [Dinh
et al. 2001].
Abbildung 1: Renin-Angiotensin-System (RAS)
Komponenten der RAS-Kaskade und deren Hauptwirkungen mit Augenmerk auf die
Regulation von Blutdruck, Wasser- und Salzhaushalt, sowie einigen durch AT2-Rezeptoren
vermittelten Effekten.
Der Stellenwert des RAS, nicht nur in der Grundlagenforschung, sondern im
angewandten, klinischen Kontext wird insbesondere durch dessen Rolle als
Angriffspunkt für die Behandlung von Bluthochdruck deutlich. Bluthochdruck selbst ist
7eine der häufigsten Erkrankungen weltweit und stellt einen wesentlichen Risikofaktor
für eine Reihe chronischer und häufig auch letal verlaufender Erkrankungen dar.
Darunter fallen unter anderen Herzinfarkte, Schlaganfälle und chronische
Nierenerkrankungen. In der Therapie des Bluthochdrucks ist, nach Änderungen der
persönlichen Lebensführung z.B. durch Gewichtsabnahme und Steigerung der
körperlichen Aktivität, die pharmakologische Therapie die wichtigste Maßnahme. Zu
den meistverwendeten Erstlinienmedikamenten zählen hier ACE-Hemmer und
Angiotensin-Rezeptor-Antagonisten. Der Wirkmechanismus beider Wirkstoffklassen
zielt auf eine Verminderung der durch das RAS vermittelten und beschriebenen
Haupteffekte desselbigen ab. Dadurch wird eine Senkung des Blutdrucks und eine
damit einhergehende Verminderung des Risikos für kardiovaskuläre
Folgeerkrankungen angestrebt [Weber et al. 2014].
1.2 Das lokale kardiale Renin-Angiotensin-System
Bisher wurde vor allem das konventionelle RAS thematisiert, dessen Bestandteile
das in der Leber synthetisierte Angiotensinogen, das vom juxtaglomerulären Apparat
der Niere sezernierte Renin und das an Endothelzellen, insbesondere der
Lungenkapillaren vorkommende ACE umfassen. Die oben beschriebene RAS-
Kaskade läuft somit im Blutkreislauf ab, ANG II wird mit dem Blutstrom zum AT1R
auf den Zellen der Effektororgane transportiert und dient der Blutdruckregulation. Bei
chronischer Stimulation des AT1R durch ANG II verbunden mit einer erhöhten
Vasokonstriktion und Wasserretention kann es jedoch zur Entwicklung eines
Bluthochdrucks kommen, gefolgt von einem erhöhten Risiko an einem
Myokardinfarkt, Arteriosklerose oder einer Herzinsuffizienz zu erkranken [Ferrario
1990; Agrawal et al. 2016]. Begleitet werden diese pathophysiologischen Vorgänge
u.a. durch eine verstärkte Proliferation von glatten Muskelzellen und Myozyten, durch
Förderung von pro-inflammatorischen Reaktionen oder die verstärkte Bildung
reaktiver Sauerstoffspezies (ROS) und Fibrose [Schelling et al. 1991; Leung 2004].
Darüber hinaus wurden in den letzten Jahrzehnten in verschiedenen Geweben
(Herz, Haut, Gefäße, Niere, Pankreas, Nebenniere, Gehirn) bzw. in subzellulären
Kompartimenten (Mitochondrien, Zellkern) lokal begrenzte RAS mit auto- oder
parakriner Wirkung entdeckt [Abadir et al. 2012; Paul et al. 2006]. Von einem lokalen
RAS spricht man, wenn die für die ANG II-Synthese notwendigen Komponenten lokal
8synthetisiert werden oder ihre Wirkung durch Aufnahme lokal entfalten [De Mello,
Walmor C. und Danser, A. H. Jan 2000; Abadir et al. 2011].
In Herzen von diabetischen Ratten wies die Arbeitsgruppe um Kumar [Singh et al.
2008; Kumar et al. 2008] eine erhöhte intrazelluläre Angiotensinogen- und
Reninexpression sowie ANG II Bildung nach. Die Degradation von ANG I zu ANG II
erfolgte dabei nicht ACE- sondern Chymase-abhängig. Eine erhöhte lokale kardiale
Produktion von ANG II wurde ebenfalls unter mechanischem Stress sowie nach
ischämischen Prozessen beobachtet. Hier war ANG II an post-ischämischen
kardialen Umbauprozessen beteiligt [Ihara et al. 2001; De Mello, Walmor C. und
Frohlich 2011].
Untersuchungen an Schweinen belegen, dass 75 % des kardialen ANG II im
Gewebe selbst produziert werden [van Kats, Jorge P. et al. 1998]. Dies erfordert
entweder die Translokation von sekretorischen RAS-Komponenten ins Zytosol oder
die Synthese von alternativen, nicht-sekretorischen RAS-Komponenten. Bezüglich
Angiotensinogen zeigte sich, dass das α-Globulin durch post-translationale
Modifikation in eine nicht-glykosylierte Form überführt werden kann, die dann im
Zytosol und im Kern nachweisbar ist [Singh et al. 2007]. Eine Akkumulation von
intrazellulärem Angiotensinogen kann, wie bereits beschrieben, in Herzen von
diabetischen Ratten unter pathologischen Bedingungen nachgewiesen werden
[Singh et al. 2007]. Auch beide AT-Rezeptorsubtypen konnten intrazellulär, in der
inneren mitochondrialen und der inneren Zellkern-Membran von kardialen Myozyten
und Fibroblasten durch high resolution Elektronen- und Konfokalmikroskopie sowie
Western-Blot Analysen von Zell- und Gewebeextrakten lokalisiert werden [Abadir et
al. 2011; Tadevosyan et al. 2010]. Verschiedene mitochondriale Proteine werden
nicht durch die mitochondriale, sondern durch die nukleäre DNA kodiert. Sie werden
im Zytosol synthetisiert, weisen eine N-terminale mitochondriale Erkennungssequenz
auf und werden unter Mithilfe von Chaperonen zu und in die Mitochondrien
transportiert. AT-Rezeptoren weisen, ebenso wie einige mitochondriale Proteine
(z.B. die Adeninnucleotid-Translokase) und nachfolgend dargestellt das nicht-
sekretorische Renin, diese Erkennungssequenz nicht auf. Daher ist noch unklar, wie
diese Rezeptoren in die Mitochondrien gelangen. Funktionell werden mitochondriale
AT2R über eine Stimulation der mitochondrialen NO-Synthase (mNOS) und
NO-Produktion mit einer Hemmung der mitochondrialen Atmung in Zusammenhang
9gebracht [Abadir et al. 2011]. Nukleäre AT-Rezeptoren, die ebenso wie auf der
Zellmembran mit G-Proteinen gekoppelt sind, regulieren nach Bindung von ANG II
die NFĸB-abhängige Gentranskription in Kardiomyozyten und kardialen Fibroblasten
durch Erhöhung der nukleären Calciumkonzentration und Stimulation der nukleären
NOS [Tadevosyan et al. 2010; Tadevosyan et al. 2017]. Es resultieren eine
gesteigerte Zellproliferation und Kollagensekretion, die mit pathophysiologischen
Prozessen bei der kongestiven Herzerkrankung assoziiert sind.
Insgesamt resultiert die, durch Aktivierung des lokalen RAS hervorgerufene
intrazelluläre ANG II Bildung, in einer erhöhten oxidativen Schädigung, Apoptose und
Nekrose von kardialen Zellen [Frustaci et al. 2000; Singh et al. 2007]. Dies steht im
Kontrast zu nachfolgend dargestellten Daten zur Funktion des nicht-sekretorischen
Renins.
1.3 Ein alternatives Renin-Transkript
Das klassische sekretorische Reninprotein besteht aus einem Signalpeptid
(Präfragment), das für seine Integration in den sekretorischen Pathway essentiell ist,
einem Prosegment, welches das aktive Zentrum des Renins bedeckt (Prosegment
und Renin entspricht dem Prorenin) und dem reifen enzymatisch aktiven Renin. Das
Reningen kodiert für eine mRNA, welche bei der Ratte aus neun Exons besteht. Das
Präfragment wird in einem kleinen Bereich im Exon1 des Reningens kodiert und
enthält eine Signalsequenz, welche für die Translation der mRNA an Ribosomen des
endoplasmatischen Retikulums (ER) verantwortlich ist. Der entstehende Peptidstrang
wird somit in das ER hinein synthetisiert und der sekretorische Weg des Renins vom
ER über den Golgi-Apparat bis zur Zellmembran ist gebahnt. Ein Vorgang der bei
nicht membrangebundenen Proteinen wie dem Renin zur Exozytose, d.h. zum
Ausschleusen in den Extrazellularraum, führen kann. Neben der
ER-Erkennungssequenz kodiert das Exon1 auch einen Teil des Prosegments,
welches bis zu dessen Abspaltung das aktive Zentrum bedeckt und somit die
Proteaseaktivität des Renins inhibiert [Clausmeyer et al. 1999]. Das zunächst
gebildete Prorenin wird kontinuierlich sezerniert, während das intrazellulär, in
Sekretionsvesikeln prozessierte und gespeicherte aktive Renin reguliert freigesetzt
wird [Hackenthal et al. 1990]. Die enzymatische Aktivität des Prorenins entspricht nur
1-2 % der des aktiven Renins [Peters 2017]. Dies legt die Vermutung nahe, dass
10Prorenin seine Effekte außerhalb der klassischen Proteasefunktion des Renins
ausübt.
Im Jahre 1999 wurde erstmals eine nicht-sekretorische Isoform des Renins (auch
zytosolisches Renin genannt) in der Ratte identifiziert, welche durch einen
alternativen Promotor im Intron1 des Reningens gekennzeichnet ist [Clausmeyer et
al. 1999] [Lutze et al. 2017] (Abbildung 2). In diesem Promotorbereich liegt ein
alternatives Exon1 (Exon1A), dessen genaue Funktion bislang unbekannt ist. Da das
nächste in-frame Translationsstartkodon im Exon2 liegt, werden dem Exon1A
höchstwahrscheinlich regulatorische Funktionen sowie eine eventuelle Beteiligung
bei der intrazellulären Translokation zuteil. Exon1A-Transkripte kodieren für eine
verkürzte Form des Prorenins, dem die Signalsequenz und die ersten 10 der 43
Aminosäuren des Profragments fehlen [Clausmeyer et al. 1999]. Die Translation der
alternativen mRNA zum, im Folgenden 1A-9Renin genannten, Protein erfolgt an
freien Ribosomen im Zytosol, so dass das verkürzte Prorenin intrazellulär verbleibt.
Inzwischen wurden auch für das menschliche Reningen alternative Renin-Varianten
beschrieben. Durch alternative Promotorregionen und alternatives Spleißen im
Intron1, entstehen ebenfalls Reninisoformen (Renin-b und Renin-c genannt) mit
intrazellulärer Lokalisation [Sinn und Sigmund 2000].
11Abbildung 2: Transkripte des Reningens der Ratte
Schematische Darstellung der mRNA verschiedener Reninvarianten. Die Renin-prä-mRNA
entspricht der transkribierten Basenfolge des Reningens vor dem Spleißen. Aus dieser
entsteht durch Spleißen aller Introns die reife mRNA des 1-9Renins. Durch einen
alternativen Promotor im Intron1 und alternatives Spleißen entsteht die mRNA des
1A-9Renins. Exon1 enthält die ER-Erkennungssequenz, welche für die Translation am ER
und die sukzessive Sekretion von Renin aus der Zelle notwendig ist. Das alternative erste
Renin-Exon1A befindet sich innerhalb des Intron1 der Renin-prä-mRNA.
Die verschiedenen Reninvarianten finden sich in unterschiedlich hohem Maße in
verschiedenen Geweben wieder. So konnte in Nierengewebe ausschließlich das für
dieses Gewebe typische sekretorische 1-9Renin nachgewiesen werden, wohingegen
in den meisten Geweben beide Reninvarianten vertreten sind [Clausmeyer et al.
2000]. Dies ist unter anderem in der Nebenniere, verschiedenen Regionen des
zentralen Nervensystems und der Leber der Fall. In Herzmuskelgewebe findet sich
unter physiologischen Bedingungen kein 1-9Renin und nur in geringem Maße
1A-9Renin.
Subzellulär konnten Peters et al. [Peters et al. 1996] bereits vor der genauen
Beschreibung des 1A-9Renins zeigen, dass sich in Nebennierenrindenzellen Renin
zu sogenannten „dense bodies“ (engl.: dichte Körper) innerhalb von Mitochondrien
konzentriert. Unter in-vitro Bedingungen wurde der Import verkürzter Reninvarianten,
nicht aber des konventionellen 1-9Renins, in Mitochondrien dokumentiert
[Clausmeyer et al. 1999]. Auch bei kardialen Myozyten von transgenen Ratten, die
das 2-9Renin intrazellulär in verschiedenen Geweben überexprimieren, konnte ein
erhöhtes Vorkommen von Renin im Zytosol und in Mitochondrien festgestellt werden
[Peters et al. 2008]. Übereinstimmend wurde auch in 2-9Renin-überexprimierenden,
embryonalen H9c2-Kardiomyoblasten das Renin in Mitochondrien mittels
Fluoreszenzmikroskopie detektiert [Wanka et al. 2009]. Der genaue Prozess der
12Translokation des 1A-9Renins in die Mitochondrien ist, wie bereits beschrieben, noch
unbekannt.
Nachdem Clausmeyer et al. [Clausmeyer et al. 2000] eine selektive Hochregulation
der 1A-9Reninexpression nach vorausgegangenem Myokardinfarkt aufzeigten,
wurde postuliert, dass diese Reninisoform an post-ischämischen
Reparaturprozessen beteiligt sein könnte. Unterstützt wurde diese Hypothese
zunächst durch den Sachverhalt, dass Herzen von 2-9Renin-transgenen Ratten
keine Anzeichen einer Hypertrophie, Entzündung oder Fibrose aufwiesen [Peters et
al. 2008]. Bestätigt wurde die kardioprotektive Rolle des 1A-9Renins dann in
Langendorff-Versuchen, bei denen isolierte Herzen einer Ischämie und
anschließenden Reperfusion ausgesetzt wurden. Die Infarktareale waren bei den
transgenen Rattenlinien etwa halb so groß wie bei den beiden Kontrolllinien [Wanka
et al. 2016]. Auch in-vitro konnte die protektive Wirkung bei der Kardiomyoblasten-
Zelllinie H9c2 bestätigt werden. Nicht-sekretorisches Renin reduzierte unter
Glukosemangel das Auftreten von nekrotischem Zelluntergang [Wanka et al. 2016].
Die Entdeckung des 1A-9Renins und seiner Effekte, besonders unter ischämischen
Bedingungen, hat Fragen nach dessen genauer intrazellulärer Wirkungsweise
aufgeworfen. Das Konzept eines intrazellulären RAS, in welchem Renin zur
intrazellulären Bildung von ANG II beiträgt, ist Gegenstand aktueller Forschung
[Abadir et al. 2012]. Aber auch dessen Einbindung in ANG II-unabhängige
Signalwege gelten als wahrscheinlich. In diese Richtung weisen unter anderem das
Vorhandensein von Bindungspartnern, wie dem (Pro)Renin-Rezeptor oder dem
zytosolisch lokalisierten Renin-Bindungsprotein [Peters 2012].
Das Auffinden des 1A-9Renins vor allem in den Mitochondrien und die zentrale Rolle
dieser Zellorganellen bei Adaptationsprozessen an Mangelsituationen, wie sie bei
ischämischen Ereignissen auftreten, legen die Vermutung einer Beeinflussung der
mitochondrialen Aktivität durch das 1A-9Renin nahe.
1.4 Mitochondrien als zentrale Zellorganellen
Aufgrund der zuvor beschriebenen Lokalisation des 1A-9Renins in Mitochondrien,
aber nicht zuletzt auch wegen der vielfältigen Verflechtung von Mitochondrien mit
13zahlreichen zellulären Prozessen, wurde diesen Organellen ein großer Teil der hier
durchgeführten Untersuchungen gewidmet.
Mitochondrien haben eine faszinierende Entstehungsgeschichte. Der
Endosymbiontenhypothese folgend, stammen Mitochondrien von Bakterien ab,
welche in eukaryotische Zellen aufgenommen und mit diesen eine symbiotische
Beziehung eingegangen sind. Es wird vermutet, dass zu einer Zeit, in welcher sich
Eukaryoten in einem höheren Maße mit einer sauerstoffhaltigen Umgebung
auseinandersetzen mussten, diese Symbiose einen evolutionären Vorteil darstellte
[Sagan 1967]. Daraus leitet sich auch direkt eine der Hauptfunktionen von
Mitochondrien ab, nämlich die Energiegewinnung durch oxidative Phosphorylierung.
Bei diesem Vorgang wird durch Einspeisung von Elektronen in die Atmungskette und
deren Übertragung auf Sauerstoff ein elektrochemischer Gradient erzeugt, der zur
Bildung von Adenosintriphosphat (ATP) führt. ATP ist ein universaler Energieträger
und die Rolle bei dessen Synthese hat Mitochondrien auch den Beinamen
„Kraftwerke der Zelle“ eingebracht [Roger et al. 2017]. Jedes Mitochondrium ist in
mehrere Kompartimente unterteilbar. Es besteht, wie in Abbildung 3 dargestellt, aus
zwei Membranen, dem Intermembranraum, sowie der inneren Matrix [Nunnari und
Suomalainen 2012]. In der inneren Membran befindet sich die zuvor erwähnte
Atmungskette, welche aus fünf Komplexen besteht. Die mitochondrialen Proteine
werden zum Teil von einem eigenen mitochondrialen und zum Teil vom
eukaryotischen Genom kodiert [Friedman und Nunnari 2014]. Der Import der
mitochondrialen Proteine, welche im Zellkern exprimiert und anschließend im Zytosol
translatiert werden, wird durch Rezeptoren an der Oberfläche der Mitochondrien
vermittelt. Die Verteilung der Proteine in die entsprechenden Kompartimente der
Mitochondrien erfolgt durch Translokasen. Beide Vorgänge werden von der Höhe
des mitochondrialen Membranpotenzials beeinflusst [Neupert und Herrmann 2007].
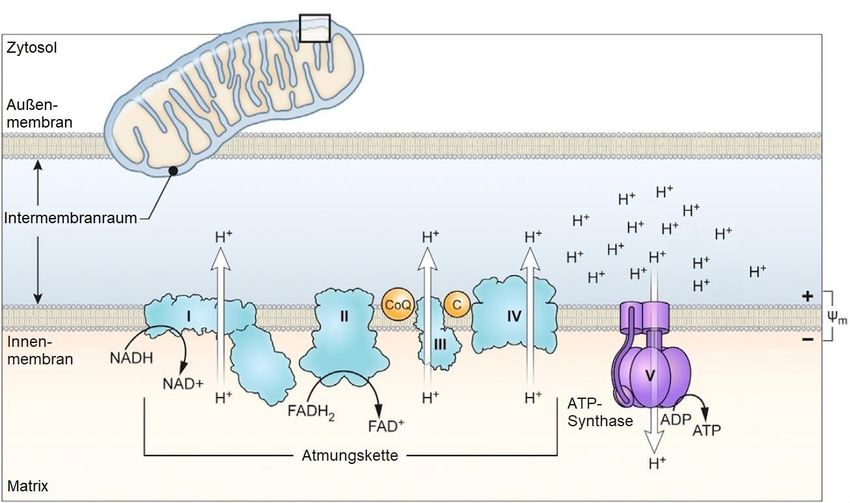
14Abbildung 3: Mitochondriale Kompartimente und Atmungskette
Ausgehend vom Zytosol folgen die Außenmembran, der Intermembranraum, die
Innenmembran und schließlich die Matrix. Die Innenmembran beherbergt die Komplexe I-V
der Atmungskette. Durch Elektronenübertragungen von NADH und FADH 2 auf die Komplexe
I und II wird durch Protonentranslokation in den Innenmembranraum das mitochondriale
Membranpotenzial (Ψm) aufgebaut. Coenzym Q (CoQ) und Cytochrom C (C) übertragen
Elektronen zwischen den Komplexen. Die ATP-Synthase (Komplex V) nutzt den
elektrochemischen Protonengradienten um ADP zu ATP zu phosphorylieren. Modifiziert
nach: Sukumar et al. 2016.
Die Atmungskette vermittelt die Synthese des ATPs durch oxidative
Phosphorylierung. Unter Verbrauch von Sauerstoff wird durch schrittweise
Übertragung von Elektronen ein elektrochemischer Gradient über die innere
mitochondriale Membran aufgebaut (Abbildung 3). Dieser treibt schließlich den
Komplex V der Atmungskette an, welcher ADP zu ATP phosphoryliert. Die Komplexe
I, III und IV bilden eine funktionelle Einheit. Im Einzelnen übernimmt Komplex I
Elektronen von NADH und Komplex II von FADH2, wobei dies im Falle von Komplex I
unter Transport von Protonen über die innere mitochondriale Membran erfolgt. Beide
übertragen die Elektronen weiter über Coenzym Q auf Komplex III. Komplex III
transferiert dann die Elektronen über Cytochrom C auf Komplex IV. Komplex IV
oxidiert schließlich molekularen Sauerstoff zu H2O. Komplex III und Komplex IV
15nutzen die in der Übertagung der Elektronen frei werdende Energie, um Protonen
entgegen ihrem Konzentrationsgradienten über die innere mitochondriale Membran
zu transportieren und so das mitochondriale Membranpotenzial aufzubauen.
Komplex V nutzt diese Energie um ADP zu ATP zu phosphorylieren [van der Bliek,
Alexander M et al. 2017].
Reduktionsmittel für die Einspeisung von Elektronen in die Atmungskette sind neben
Succinat und Malat vor allem NADH und FADH2. Diese werden in der
mitochondrialen Matrix durch den Citratzyklus synthetisiert. Der Citratzyklus besteht
aus einer Serie von acht enzymatischen Schritten, bei welchen Citrat verbraucht und
anschließend wieder regeneriert wird. Dadurch verbindet es den Katabolismus von
Kohlenhydraten, Proteinen und Fetten miteinander. Bei deren Abbau wird das Acetyl-
Coenzym A (Acetyl-CoA) gebildet. Beim Eintritt von Acetyl-CoA in den Citratzyklus
wird es oxidiert, wodurch NADH und FADH2 entstehen [van der Bliek, Alexander M et
al. 2017]. Auch anabole Stoffwechselvorgänge wie z.B. die Synthese von Lipiden,
Pyrimidinen, Häm und Eisen-Schwefel-Clustern laufen unter Beteiligung der
Mitochondrien ab [Nunnari und Suomalainen 2012].
Neben ihrer zentralen Stellung im Stoffwechsel sind Mitochondrien ebenso komplex
und vielfältig in zelluläre Signalwege eingebunden. Durch die Bereitstellung zellulärer
Metaboliten, Aminosäuren und Cofaktoren nehmen sie Einfluss auf eine Reihe von
regulatorischen Enzymen, darunter die Histon-Deacetylasen, welche ihrerseits die
Genexpression beeinflussen. Der zelluläre Calciumhaushalt wird ebenfalls von
Mitochondrien mitbestimmt. Sie beteiligen sich an der Koordination der räumlich-
zeitlichen Verteilung dieses sekundären Botenstoffs in der Zelle und puffern den
Calciumeinstrom über die Zellmembran oder aus dem ER ab [Nunnari und
Suomalainen 2012]. Ein für die einzelne Zelle besonders folgenreicher Prozess,
welcher zu einem großen Teil von Mitochondrien gesteuert wird und durch
Calciumsignale ausgelöst werden kann, ist die Apoptose, der kontrollierte
Zelluntergang. Apoptose kann vereinfacht betrachtet auf zwei verschiedenen Wegen
ausgelöst werden: extrinsisch (von „außen“) durch Bindung von extrazellulären
Liganden an sogenannte Todesrezeptoren, wie den Fas-Rezeptor, oder intrinsisch
(von „innen“) insbesondere nach vorheriger Schädigung der mitochondrialen
Funktion oder des Erbguts, aber auch physiologischerweise während der
Embryonalentwicklung oder in sich regenerierenden Geweben. Die intrinsische
16Apoptose wird durch ein Überwiegen pro-apoptotischer Faktoren in der Zelle und
deren Wirkung auf die Mitochondrien initiiert. Aus dem Intermembranraum der
Mitochondrien werden Signalmoleküle wie das Cytochrom C und die Cysteinyl-
Aspartat-spezifischen Proteasen (Caspasen) in das Zytosol freigesetzt. Hier kommt
es zu einer kaskadenartigen Aktivierung weiterer Caspasen und anderer Enzyme,
welche in Summe den Untergang der Zelle zur Folge haben [Jeong und Seol 2008].
Im Gegensatz zur Apoptose kommt es in Folge vieler pathologischer Vorgänge, wie
z.B. bei traumatischen Verletzungen, Infektionen oder nach Ischämie, zu einem
ungeregelten Absterben von Zellen. Diese Form des Zelluntergangs ist durch den
Verlust der Integrität der Zellmembran gekennzeichnet und geht mit einer
ausgeprägten Entzündungsreaktion einher. Der Schaden für das umliegende
Gewebe ist bei diesem nekrotischen Zelluntergang um ein Vielfaches größer als
nach dem kontrollierten Zelltod mittels Apoptose [Bohle et al. 2010].
Die im nächsten Abschnitt dargestellten Fragestellungen der Arbeit integrieren die
Lokalisation und Funktion des 1A-9Renins in den Mitochondrien mit potenziellen
Veränderungen der mitochondrialen Aktivität und Stabilität sowie dem Zellüberleben
von H9c2-Kardiomyoblasten nach einer ischämischen Phase.
171.5 Fragestellungen
Um zu überprüfen, ob die in dieser Arbeit verwendeten ischämischen Bedingungen
Einfluss auf die Expression des nicht-sekretorischen Renins nehmen, wurde
folgender Frage nachgegangen:
1. Kommt es in embryonalen H9c2-Kardiomyoblasten unter den Ischämie-
relevanten Bedingungen bestehend aus Sauerstoff- und Glukosedepletion
(OGD) zu einer selektiven Hochregulation des 1A-9Renins?
Kardioprotektive Effekte konnten bereits in transgenen Ratten sowie in embryonalen
H9c2-Zellen, die das nicht-sekretorische 2-9Renin überexprimieren, nachgewiesen
werden. Bisher ist jedoch zur Funktion des alternativen Exon1 (Exon1A) nichts
bekannt. Daraus resultierten folgende Fragen:
2. Welchen Einfluss hat unter Basalbedingungen eine moderate Überexpression
das 1A-9Renins in H9c2-Zellen auf deren Überlebensrate im Vergleich zu
Kontrollvektor-transfizierten Zellen?
3. Sind unter OGD in 1A-9Renin-überexprimierenden Zellen im Vergleich zu
Kontrollzellen ähnliche protektive Effekte auf das Zellüberleben und die Art
des Zelltodes zu beobachten?
Eine mögliche Ursache der protektiven Effekte könnte im Bereich der
mitochondrialen Funktion zu finden sein. Auch hierzu gibt es bislang keine Daten.
Daher wurde folgenden Fragen nachgegangen:
4. Kommt es in 1A-9Renin-überexprimierenden Zellen unter basalen
Bedingungen zur Umstellung mitochondrialer Funktionen und/oder des
Metabolismus?
5. Kann diese Umstellung im Sinne einer Präkonditionierung für die protektiven
Effekte unter OGD verantwortlich sein?
182 Material und Methoden
2.1 Material und Geräte
2.1.1 Chemikalien und Reagenzien
Tabelle 1: Chemikalien und Reagenzien
Substanz Hersteller
Aqua B. Braun (A. dest.) B. Braun Melsungen
Calciumchlorid (CaCl2) Carl Roth
CaspACETM FITC-VAD-FMK in Situ Marker Promega
di-Natriumhydrogenphosphat (Na2HPO4) Carl Roth
Dulbecco's Modified Eagle Medium (DMEM) Lonza
DMEM, no glucose Gibco
Ethanol absolut Th.Geyer
FACSFlow™ Sheath Fluid BD
Fetales Kälberserum (FKS) PAN Biotech
FITC-Annexin V BD Pharmingen
Geneticindisulphat (G418) Carl Roth
Glukose Carl Roth
Glutamin Carl Roth
HEPES-Puffer Serva
JC-1 invitrogen
Kaliumchlorid (KCl) Carl Roth
Kaliumdihydrogenphosphat (KH2PO4) Carl Roth
MitoTracker RedCMXRos invitrogen
Natriumazid (NaN3) Carl Roth
Natriumchlorid (NaCl) Carl Roth
Natrium-Pyruvat Carl Roth
Penicillin/Streptomycin Thermo Fisher
Phosphate-buffered-saline (PBS) PAN Biotech
Propidiumiodid Sigma
Salzsäure (HCl) 32% Carl Roth
Triton X-100 Carl Roth
Trypanblau Lösung 0,5 % Serva
19Trypsin Sigma-Aldrich
XF base medium Agilent Technologies
2.1.2 Weitere Materialien
Tabelle 2: Kommerziell erhältliche Kits
Produkt Hersteller
CellTiter-Glo Luminescent Promega
Cell Viability Assay
Cytotoxicity Detection Kit (LDH) Roche
High Capacity cDNA Kit Life Technologies
In vitro Toxicology Assay Kit (XTT based) Sigma-Aldrich
Quick-RNA Miniprep Zymo Research
Rotor-Gene SYBR Green PCR Kit QIAGEN
XF Cell Mito Stress Test Kit Agilent Technologies
Tabelle 3: Weitere Produkte
Produkt Hersteller
AnaeroPack Mitsubishi Gas Chemical Company
pIRES/Neo3-Plasmid BD Biosciences Clontech
XF96-well Platte Agilent Technologies
Zellkultur Mikroplatte, 96-well Greiner Bio-One
202.1.3 Puffer
Tabelle 4: FACS-Puffer
Substanz Endkonzentration
FKS 1%
KCl 2,7 mM
KH2PO4 1,5 mM
NaCl 140 mM
Na2HPO4 (Dihydrat) 1,1 mM
NaN3 15,4 mM
in A. dest. gelöst
Tabelle 5: Annexin-Bindungspuffer
Einstellung des pH auf 7,4 mit HCl
Substanz Endkonzentration
HEPES 10 mM
NaCl 140 mM
CaCl2 2,5 mM
in A. dest. gelöst
2.1.4 Zelllinie
Tabelle 6: Zelllinie
H9c2 ATCC, Manassas, VA, USA
2.1.5 Antikörper
Tabelle 7: Antikörper
Antigen Hersteller Wirt
FasR (CD95) Enzo, Life Science Kaninchen
Kaninchen-IgG1 (Fluorescein Dianova Ziege
isothiocyanate (FITC)-gebunden)
212.1.6 Geräte und Software
Tabelle 8: Geräte
Name Hersteller
DS-11+ Spectrophotometer DeNovix Inc
Fluorescence Activated Cell Sorting (FACS) BD
Calibur flow cytometer
GENbox Biomerieux
HERAcell 240i CO2-Inkubator Thermo Fisher
HERAsafe Sterilwerkbank Heraeus Instruments
Microplate Luminometer LB96V EG&G Berthold
MRX Microplate Reader Dynatech Laboratories
pH 211 Microprocessor pH Meter Hanna Instruments
Rotor-Gene Q QIAGEN GmbH
SureCycler 8800 Agilent Technologies
Seahorse XF Analyzer Agilent Technologies,
Seahorse Bioscience
Tabelle 9: Software
Name Hersteller
GraphPad Prism 7 GraphPad Software
222.2 Methoden
2.2.1 Zellkultur
In der vorliegenden Arbeit erfolgten alle Versuche mit embryonalen
Kardiomyoblasten der Zelllinie H9c2, welche ursprünglich aus Ratten isoliert wurden.
In flüssigem Stickstoff kryokonservierte Zellen wurden aufgetaut und in
Zellkulturflaschen in DMEM-Zellkulturmedium mit 10 % fetalem Kälberserum (FKS)
sowie 100 IU/ml Penicillin und 100 µg/ml Streptomycin bei 37 °C, 95%iger
Luftfeuchtigkeit und 5 % CO2 inkubiert. Ein Medienwechsel der Erhaltungskultur
erfolgte unter sterilen Bedingungen alle 72 h, die Trypsinierung nach 7 d. Dazu
wurden nach dem Waschen mit phosphatgepufferter Salzlösung (PBS) die
Plaste-adhärenten Zellen durch Inkubation in einer 1:10 mit PBS verdünnten
Trypsin/EDTA-Lösung für 5 min vom Boden des Kulturgefäßes abgelöst. Durch
wiederholtes Pipettieren wurden die Zellen zusätzlich mechanisch vereinzelt, in ein
Zentrifugenröhrchen überführt und anschließend zum Abstoppen der Trypsinierung in
kaltem Medium gewaschen (Zentrifugation bei 1000 U/min für 4 min). Durch Einsatz
von 1:3 in PBS verdünnter Trypanblaulösung wurde der Anteil toter Zellen an der
Gesamtzellzahl der Zellsuspension bestimmt.
In Abhängigkeit von den Versuchen wurde eine definierte Anzahl von Zellen pro
Kavität in 96-Well- oder 6-Well-Kulturplatten pipettiert. Die Anwachsphase bei 37 oC
und 5 % CO2 betrug 3 d.
2.2.2 Transfektion der Zelllinie
Die H9c2-Zellen wurden entweder ohne molekularbiologische Modifikation verwendet
oder mittels transienter Transfektion des pIRES/Neo3-Vektors genetisch verändert.
Die Transfektion und Selektion der verschiedenen Zelllinien erfolgte wie zuvor durch
Clausmeyer [Clausmeyer et al. 1999] beschrieben. Als Selektionsantibiotikum, das
während der Erhaltungskultur aber nicht während des eigentlichen
Versuchsansatzes im Kulturmedium enthalten war, kam Geneticindisulphat (G418)
zum Einsatz. Für die Untersuchungen wurden mir folgende gentechnisch-veränderte
Zelllinien zur Verfügung gestellt:
Eine pIRES/Neo3-Vektor-transfizierte Zelllinie, in die der Leervektor mit der
Antibiotikaresistenz integriert wurde. Sie stellte die primäre Kontrolle dar und aus
23dieser stammende Zellen werden im Folgenden der Einfachheit halber als
pIRES-Zellen bezeichnet.
Durch Einbringen des pIRES/Neo3-Vektors mit der Exon(1A-9)Renin-cDNA und der
Antibiotikaresistenz wurde eine Zelllinie erzeugt, die eine 4fache Überexpression des
nicht-sekretorischen 1A-9Renins aufweist.
Alle nachfolgend beschriebenen Versuche wurden im Vergleich zwischen
unbehandelten H9c2-Zellen, Leervektor-transfizierten Kontrollzellen (pIRES-Zellen)
und Exon(1A-9)Renin-transfizierten Zellen (1A-9Renin-Zellen) durchgeführt. Da
zwischen unbehandelten H9c2-Zellen und Leervektor-transfizierten Kontrollzellen
keine signifikanten Unterschiede auftraten, wurde zur besseren Übersichtlichkeit auf
die Darstellung der Ergebnisse der unbehandelten H9c2-Zellen verzichtet.
2.2.3 Ischämie-relevante Bedingungen
Zur Simulation einer Ischämie des Herzmuskels, wie z.B. im Rahmen eines
Myokardinfarktes, wurden die Zelllinien einer Sauerstoff- und Glukose-
Depletion (OGD) ausgesetzt.
Für die funktionellen Analysen wurden 1*104 Zellen pro Kavität einer 96-Well bzw.
1*105 Zellen pro Kavität einer 6-Well Kulturplatte ausgesät, für 3 d unter
Kontrollbedingungen inkubiert und anschließend für 24 h einer OGD ausgesetzt.
Glukosemangel wurde durch die Kultivierung in glukosefreiem DMEM-Nährmedium
nachgestellt. Anoxie wurde durch Einbringen von AnaeroPacks (enthalten stark
oxidierendes Material) in luftdicht verschließbaren Behältern erzeugt. Die Inkubation
der Zellen unter diesen Bedingungen wird im Folgenden als Inkubation unter
ischämischen Bedingungen bzw. OGD bezeichnet. Im Gegensatz dazu wird die
Inkubation unter Fortführung der zuvor beschriebenen Bedingungen, als Inkubation
unter basalen bzw. Kontrollbedingungen bezeichnet.
Neben den Untersuchungen zum Einfluss der OGD auf die Überlebensrate wurde
auch der Einfluss einer alleinigen Glukosedepletion bzw. Anoxie analysiert, um die
idealen Bedingungen für die Nachahmung eines Ischämie-relevanten Geschehens
zu erstellen. Da die beobachteten Effekte nur minimal ausfielen, wurden diese
beiden Interventionsgruppen nicht in die endgültige Arbeit aufgenommen. Als Folge
resultierte teils eine Abweichung der n-Zahlen zwischen den Kontroll- und den OGD-
behandelten Gruppen. Dies stellt eine Schwäche der vorliegenden Arbeit dar.
24Aufgrund der technischen Rahmenbedingungen konnten die, sich der Inkubation
unter ischämischen Bedingungen, anschließenden Analysen nicht unter
ischämischen Bedingungen fortgesetzt werden. Dies hat zur Folge, dass die hier als
„ischämischen Bedingungen“ bezeichneten Bedingungen eher einer „Ischämie mit
anschließender Reperfusion“ entsprechen. Zur Einschränkung der potenziellen
Reperfusionsphase wurden alle Analysen möglichst direkt nach Beendigung der
Inkubationsbedingungen durchgeführt. Bei einigen Versuchen, insbesondere bei der
Messung der Sauerstoffverbrauchsrate, war jedoch die Wiedereinführung von
Glukose und von Sauerstoff, nicht nur technisch unausweichlich, sondern sogar für
die Durchführung der sich anschließenden Messungen notwendig.
2.2.4 RNA-Quantifizierung
Um die Expression spezifischer Gene quantitativ untersuchen zu können, bedient
man sich der reverse-Transkriptase-Polymerase-Kettenreaktion (rtPCR) und
quantitativen-PCR (qPCR). Bei dieser Methode wurde zunächst RNA aus den zu
untersuchenden Zellen isoliert, diese mittels reverser Transkription in die
komplementäre DNA (cDNA) überführt und dann in sich wiederholenden PCR-Zyklen
vervielfältigt.
Die RNA wurde aus den untersuchten Zellen mittels Quick-RNA Miniprep-Kit
entsprechend den Herstellerangaben isoliert. Die in der jeweiligen Probe vorhandene
RNA-Konzentration wurde photometrisch gemessen, um anschließend annähernd
gleiche RNA-Mengen für die reverse Transkription einsetzen zu können. Die reverse
Transkription, also das Enzym-vermittelte Überschreiben der RNA in cDNA, wurde
mittels High Capacity cDNA-Kit nach Herstellerangaben durchgeführt. Dabei wurden
jeweils 1.000 ng RNA mit einem Gemisch zufälliger Primer, dem Enzym reverse
Transkriptase, Nukleinbasen, und einem Reaktionspuffer gemischt. Die rtPCR wurde
im Thermocycler temperaturkontrolliert bei 25 °C für 10 min, bei 37 °C für 120 min,
bei 85 °C für 5 min und anschließendem Abkühlen auf 4 °C durchgeführt. Die so
erhaltene cDNA wurde daraufhin bis zu ihrer weiteren Untersuchung bei -70 °C
gelagert.
Für die qPCR wurden Duplikate von jeweils 20 ng cDNA der entsprechenden Probe,
verdünnt in nukleasefreiem Wasser, hergestellt. Diese wurden mit SYBR® FAST
Universal 2X Master Mix vermischte, welcher den an DNA bindenden Farbstoff
SYBR®-green enthält. Außerdem wurden Gen-spezifische, optimierte Primerpaare
25bestehend aus den entsprechenden Vorwärts- und Rückwärtsprimern hinzugefügt
(Tabelle 10). Neben der Expression der verschiedenen Reninvarianten, wurde auch
die Expression des Haushaltsgens Tyrosin-3-Monooxygenase/Tryptophan-5-
Monooxygenase aktivierendes Protein, ζ (Zeta)-Polypeptid (YWHAZ) analysiert. Die
Expression dieses Gens sollte unter allen Versuchsbedingungen konstant, d.h. nicht
reguliert sein, um eine Quantifizierung der Reninexpression vornehmen zu können.
Bei der qPCR laufen, nacheinander durch Veränderung der Temperatur und
Enzymaktivität die Schritte der Denaturierung, der Primerhybridisierung und der
Elongation der cDNA ab. Theoretisch verdoppelt sich also die Menge der cDNA in
jedem Zyklus, so dass es zu einer exponentiellen Zunahme der cDNA-Menge mit
steigender Zyklenanzahl kommt. Das proportional zur vermehrten cDNA-Menge
detektierte Farbsignal der Probe erlaubt die Registrierung des Zeitpunktes d.h. der
Zyklenanzahl bei dem ein zuvor gesetzter Schwellenwert (CT; cycle threshold)
überschritten wird. Die Differenz der CT-Werte (∆CT) der beiden Renine im Vergleich
zu YWHAZ ermöglicht die Darstellung der Ergebnisse in logarithmischer Form
als 2-∆CT. Die Ergebnisse erlauben letztlich Rückschlüsse auf die Ausprägung der
jeweiligen Genexpression in den untersuchten Zellen unter Kontroll- und
ischämischen Bedingungen.
Tabelle 10: Verwendete Primer
Für die qPCR verwendete Vorwärts- und Rückwärtsprimer. Basensequenzen in
entsprechender Reihenfolge. Abkürzungen der Nukleinbasen: Adenin (A), Cytosin (C),
Guanin (G) und Thymin (T).
mRNA Vorwärtsprimer Rückwärtsprimer
YWHAZ CATCTGCAACGACGTACTGT GACTGGTCCACAATTCCTTTCTTG
CTCT
1-9 Renin ATGAATTCACCCCATTCAGC CCAGATGGGCGGGAGGAGGATG
1A-9Renin TGAATTTCCCCAGTCAGTGAT GAATTCACCCCAT TCAGCAC
2.2.5 Vitalitätsbestimmung
Nach dem Trypsinieren und zweimaligen Waschen wurden die Zellen mit 1:3 in PBS
verdünnter Trypanblaulösung gefärbt und in einer Thoma-Zählkammer
lichtmikroskopisch vergrößert gezählt. Die Trypanblau-gefärbten Zellen entsprechen
dabei den toten Zellen mit einer gestörten Membranintegrität. Der prozentuale Anteil
26ungefärbter Zellen relativiert auf die Gesamtzahl der Zellen der Zellsuspension
reflektiert die Vitalität der Zellen.
2.2.6 Durchflusszytometrie
Für die durchflusszytometrische Untersuchung, auch Fluorescence Activated Cell
Sorting (FACS) genannt, wurden in Pufferlösung suspendierte, vereinzelte Zellen
zunächst mit einem oder mehreren fluoreszenten Farbstoffen markiert. Die Bindung
der Farbstoffe an die Zellen kann durch Antikörper an spezifische subzelluläre
Strukturen vermittelt werden oder unspezifisch durch Penetration der Zellmembran
und Anreicherung des Fluorophors in subzellulären Zellkompartimenten erfolgen.
Nach Inkubation mit den entsprechenden Farbstoffen, wurden die Zellen mittels
FACS einzeln entsprechend ihrer Fluoreszenzeigenschaften analysiert. Dazu wurden
die Farbstoffe der beladenen Zellen mit einem Laser angeregt und die Emissionen
typischerweise in zwei Fluoreszenzbereiche (FITC-gebundene Marker: grün;
Propidiumiodid (PI): rot) detektiert. Die resultierenden Daten wurden in einem
Histogramm dargestellt. Durch das Setzen von definierten Fenstern und Grenzwerten
ließen sich die Zellen in Subgruppen eingrenzen. Für jeden gemessenen Parameter
konnten die Zellen dann entsprechend ihres Fluoreszenzsignals als positiv oder
negativ, oder entsprechend ihrer Lage als ober- oder unterhalb des gesetzten
Grenzwertes liegend eingeteilt werden. Die Daten von 5000 Zellen wurden bezüglich
des prozentualen Anteils fluoreszierender Zellen und der Fluoreszenzintensität der
markierten Zellen erfasst. Durch entsprechendes Setzen des Analysefensters
wurden einerseits intakte Zellen von apoptotischen Zellen differenziert (andere
Eigenfluoreszenz) und Zellbruchstücke von den Bestimmungen ausgeschlossen.
Die Bestimmung des Anteils apoptotischer Zellen erfolgte durch drei Indikatoren:
FITC-Annexin V (Annexin V), CaspACE FITC-VAD-FMK in situ Marker (CaspACE),
und den Fas-Rezeptor (FasR)-Antikörper. Annexin V ist ein Phospholipid-
Bindungsprotein und bindet an Phosphatidylserinreste, welche sich normalerweise
an der Zellmembraninnenseite befinden und bei der intrinsischen Form der Apoptose
auf die Außenseite der Zellmembran transloziert werden [Cederholm und Frostegård
2007]. CaspACE bindet nach Penetration der Zellmembran als Inhibitor an die, in der
späten Phase der Apoptose aktivierten, Caspasen. Der FasR-Antikörper bindet an
den Fas-Rezeptor, welcher besonders bei Zellen die sich in extrinsischer Apoptose
befinden, auf der Zelloberfläche exprimiert wird [Jeong und Seol 2008]. Parallel zu
27den Apoptosemarkern wurden die Zellen mit Propidiumiodid (PI) inkubiert, um zeitige
und späte Formen des apoptotischen Zelluntergangs differenzieren zu können. Erst
bei später Apoptose geht die Membranintegrität verloren, so dass PI in die Zellen
diffundieren und dort an Nukleinsäuren binden kann. [Bertuzzi et al. 1990]
Im Hinblick auf den unkontrollierten Zelltod, die Nekrose, lassen sich mittels
Durchflusszytometrie lediglich frühe Formen untersuchen, bei denen zwar die
Membranintegrität gestört, die Zellstruktur aber noch erhalten ist. Späte Formen der
Nekrose sind dagegen durch den Verlust der Zellintegrität nicht mehr als Einzelzellen
im Durchflusszytometer erfassbar.
Es wurden jeweils 1*105 Zellen mit den entsprechenden Fluorophoren inkubiert. Der
Nachweis der Gesamt-Apoptose erfolgte mit 5 µl des 1:100 in FACS-Puffer
verdünnten CaspACE FITC-VAD-FMK-Farbstoffs über 20 min bei 37°C in 50 µl
FACS-Puffer im CO2 Inkubator. Zur Differenzierung der intrinsischen Apoptose
wurden die Zellen mit 5 µl FITC-Annexin V in 100 µl Annexin-Bindungspuffer über
15 min bei Raumtemperatur in Dunkelheit inkubiert. Der Anteil extrinsischer
Apoptose wurde durch Inkubation der Zellen mit 1,5 µl des primären nicht-markierten
anti-Fas-Rezeptor-Antikörpers in 100 µl FACS-Puffer über 15 min bei 4°C ermittelt.
Anschließend folgte ein zweiter Inkubationsschritt mit 10 µl des 1:10 verdünnten
anti-Kaninchen-IgG-FITC-Antikörpers über 15 min bei 4°C. Nach jedem
Inkubationsschritt wurden die Zellen mit 3 ml FACS-Puffer, respektive 3 ml Annexin-
Bindungspuffer gewaschen, um ungebundenen Farbstoff oder Antikörper zu
entfernen. Direkt vor der Messung wurden den Zellen zusätzlich 500 ng/ml PI für
5 min zugesetzt.
Um Aussagen über den Zustand der Mitochondrien treffen zu können, wurden zwei
Farbstoffe verwendet, der MitoTrackerRed CMXRos (kurz MitoTracker genannt) und
der JC1-Farbstoff (5,5’,6,6’-Tetrachloro-1,1’,3,3’-Tetraethylbenzimidazolylcarbo-
cyaniniodid). MitoTracker wird passiv über die Zellmembran aufgenommen und
besitzt eine Thiol-reaktive Chloromethylgruppe, welche für die Interaktion mit
Mitochondrien verantwortlich ist. Die Intensität der Färbung ist ein grober Indikator für
das mitochondriale Membranpotenzial und den Funktionszustand der Mitochondrien
allgemein [Poot et al. 1996]. JC-1 lässt spezifischere Aussagen über die Höhe des
mitochondrialen Membranpotenzials zu. Er wird selektiv durch Mitochondrien
aufgenommen und weist als Monomer eine grüne Fluoreszenz auf. In niedriger
28Konzentration sowie bei niedrigem mitochondrialen Membranpotenzial liegt JC-1 als
Monomer vor. Steigt die Konzentration über 0,1 µM an oder liegt ein hohes
mitochondriales Membranpotenzial vor, kommt es zur Dimerisierung von JC-1. Diese
als J-Aggregate bezeichneten Dimere weisen eine rote Fluoreszenz auf. Durch
Bestimmung des Verhältnisses zwischen roter und grüner Fluoreszenz können somit
Rückschlüsse auf die Höhe des mitochondrialen Membranpotenzials gezogen
werden [Smiley et al. 1991]. Da Veränderungen des mitochondrialen
Membranpotenzials mit massiven Konsequenzen für die mitochondriale Aktivität und
die zelluläre Vitalität verbunden sind, eignet sich JC-1 besonders gut als Marker.
Die vorbehandelten Zellen wurden in 500 µl DMEM-Kulturmedium mit 5 µl des 1:100
verdünnten MitoTracker Farbstoffes für 45 min bei 37 °C im CO2-Inkubator inkubiert.
Für die Färbung mit JC-1 wurden die Zellen in 500 µl DMEM-Medium mit 5 µl der
1:10 verdünnten JC-1-Lösung für 10 min bei 37 °C in Dunkelheit inkubiert.
Anschließend wurde der nicht in die Mitochondrien aufgenommene, überschüssige
Farbstoff durch Waschen mit FACS-Puffer entfernt. Nach der Zentrifugation und
Resuspension in 500 µl Medium wurden die Zellen am Durchflusszytometer
analysiert.
2.2.7 Photometrie
Die Photometrie ist ein Messverfahren, bei welchem die Extinktion von Licht
bestimmter Wellenlängen gemessen wird. Damit erlaubt sie eine Quantifizierung des
Farbstoffgehaltes von Lösungen und lässt Rückschlüsse z.B. auf Proteingehalte oder
Enzymaktivitäten zu. Für die Untersuchungen wurden jeweils 1*104 Zellen in eine
Kavität einer 96 Well-Zellkulturplatte pipettiert und für 24 h zum Anwachsen im
CO2-Inkubator inkubiert. Anschließend wurden die vorkultivierten Zellen Kontroll-
bzw. ischämischen Bedingungen ausgesetzt. Als Negativkontrollen für die
photometrischen Analysen dienten Kavitäten, in denen das Medium und alle
Komponenten des eingesetzten Kits aber keine Zellen enthalten waren.
Beim Zelluntergang durch Nekrose kommt es zu einem Verlust der Integrität der
Zellmembran und damit zu deren Perforation. Die Zelle verliert letztlich ihre Struktur
und ist nicht mehr als abgrenzbare Einheit vorhanden. Im Rahmen dieser Vorgänge
kommt es in-vitro zur Freisetzung von intrazellulären Bestandteilen ins Medium, z.B.
der im Zytosol lokalisierten Laktatdehydrogenase (LDH).
29Sie können auch lesen

















































