BLAST Basic Local Alignment Search Tool - Martin Winkels 21.12.2012 - Medizinische Fakultät ...
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
BLAST Basic Local Alignment Search Tool Martin Winkels 21.12.2012 wissen leben Institut für WWU Münster Medizinische Informatik
Inhaltsverzeichnis Inhaltsverzeichnis
Inhaltsverzeichnis
1 Einleitung 2
2 Grundlagen 3
2.1 DNA - Desoxyribonukleinsäure . . . . . . . . . . . . . . . . . . . . . . . . 3
2.2 Proteine - Aminosäuren . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2.3 DNA/Protein-Sequenzalignment . . . . . . . . . . . . . . . . . . . . . . . . 5
2.3.1 Beispiel: Globales Alignment . . . . . . . . . . . . . . . . . . . . . . 6
2.3.2 Beispiel: Lokales Alignment . . . . . . . . . . . . . . . . . . . . . . 7
2.4 Maximal Seqment Pair Measure . . . . . . . . . . . . . . . . . . . . . . . . 8
2.5 Dynamische Programmierung . . . . . . . . . . . . . . . . . . . . . . . . . 8
2.6 Heuristische Algorithmen . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
3 BLAST 11
3.1 Konzept . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
3.1.1 Beispiel BLAST . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
3.1.2 Besonderheiten der Analyse von DNA-Sequenzen . . . . . . . . . . 14
3.1.3 Signifikante Ähnlichkeiten von Sequenzen . . . . . . . . . . . . . . . 14
3.2 Performance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
3.2.1 Wortlänge und Schwellwerte . . . . . . . . . . . . . . . . . . . . . . 15
3.2.2 Proteine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
3.2.3 DNA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
4 Fazit 18
5 Literaturverzeichnis 19
6 Anhang 21
1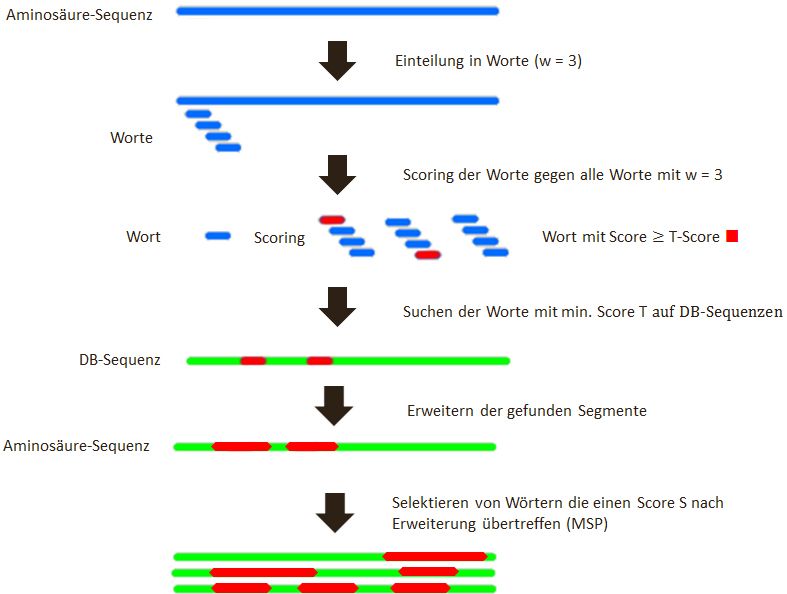
1 EINLEITUNG
1 Einleitung
Der Mensch besitzt etwa 20.000-25.000 Gene, die für die Produktion von Proteinen
verantwortlich sind. Daraus werden 500.000-1.000.000 Proteine gebildet. Das Genom des
Menschen ist komplett entschlüsselt, die Funktion jedoch größtenteils unbekannt [9].
Der Vergleich von neu sequenzierten Proteinen oder DNA mit bekannten Samples aus
einer Datenbank kann einen ersten Hinweis auf die Funktion dieser geben [2]. Die Sample-
Datenbanken sind jedoch groß und wachsen immer schneller. Mehr und mehr Institutionen
machen ihre Samples verfügbar oder speisen neue Samples in die Datenbanken ein [6, 13].
Es wird viel Rechnerkapazität benötigt, diese zu durchsuchen, um neu sequenzierte
Samples mit entsprechenden Einträgen in den Datenbanken zu vergleichen. Weiterhin
steigt die benötigte Rechenzeit für den Vergleich mit zunehmender Größe der Datenbank
stark an. Algorithmen der dynamischen Programmierung“ besitzen einen Aufwand von
”
O(n2 ) − O(n3 ) [8, 12, 14]. Eine akzeptable Laufzeit für diese Algorithmen wird nur mit
Supercomputern oder spezieller Hardware erreicht. Dynamische Programmierung ist so-
mit kein praktikabler Ansatz große Datenmengen zu vergleichen, auch wenn ihre Lösung
optimal ist.
Einen Lösungsansatz für dieses Problem bietet jedoch die Heuristik. Heuristische Ansätze
können das Problem aufgrund ihres systematischen Ansatzes nach einer bestimmten Stra-
tegie lösen. Durch Annahmen und Vorentscheidungen wird die Laufzeit um ein vielfaches
reduziert. Der Rechenaufwand verringert sich mindestens um eine Potenz [2, 3]. Der heu-
ristische Ansatz kann die optimale Lösung allerdings nicht garantieren.
Die von Altschul et al. vorgestellte Arbeit Basic Local Alignment Search Tool“ - kurz
”
BLAST [2] beschäftigt sich mit einem Algorithmus, der es mit Methoden der Heuristik
möglich macht diese Samples mit entsprechend weniger Rechenaufwand zu vergleichen.
Diese Arbeit wird sich mit den Hintergründen, den Vorteilen, den Nachteilen und den
Alternativen zu diesem Algorithmus beschäftigen. Weiterhin werden die Grundlagen von
BLAST erläutert.
Das Paper ist 12 Jahre alt, gehört aber immer noch zu der wichtigsten Literatur für
diesen Algorithmus. BLAST ist heutzutage in vielen Variationen im Einsatz [4, 7, 3]. Es
wird jedoch nicht auf die neueren Ansätze zu diesem Themengebiet oder die Weiterent-
wicklungen von BLAST eingegangen. Vielmehr soll nur ein erster Einblick in das Thema
gegeben werden und der BLAST-Algorithmus in seinen Einzelheiten und Eigenschaften
vorgestellt werden.
2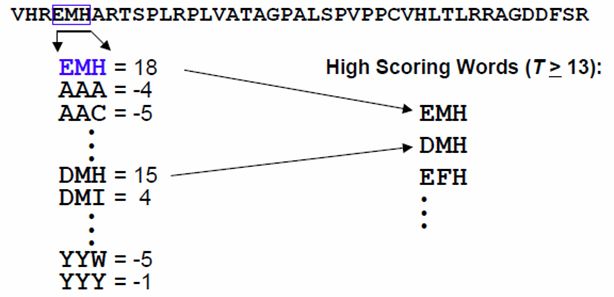
2 GRUNDLAGEN
2 Grundlagen
Im Folgenden werden biologische und technische Grundlagen erläutert, um die Pro-
blemstellung zu verdeutlichen.
2.1 DNA - Desoxyribonukleinsäure
Die Desoxyribonukleinsäure, kurz DNS, ist Träger der Erbinformationen, der Gene.
Die DNS wird häufig als DNA abgekürzt nach ihrem englischen Namen ”deoxyribonucleic
acid”. Bausteine der DNA sind die Nukleotide. Nukleotide sind aufgebaut aus einem
Zucker, einem Phosphat-Rest sowie einer von vier Basen. Abb. 1 zeigt eine solche DNA-
Doppelhelix aus zwei Nukleotidsträngen.
Abbildung 1: DNA Doppelhelix als Riesenmolekül aus zwei Nukleotidsträngen mit Basen
sowie dem gelben Phosphatrückgrat
Die DNA ist ein in allen Lebewesen vorkommendes Biomolekül, welches die Informati-
onen für die Herstellung von Ribonnukleinsäuren, kurz RNA, enthält. Es gibt verschiedene
Gruppen von RNAs, die für diesen Artikel wichtige Gruppe, die messanger RNA - mRNA,
enthält die Informationen, um Proteine zu synthetisieren. Welches Protein erzeugt wird
hängt von der Reihenfolge, der Sequenz der Basen ab. Es stehen jeweils drei Basen für
eine bestimmte Aminosäure. Die RNA unterscheidet sich von der DNA nur durch eine
Base. Thymin wird bei der RNS durch Uracil ersetzt.
Die mRNA transkribiert einen zu einem Gen gehörenden Abschnitt auf der DNA.
Anschließend wird die RNA noch durch Splicen weiter verändert. Nachfolgend wird die
3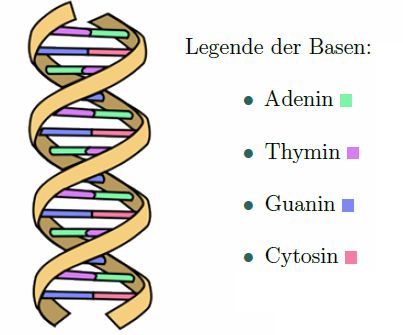
2.2 Proteine - Aminosäuren 2 GRUNDLAGEN
prozessierte mRNA in das Cytoplasma transportiert und an den Ribosomen für die
Proteinsynthese translatiert [10].
2.2 Proteine - Aminosäuren
Proteine sind Makromoleküle, die aus bis zu 20 unterschiedlichen Aminosäuretypen
aufgebaut sind. Die Sequenz der Aminosäuren ist durch die Reihenfolge der Basen in
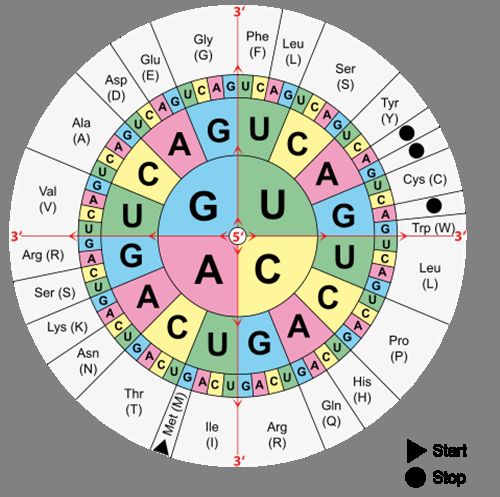
der DNA vorgegeben. Abb. 2 zeigt die Reihenfolge von Basen der mRNA sowie deren
Übersetzung in eine Aminosäure. Start und Stopp bezeichnen dabei jeweils den Anfang
bzw. das Ende einer Aminosäuresequenz. Proteine können aus mehreren zehntausend
Aminosäuren bestehen oder aus nur zwei bis drei. Die meisten Proteine bestehen aus
100-300 Aminosäuren. Proteine können weiterhin noch nach ihrer Synthese durch Enzyme
verändert werden.
Abbildung 2: Code-Sonne: Zeigt die Codierung der Aminosäuren (außen) durch die Ba-
sentripletts auf der mRNA. Sie wird von innen (5’) nach außen (3’) gelesen.
Die Funktion von Proteinen im Körper ist vielfältig. Sie kommen in jeder Zelle vor
und verleihen ihnen neben einer Struktur auch weitere Funktionen. Sie werden auch als
”Molekulare Maschinen”bezeichnet. Aufgrund der überaus vielfältigen Aufgaben die Pro-
teine im Organismus haben können, sollen hier nur einige beispielhaft aufgezählt werden.
4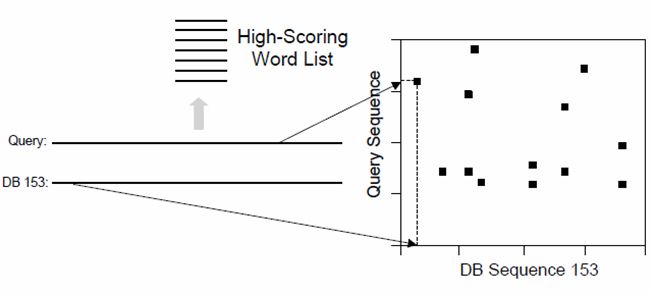
2.3 DNA/Protein-Sequenzalignment 2 GRUNDLAGEN
Sie sind als Antikörper daran beteiligt, Infektionen abzuwehren sowie als Strukturproteine
für den Aufbau einer Zelle verantwortlich. Ebenfalls sind sie als Enzyme am Metabolismus
beteiligt [10].
2.3 DNA/Protein-Sequenzalignment
Sequenzalignment bezeichnet das Vergleichen zweier oder mehrerer Strings. Sie wird
verwendet, um funktionale oder evolutionäre Homologie zwischen Nukleotidsequenzen
(DNA) oder Aminosäuresequenzen (Proteine) zu bestimmen. Sequenzen liefern Informa-
tionen über die Abfolge und Position der Basenpaare. Gesucht werden charakteristische
Abschnitte auf einer Sequenz. Werden zwei Sequenzen verglichen, nennt sich dies paarwei-
ses Alignment, bei mehreren Sequenzen multiples Alignment. Gesucht werden Sequenzen,
die unter Verwendung einer Scoring-Kostenfunktion einen optimalen Score liefern. Wie die
Kostenfunktion aussieht, kann unterschiedlich sein. So können Matches stärker belohnt
oder Missmachtes stärker bestraft werden. Auch das Einfügen von Gaps (Lücken) in das
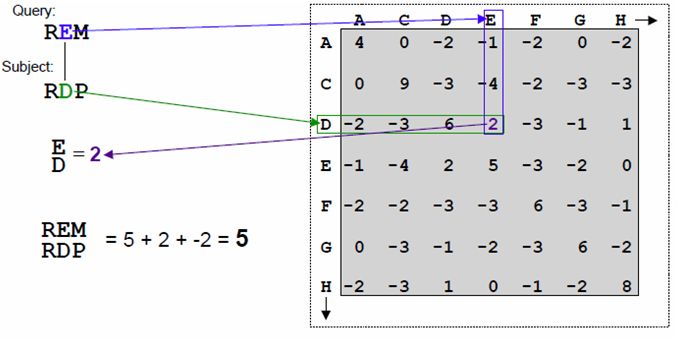
Alignment kann in die Kostenfunktion einbezogen werden. Formel (1) zeigt ein Beispiel für
solch eine Kostenfunktion. Ebenfalls können ähnliche Paarungen berücksichtigt werden.
Sie haben einen positiven Wert. Nur schwach ähnliche oder gar nicht ähnliche Paarungen
haben einen negativen Wert [5, 12].
Zu unterscheiden sind automatisierte und manuelle Alignment-Methoden. Manuelles
Alignment findet nur bei kleinen Datensätzen statt und bietet den Vorteil der höheren
Genauigkeit. Automatisierte Methoden sind wesentlich schneller, können aber aufgrund
ihres Charakters nicht oder nur begrenzt auf individuelle Besonderheiten in Sequenzen
eingehen.
+5 a = b match
δ(a, b) = (1)
−4 a =
/ b missmatch
Das Scoring von DNA und Proteinen unterliegt unterschiedlichen Ansätzen. Beiden
gleich ist, dass sie eine Scoring-Matrix oder Kostenfunktion nutzen, die für alle möglichen
Paarungen einen gewissen Wert (Score) ausweist. Diese Matrix nennt sich Ähnlichkeits-
matrix oder matrix of similiarity [2].
Für Proteine wird oft eine PAM-120 oder PAM-250 (Point Accepted Mutation Matrix)
Matrix genutzt, um alle möglichen Paarungen mit einem Wert zu versehen. Die Matrix
wird verwendet, da sich mit zunehmender Auseinanderentwicklung der Sequenzen im-
mer mehr Mutationen ansammeln. Allerdings mutieren nicht alle Aminosäuren mit der
52.3 DNA/Protein-Sequenzalignment 2 GRUNDLAGEN
gleichen Wahrscheinlichkeit zu einer anderen Aminosäure. Eine saure Aminosäure wird
wahrscheinlich mit einer anderen sauren Aminosäure getauscht. Dies wird bei der PAM-
Matrix berücksichtigt. Die Zahl gibt an, in welcher Höhe Mutationen akzeptiert werden.
Eine PAM-120 Matrix akzeptiert eine Mutation bis zu 40%. Eine PAM-250 akzeptiert
sogar Mutationen bis zu 80% [1]. Bei noch höheren Werten spricht man nicht mehr von
Ähnlichkeiten. Im Gegensatz dazu ist die DNA stark zufallsbasiert.
Es können globale oder lokale Homologien ausgewiesen werden. Globale Homologie be-
zeichnet die Ähnlichkeit über die komplette Sequenz worin Strecken von geringer Ähnlich-
keit enthalten sein können. Bei globalem Alignment dürfen Gaps (Lücken) hinzugefügt
werden. Lokale Homologie bezeichnet das Suchen von Homologien in kleineren Abschnit-
ten auf der gesamten Sequenz. Ausgegeben werden dann mehrere kleinere Subsequenzen
pro Sequenz die eine Ähnlichkeit aufweisen. Ob bei lokalem Alignment Gaps hinzugefügt
werden dürfen, kommt auf die Methode an. Wird nach cDNA (complementary DNA)
gesucht werden lokal-basierte Ansätze genutzt, da zugeordnete Proteine nur einzelne, iso-
lierte Ähnlichkeiten aufweisen. Kombinierte Ansätze werden ebenfalls genutzt um die
Nachteile beider Methoden auszugleichen [5]. Die Unterschiede von globalem und lokalem
Alignment sind in Abschnitt 2.3.1 und 2.3.2 in einem Beispiel dargestellt.
Für das Alignment werden die Elemente der zu untersuchenden Sequenz so angeordnet,
dass die Reihenfolge erhalten bleibt und jedem Element ein anderes Element oder ein Gap
zugeordnet werden kann. Eine Fehlpaarung entspricht einer Mutation. Vorhandene Gaps
hingegen weisen auf Deletion oder Insertion hin. Gesucht wird in den einander zugeord-
neten, also alignierten, Elementen nach identischen oder möglichst ähnlichen Elementen,
da viele gleiche bzw. ähnliche Elemente in gleicher Reihenfolge auf eine Verwandtschaft
hinweisen.
2.3.1 Beispiel: Globales Alignment
Gegeben sind die beiden Sequenzen S = ACG und T = CG
Kostenfunktion:
+5 a = b match
δ(a, b) = (2)
−4 a =
/ b missmatch
Gesucht ist die Lösung mit dem höchsten Score. Es dürfen Gaps eingefügt werden, die
Reihenfolge der Basen darf aber nicht verändert werden. Für jedes richtige Alignment
werden nach der Kostenfunktion fünf Punkte addiert, für jedes falsche Alignment vier
62.3 DNA/Protein-Sequenzalignment 2 GRUNDLAGEN
Punkte abgezogen.
Abbildung 3: Möglichkeiten des Alignments. Möglichkeit drei bietet die beste Lösung
2.3.2 Beispiel: Lokales Alignment
Gegeben sind die beiden Sequenzen S = AAAACCG und T = CCG
Kostenfunktion:
+5 a = b match
δ(a, b) = (3)
−4 a =
/ b missmatch
Gesucht ist die Lösung mit dem höchsten Score. Es dürfen keine Gaps eingefügt werden,
die Reihenfolge der Basen darf nicht verändert werden. Die zweite Sequenz wird nur durch-
geschoben. Für jedes richtige Alignment werden nach der Kostenfunktion fünf Punkte
addiert, für jedes falsche Alignment vier Punkte abgezogen.
72.4 Maximal Seqment Pair Measure 2 GRUNDLAGEN
Abbildung 4: Möglichkeiten des Alignments. Möglichkeit drei bietet die beste Lösung
2.4 Maximal Seqment Pair Measure
Methoden, um Ähnlichkeiten in Sequenzen zu finden und zu bewerten, werden ”similarity
measures”genannt. Die Methode, die BLAST nutzt, nennt sich Maximal Seqment Pair
”
Measure“ und basiert auf dem lokalen Alignment. Sie wird bei automatisierten Alignment
häufig verwendet [15].
Ein Maximal Seqment Pair ist ein aligntes Paar von zwei Sequenzen, die nach Anwen-
dung der Kostenfunktion einen Score aufweisen, der über einem vorher festgelegten Score
S liegt. Das hat den Hintergrund das Ähnlichkeiten in Sequenzen meist zu klein sind, um
biologisch relevant zu sein. Erst Worte die einen Wert größer einem Score S erzielen sind
deshalb für den Forscher interessant [11].
Die Länge eines MSP wird so gewählt, dass der lokale Score maximiert wird. Weiterhin
wird definiert, dass ein Segmentpaar dann lokal maximal ist, wenn ihr Score nicht mehr
durch Erweitern des Segments auf der Sequenz erhöht werden kann. Der MSP Score
kann sowohl von dynamischer Programmierung als auch von heuristischen Algorithmen
berechnet werden [15, 5].
2.5 Dynamische Programmierung
Bewertungsalgorithmen der ”Dynamischen Programmierung”nutzen verschiedene Va-
riationen dynamischer Algorithmen [2]. Die Algorithmen haben gemeinsam, dass sie einen
82.6 Heuristische Algorithmen 2 GRUNDLAGEN
Scorewert erstellen der auf Einfügen, Löschen und Ersetzen von zwei Sequenzen ba-
siert. Die Variante mit dem höchsten Score aller Permutationen zweier Sequenzen wird
dann ausgegeben. Dadurch wird die Ähnlichkeit zweier Sequenzen berechnet. Dieser An-
satz ist sehr rechenaufwendig, aber durch seine Einfachheit auch leicht nachvollzieh-
bar und überprüfbar. Das Finden der optimalen Lösung ist garantiert. Algorithmen der
”dynamischen Programmierung”besitzen einen hohen Aufwand, der im Bereich des qua-
dratischen oder kubischen Wachstums angesiedelt ist.
Der Needleman-Wunsch- und der Earley-Algorithmus besitzen eine Komplexität von
O(n3 ) [8, 12]. Der Smith-Waltermann-Algorithmus eine Komplexität von O(n2 ) [14].
Aufgrund der Anforderungen an die Rechenkapazität sind sie nicht für große Daten-
banken nutzbar, falls nicht Supercomputer oder spezielle Hardware verwendet werden.
2.6 Heuristische Algorithmen
Heuristik bedeutet, die Komplexität eines gegebenen Problems durch Mutmaßungen,
die durch Kenntnis des Systems erlangt werden, zu reduzieren. Der Rechenaufwand wird
also durch Hypothesen reduziert. Mit begrenztem Wissen und geringem Zeitaufwand wird
durch analytisches Vorgehen eine Lösung berechnet. Während die dynamische Program-
mierung jede Permutation berechnet, geht der heuristische Algorithmus nach einem Sys-
tem vor, das auf die Problemstellung zugeschnitten wird.
Zur Verdeutlichung dieses Sachverhalts wird im Folgenden ein Beispiel skizziert:
• Beispiel: Bilde aus den Buchstaben A B E E K N T T T ein Wort.“
”
• Dynamischer Algorithmus:
∗ Jegliche Buchstabenkombinationen berechnen und vergleichen
• Heuristischer Algorithmus:
∗ Ansatz: Das Wort besteht aus Teilwörtern die sich zu einem Wort
zusammensetzen lassen
∗ Die Buchstabenkombination wird zuerst nach Teilwörtern durchsucht
∗ Es werden entsprechnde Teilwörter gebildet und zusammengesetzt
∗ Beispiele für Teilwörter: Tee, Beet, Bett, Tante, Kante
∗ Zusammensetzung der Teilwörter zu einem ganzen Wort. Bett + Kante
∗ Systematisch zur Lösung: ”Bettkante”
92.6 Heuristische Algorithmen 2 GRUNDLAGEN
Durch die Vorannahmen und den Einsatz eines Wörterbuchs lässt sich der Rechenauf-
wand reduzieren. Das resultiert in einem schnelleren Ansatz, da viele Berechnungen die
nicht für das Endresultat notwendig sind, ausgelassen werden. Ist die Strategie allerdings
nicht perfekt, was sie in der Praxis nie sein kann, kann es auch darin münden, dass gar kein
oder nur ein suboptimales Ergebnis berechnet wird. Durch den Vergleich einer Heuristik
mit der optimalen Lösung kann die Güte dieser Heuristik bestimmt werden.
103 BLAST
3 Basic Local Alignment Search Tool
3.1 Konzept
BLAST ist ein heuristischer Algorithmus der ausgehend von der Tatsache, dass Ähnlich-
keiten in Wörtern erst ab einem Score S (MSP) relevant sind. Datenbanksequenzen werden
erst mit kleinen Teilwörtern der Größe w und des Score T durchsucht, um sie anschließend
lokal zu erweitern. Lokal erweiterte Worte müssten anschließend den Score S übertreffen,
um ausgegeben zu werden [2]. Dies wird im nachfolgendem Absatz schematisch sowie in
einem Beispiel praktisch erläutert. Die Laufzeitkomplexität von O(n · m) ergbit sich aus
der Länge der Eingangssequenz multipliziert mit der gesamten Länge der Datenbank. [3]
Abbildung 5: Schematischer Ablauf des BLAST-Prozesses
Abb. 6 zeigt den schematischen Ablauf des BLAST Algorithmus bei Aminosäuresequenzen.
Die Eingangssequenz (blau) wird zuerst in Worte mit w = 3 eingeteilt. Anschließend wer-
den diese Worte jeweils einzeln gegen alle Worte mit w = 3 gescort. Worte deren Score
113.1 Konzept 3 BLAST
einen Score T überschreitet werden übernommen und anschließend auf den Sequenzen
in der Datenbank gesucht. Hits, also Treffer auf der Datenbank werden anschließend er-
weitert bis zu einem Cut-Off Score. Anschließend wird der beste Score des Hits darauf
überprüft, ob er den Score S erreicht hat. Wird der Score S erreicht, wird dieses Wort
ausgegeben. Das wird mit allen Wörtern mit einem Score T wiederholt. Anschließend
erhält man eine Liste mit Wörtern, die den Score S überschreiten [2].
3.1.1 Beispiel BLAST
Die Eingangssequenz wird in Wörter der Größe w = 3 geteilt.
Durch die PAM-Socring Matrix werden die Wörter gewertert (gescored)
Alle Worte der Größe w = 3 werden gegen jeden Wort der Eingangssequenz mit w = 3
gescored. Anschließend werden alle Wörter die einen empirisch ermittelten Score T errei-
chen oder überschreiten ausgewählt und die ”High Scoring Words List”übernommen.
123.1 Konzept 3 BLAST
Jedes Wort der High Scoring Words List wird mit allen Datenbanksequenzen verglichen.
Das Wort wird dabei durch die Datenbanksequenz durchgeschoben. Hits, also Sequenzen
auf der Datenbank die einem Wort der Liste gleichen, werden markiert.
Die gefundenen Hits werden erweitert bis zu einem Cut-Off Score. Anschließend wird
das Segment des erweiterten Teilworts mit dem höchsten Score ausgewählt. Segmente, die
nach der Erweiterung mindestens den Score S erreichen, werden ausgegeben.
133.1 Konzept 3 BLAST
3.1.2 Besonderheiten der Analyse von DNA-Sequenzen
Die Verwendung von BLAST bei Aminosäuren und DNA unterscheidet sich im Detail.
Bei der DNA werden alle aufeinanderfolgenden Worte der Wortgröße w = 12 gewählt.
Damit enthält die Wortliste n - w + 1 Worte. DNA-Sequenzen sind stark zufallsbasiert
und in der Regel wesentlich länger als Aminosäureketten. Daraus Ergibt sich die längere
Wortliste sowie der Verzicht auf ein T-Scoring für die Vorauswahl.
DNA-Sequenzen werden weiterhin noch nach zwei häufig auftretenden Merkmalen ge-
filtert, die die Analyse negativ beeinflussen. Diese sind lokal fehlerbehaftete Basen durch
A+T reiche Regionen und sich wiederholende Sequenzelemente mit Alu Sequenzen. Durch
Entfernung dieser Worte mittels Frequenzfiltern wird die Wortliste verkleinert.
3.1.3 Signifikante Ähnlichkeiten von Sequenzen
Eines der wichtigsten Features von BLAST ist es, dass nicht nur Paare mit einem Score
S in akzeptabler Zeit gefunden werden können, sondern auch, dass die Signifikanz eines
Paars oder einer Gruppe von Paaren ausgegeben werden kann. Signifikanz bedeutet, dass
die Beobachtung nicht zufällig ist. Werden zwei Sequenzen verglichen, werden allein durch
Zufall schon entsprechend viele Paare mit einem Score S gefunden. Wird ein Signifikanz-
niveau von α = 0.05 gewählt, kann mit 95% Sicherheit davon ausgegangen werden, das
die Beobachtung nicht zufällig war [2, 11].
Folgende Formeln werden von BLAST für die Berechnung der Signifikanz genutzt:
1 − ey (4)
y = K · m · n · e−λS (5)
Hierbei bezeichnen K und λ den geschätzten Parameter [11], m die Gesamte Länge
der Sequenz in der Datenbank, n die Länge der Query Sequenz sowie S den minimalen
MSP-Score.
Mit (4) lässt sich berechnen wie wahrscheinlich es ist, ein Paar mit einem Score ≥ S zu
finden. Formel (5) gibt an, wie viele Paare mit einem Score ≥ S durch Zufall gefunden
werden.
143.2 Performance 3 BLAST
c−1 i
X y
1 − ey (6)
i=0
i!
Die dritte Formel (6) gibt die Wahrscheinlichkeit an, c oder mehr Paare ≥ S zu finden.
Auch wenn einzelne Paare keine signifikante Abhängigkeit besitzen, können mehrere Paare
in ihrer Gesamtheit dennoch signifikant sein. Das BLAST diese Abhängigkeiten berechnen
und ausgeben kann ist eines der wichtigsten Features von BLAST.
3.2 Performance
3.2.1 Wortlänge und Schwellwerte
Um eine geeignete Wortlänge sowie Schwellwerte zu bestimmen, wurde eine randomi-
sierte Datenbank von 16.000 Proteinsequenzen mit einer Länge von 250 erstellt. Die Zahl
von 250 wurde gewählt, da sie der durchschnittlichen Länge eines Proteins entspricht.
Die Ergebnisse von BLAST auf dieser Datenbank ergaben, dass MSPs ab einem Wert
von 55 signifikant waren (Anhang - Table 1, Anhang - Figure 1). Mit einer Wortlänge von
w = 4 und einem HSP-Wert von T = 17 werden rund 19% der MSPs nicht erkannt. Ab
einem MSP von 70 waren es 11%. Den Erwartungen entsprechend kann der Wert w oder T
verringert werden, um eine höhere Genauigkeit zu erzielen. Für w = 4 und T = 14 sinkt
der Wert auf 2% (S = 55) bzw. 0% (S = 70). Die Parameterpaare (w = 3, T = 14),
(w = 4, T = 16) sowie (w = 5, T= 18) liefern ähnliche Ergebnisse.
Wird der Parameter w erhöht, kann die Rechenzeit bei der Erweiterung der Wörter
reduziert werden. Allerdings werden für Proteine 20w mögliche Wörter erzeugt. Dement-
sprechend werden mit höherem w auch wesentlich mehr Worte erzeugt (w = 3 = 296,
w = 4 = 3561, w = 5 = 40.939). Dies erhöht die Rechenzeit bei der Generierung der
Wörter exponentiell (Anhang - Figure 2). In der Praxis hat sich deshalb der Wert von
w = 4 als am besten geeignet erwiesen.
Eine Reduzierung des Parameters T erhöht die Sensitivität, allerdings erhöht es eben-
falls die Rechenzeit, da mehr Wörter und mehr Hits generiert werden. Für eine genaue-
re Überprüfung wurde eine Sequenz der Länge 250 mit der PIR Datenbank verglichen
(Anhang - Table 2, Anhang - Figure 2). Der Wert T reichte hier von 13 bis 20. Die Anzahl
der Worte erhöht sich exponentiell mit fallendem T. Während die Worte w in Abhängigkeit
von T exponentiell wächst, erhöht sich die Anzahl der Worte w lediglich linear mit der
Länge der Eingangssequenz. Eine Verdopplung der Länge der Eingangssequenz resultiert
also lediglich in einer Verdopplung der generierten Worte. In der Praxis wurde ein Wert
153.2 Performance 3 BLAST
von T = 17 gewählt, da eine Verringerung des Parameters nur in einem kleinen Anstieg
der Sensitivität mündete. Für eine Genauigkeit von 90% ist eine Rechenzeit von 9s nötig.
Um die Genauigkeit auf 98% zu erhöhen, muss der Parameter T verringert werden, was
in einer Verdopplung der Rechenzeit resultiert. Hervorzuheben ist das für einen höheren
MSP, und damit statistisch signifikanteren MSP eine geringere Rechenzeit nötig ist.
3.2.2 Proteine
Die Performance von BLAST auf der PIR Datenbank im Zusammenhang mit der Zu-
ordnung von Eingangssequenzen zu Proteinfamilien (Anhang - Table 3) wurde ebenfalls
evaluiert. Die Ergebnisse waren im Einzelnen unterschiedlich. Teilweise war BLAST bes-
ser als vorhergesagt, teilweise schlechter. Die Ursache für die schlechten Ergebnisse liegt in
den gleichartigen Mustern der Globine begründet, welche in relativ wenig MSP-Wörtern
resultieren. Von den MSPs zwischen 45 und 65 wurden lediglich zwei nicht erkannt, im
gegensatz zu acht vorhergesagten. Insgesamt sollte BLAST auf diesen Daten bessere Er-
gebnisse liefern, da die Verteilung der Mutationen stärker geclustert ist, als von dem
Poission Prozess [11] vorhergesagt.
BLAST zeigt hier seine Stärke, MSPs mit hohen Werten schnell zu finden. Im erwähnten
Beispiel wurden 89 von 90 Globinen mit einem Score über 80 sowie alle 125 mit einem
Score über 50 gefunden. Vergleicht man BLAST (w = 4, T = 17) mit FASTP ergibt
sich das BLAST ähnlich sensitiv, mit weniger falsch positiven Ergebnissen und über eine
Größenordnung schneller ist [2].
3.2.3 DNA
Für den Vergleich wurden 73.460 Basenpaare des Menschen sowie 44.595 Basenpaare
des Hasen, die ß-ähnliche Globingencluster aufweisen, herangezogen (Anhang - Table 4).
Diese weisen drei Hauptklassen ähnlicher Regionen auf. Die Klassen teilen sich auf in Gene,
lange mit Wiederholungen durchsetzte Regionen und Regionen geringerer Ähnlichkeiten.
BLAST wurde genutzt um lokale, ähnliche Regionen zu finden, die ohne das Einfügen von
Gaps alignt werden konnten. Gewertet wurden Treffer mit +5 und Fehler mit -4, sowie
einem anfänglichen w = 12. Es wurden 98 Matches mit einem Score von 200 gefunden.
Der höchste Score betrug 1301. Es konnten damit drei Klassen identifiziert werden. Von
den 57 Alignments mit einem Score über 350 waren 45 gepaarte Gene und 12 gehörten
zu L1 Sequenzen. Bei Alignments von einem Score von unter 350 wurden Zwischengenbe-
ziehungen deutlich. Zwei Alignments mit einem Score zwischen 200-350 passten in keine
der erwarteten Klassen. Das erste Alignment weisst auf eine neu entdeckte Sektion einer
163.2 Performance 3 BLAST
L1 Sequenz hin. Das andere Alignment zeigt die Verbindung eines menschlichen Gens mit
dem des Hasen.
Mit einem kleineren w = 8 wurden 32 zusätzliche Alignments gefunden die aber kei-
nerlei neue Informationen enthielten. Dafür wurde die Rechenzeit von 3,2 auf 15,9 fast
verfünffacht.
174 FAZIT
4 Fazit
BLAST ist ein einfacher, robuster Algorithmus der mit einer Laufzeitkomplexität von
O(n · m) mindestens eine Größenordnung schneller ist als dynamische Programmierung
[3]. Er ist einfach zu implementieren und kann in vielfacher Weise erweitert werden. Eine
mögliche Erweiterung wäre zum Beispiel das Einfügen von Gaps. Weiterhin kann durch
das Laden von Sequenzen in den Hauptspeicher die Rechenzeit weiter verringert werden.
BLAST zeichnet sich auch durch seine statistische Komponente aus. Die Fähigkeit
anzugeben, ob Zusammenhänge signifikant oder bloß zufällig sind ist ein sehr wichtiges
Feature, um Fehlannahmen auszuschließen.
Das Paper und die vorliegende Arbeit behandeln den originalen BLAST-Algorithmus
aus dem Jahr 1990. BLAST war zu diesem Zeitpunkt ein Algorithmus dessen Zeitkomple-
xität in Zusammenhang mit seiner Sensitivtät nahezu unerreicht war. BLAST ist Grund-
lage für viele neuere Entwicklungen wie BLASTP oder BLASTN [4, 7, 3]. Er ist heutzutage
in vielen Varianten im Einsatz, die alle für bestimmte Einsatzzwecke optimiert sind.
Der größte Nachteil von BLAST ist der, dass nicht garantiert werden kann, dass die
besten Alignments gefunden werden. Dies ist ein Problem aller heuristischen Algorithmen
und kann nie mit absoluter Sicherheit ausgeschlossen werden.
Ein weiterer Nachteil liegt in der Abschätzung der Eingangsparameter des Algorithmus.
Werden die Wortlänge w und der Schwellwert T nicht richtig gewählt, kann das die Ergeb-
nisse des Algorithmus so stark beeinflussen, dass die Ergebnisse nicht repräsentativ sind.
Ein erfahrener Benutzer ist erforderlich, um diese abhängig von der Datenlage richtig zu
wählen. Abzuschätzen, welche Parameter die Richtigen sind, kann eine längere Testphase
erfordern. Besonderheiten in den Daten können nur berücksichtigt werden, wenn sie auch
bekannt sind.
Es konnte gezeigt werden, das eine höhere Genauigkeit/Sensitivität die Laufzeit ent-
sprechend erhöht. Ein Kompromiss zwischen Laufzeit und Genauigkeit ist notwendig.
Unter dem Strich bietet BLAST einen sehr viel schnelleren Ansatz als alle vorherigen
Ansätze. Dieser Vorteil wird allerdings durch eine geringere Genauigkeit beeinträchtigt.
Will man die Genauigkeit erhöhen, wird damit wiederum die Laufzeit erhöht.
18Literatur
5 Literaturverzeichnis
Literatur
[1] Altschul, S.F. u. a.: Amino acid substitution matrices from an information theo-
retic perspective. In: Journal of molecular biology 219 (1991), Nr. 3, S. 555–565
[2] Altschul, S.F. ; Gish, W. ; Miller, W. ; Myers, E.W. ; Lipman, D.J. u. a.:
Basic local alignment search tool. In: Journal of molecular biology 215 (1990), Nr. 3,
S. 403–410
[3] Altschul, S.F. ; Madden, T.L. ; Schäffer, A.A. ; Zhang, J. ; Zhang, Z. ;
Miller, W. ; Lipman, D.J.: Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs. In: Nucleic acids research 25 (1997), Nr. 17, S.
3389–3402
[4] Balakrishnan, R. ; Christie, K.R. ; Costanzo, M.C. ; Dolinski, K. ; Dwight,
S.S. ; Engel, S.R. ; Fisk, D.G. ; Hirschman, J.E. ; Hong, E.L. ; Nash, R. u. a.:
Fungal BLAST and model organism BLASTP best hits: new comparison resources at
the Saccharomyces Genome Database (SGD). In: Nucleic acids research 33 (2005),
Nr. suppl 1, S. D374–D377
[5] Baxevanis, A.D. ; Ouellette, B.F.F.: Bioinformatics: a practical guide to the
analysis of genes and proteins. Wiley-Interscience, 2004
[6] Benson, D.A. ; Boguski, M.S. ; Lipman, D.J. ; Ostell, J.: GenBank. In: Nucleic
acids research 25 (1997), Nr. 1, S. 1–6
[7] Buhler, J.D. ; Lancaster, J.M. ; Jacob, A.C. ; Chamberlain, R.D. u. a.: Mer-
cury BLASTN: Faster DNA sequence comparison using a streaming hardware archi-
tecture. In: Proc. of Reconfigurable Systems Summer Institute (2007)
[8] Earley, J.: An efficient context-free parsing algorithm. In: Communications of the
ACM 13 (1970), Nr. 2, S. 94–102
[9] Gregory, SG ; Barlow, KF ; McLay, KE ; Kaul, R. ; Swarbreck, D. ; Dun-
ham, A. ; Scott, CE ; Howe, KL ; Woodfine, K. ; Spencer, CCA u. a.: The
DNA sequence and biological annotation of human chromosome 1. In: Nature 441
(2006), Nr. 7091, S. 315–321
19Literatur Literatur
[10] Jochen Graw, Wolfgang H.: Genetik. 5th. Springer Berlin Heidelberg : Springer,
2010
[11] Karlin, S. ; Altschul, S.F.: Methods for assessing the statistical significance of
molecular sequence features by using general scoring schemes. In: Proceedings of the
National Academy of Sciences 87 (1990), Nr. 6, S. 2264–2268
[12] Needleman, S.B. ; Wunsch, C.D. u. a.: A general method applicable to the search
for similarities in the amino acid sequence of two proteins. In: Journal of molecular
biology 48 (1970), Nr. 3, S. 443–453
[13] Salwinski, L. ; Miller, C.S. ; Smith, A.J. ; Pettit, F.K. ; Bowie, J.U. ; Ei-
senberg, D.: The database of interacting proteins: 2004 update. In: Nucleic acids
research 32 (2004), Nr. suppl 1, S. D449–D451
[14] Smith, T. F. ; Waterman, M. S.: Identification of common molecular subsequences.
In: Journal of molecular biology 147 (1981), März, Nr. 1, 195–197. http://view.
ncbi.nlm.nih.gov/pubmed/7265238. – ISSN 0022–2836
[15] Swanson, R.: A unifying concept for the amino acid code. In: Bulletin of mathe-
matical biology 46 (1984), Nr. 2, S. 187–203
206 ANHANG
6 Anhang
216 ANHANG 22
6 ANHANG 23
6 ANHANG 24
Sie können auch lesen

















































