Die 1001 Genome der Ackerschmalwand - Pfl anzenforschung
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

Weigel, Detlef | Die 1001 Genome der Ackerschmalwand Tätigkeitsbericht 2008 Pflanzenforschung Die 1001 Genome der Ackerschmalwand Weigel, Detlef; Max-Planck-Institut für Entwicklungsbiologie, Tübingen Abteilung - Abt. 6: Molekularbiologie (Weigel) Korrespondierender Autor Weigel, Detlef, E-Mail: weigel@weigelworld.org Zusammenfassung Die Ackerschmalwand, Arabidopsis thaliana, ist das bevorzugte Studienobjekt vieler Pflanzengeneti- ker. Außer dem Menschen gibt es keine Art, über deren innerartliche Variation in der Genomsequenz man so viel weiß. In den letzten zwei Jahren ist die Technologie zur Sequenzierung von Genomen revolutioniert worden, und A. thaliana bietet sich wie kaum ein anderer vielzelliger Organismus zur Anwendung dieser neuen Methoden an. In diesem Bericht werden die Anfänge eines Projektes be- schrieben, das die komplette Entzifferung des Genoms von 1001 A. thaliana-Stämmen zum Ziel hat. Abstract Mouse ear cress, Arabidopsis thaliana, is the workhorse of plant genetics, and currently only second to humans when it comes to information about genomic variation within the species. In the past two years, there has been a revolution in sequencing technology, and A. thaliana is an ideal object for ex- ploiting the dramatic improvements in sequencing speed and cost. This report describes the beginning of the 1001 Genomes Project, which has as its goal the complete description of the genomes of 1001 wild strains of A. thaliana. Arabidopsis thaliana Die Ackerschmalwand, Arabidopsis thaliana, gehört zu einer der größten Familien der Blütenpflanzen, den Kreuzblütlern, die auch die vielen Kohl-, Senf- und Retticharten einschließt. Der Ursprung der Art liegt wahrscheinlich in Zentralasien, von wo aus sie sich über große Teile Eurasiens ausgebreitet hat. Während der letzten Eiszeit wurde sie an den südlichen Rand ihres Verbreitungsareals verdrängt. Nachdem sich das Eis zurückgezogen hatte, wurde Europa durch Populationen aus verschiedenen Rückzugsgebieten wiederbesiedelt. Heute wächst A. thaliana in vielen Bereichen der nördlichen Halb- kugel, vor allem in Gegenden mit gemäßigtem Klima, von den Bergen Nordafrikas bis zum Polarkreis. Wie viele andere Pflanzen, wurde sie von Europäern im letzten Jahrtausend nach Nordamerika einge- schleppt [1-5]. Dass A. thaliana heute eine der beliebtesten Arten für die Grundlagenforschung an Pflanzen ist, ob- wohl sie keinerlei wirtschaftliche Bedeutung hat, liegt an ihren genetischen Vorzügen. Das Genom ist relativ klein, etwa ein Zwanzigstel des menschlichen Genoms, und da sie sich normalerweise selbst bestäubt, sind die mütterlichen und väterlichen Kopien jedes Gens in einem Individuum gewöhnlich identisch. © 2008 Max-Planck-Gesellschaft www.mpg.de
Tätigkeitsbericht 2008 Weigel, Detlef | Die 1001 Genome der Ackerschmalwand
Die erste Genomsequenz
A. thaliana war die erste Pflanzenart, von der ein komplettes Erbgut entziffert wurde. Diese erste
Genomsequenz kam von einem einzigen, in-gezüchtetem Stamm. Wie erwartet, beschleunigten die
umfangreichen Informationen über die Gen-Ausstattung die Arabidopsis-Forschung in großem Maße.
Dazu gehörten auch Arbeiten, die sich mit der genetischen Grundlage der Vielfalt an Eigenschaften
beschäftigen, wie man sie bei wilden Sorten der Art findet, die in verschiedensten Gebieten verbrei-
tet sind. Zum einen konnte man viel schneller Gene identifizieren, die direkt Unterschiede wie zum
Beispiel im Blühverhalten oder in der Resistenz gegen Schädlinge bedingen. Erstaunlicherweise sind
dafür oft nicht nur subtile Änderungen in der Aktivität oder Struktur dieser Gene verantwortlich,
sondern oft auch das komplette Fehlen von Genen in dem einen oder anderen Stamm. Allerdings ist
das Auffinden letzterer Gene immer noch sehr mühsam, besonders wenn sie denn nicht im Genom des
zuerst sequenzierten Stamms vorhanden waren.
Fortschritte bei der Entdeckung von genetischen Unterschieden
Um wichtige Genvarianten schneller aufspüren zu können, wurden in den vergangenen Jahren zwei
große Studien durchgeführt. In dem ersten Projekt verschaffte man sich erst einmal ein Bild von
der insgesamt vorhandenen Variabilität [4]. Auf diese Vorarbeiten aufbauend, wurden 20 Stämme
ausgewählt, deren gesamtes Genom Erbbaustein für Erbbaustein, Basenpaar für Basenpaar auf Ab-
weichungen in der Sequenz durchforstet wurde. Ein derartig ambitioniertes Unternehmen hatte man
vorher nur beim Menschen gewagt. Für jedes der 120 Millionen Basenpaare im Genom der zuerst
sequenzierten Sorte wurden acht verschiedene Sonden synthetisiert, um Austausche der bekannten
Sequenz gegen eines der anderen drei möglichen Basenpaare an jeder Stelle des Genoms nachweisen
zu können [6, 7]. Diese Studie war nur durch eine groß angelegte Kooperation verschiedener Einrich-
tungen der Max-Planck-Gesellschaft sowie mit Kollegen inner- und außerhalb Deutschlands möglich
und wurde wesentlich durch den Innovationsfonds des Präsidenten der Max-Planck-Gesellschaft
unterstützt.
Hunderttausende von Sequenzabweichungen wurden festgestellt, wobei eine der größten Überra-
schungen war, dass etwa 4 % der Gene in mindestens einem der 20 untersuchten Stämme defekt
waren. Von zweihundert der etwa 25.000 bekannten Gene wiederum wurden Varianten gefunden, die
längere Proteine kodieren konnten als es im Referenzstamm der Fall ist, was darauf hindeutete, dass
in der Standardsorte ebenfalls viele Gene nicht normal aktiv sind. Diese Funde machten in beeindru-
ckender Weise klar, dass die Entzifferung eines einzigen individuellen Genoms zum Verständnis des
Erbguts einer Art nicht ausreicht. Zu einem ähnlichen Schluss ist man übrigens auch beim Menschen
und seinen nächsten Verwandten gekommen. Der oft zitierte Unterschied von nur einem Prozent
zwischen den Schimpansen und uns stimmt nur, wenn man außer Acht lässt, dass große Teile unseres
Erbgutes gar nicht im Schimpansen vorhanden sind, und wenn, dann in unterschiedlicher Kopienzahl;
umgekehrt ist das natürlich auch der Fall.
Das 1001 Genomes Projekt
Da immer deutlicher wurde, dass es irreleitend ist, von dem Genom einer Art zu sprechen, ist im letz-
ten Jahr begonnen worden, eine große Anzahl von Genomen bei ein und derselben Art zu sequenzie-
ren. Den Anfang, besonders wegen der Bedeutung erblicher Unterschiede für Krankheitsanfälligkeiten,
hat dabei der Mensch gemacht, und das dabei prominenteste Unterfangen ist das 1000 Genomes
Projekt (http://1000genomes.org), an dem viele große Institutionen beteiligt sind.
www.mpg.de © 2008 Max-Planck-GesellschaftWeigel, Detlef | Die 1001 Genome der Ackerschmalwand Tätigkeitsbericht 2008
Auf Grund der überragenden Stellung von A. thaliana in der Pflanzenbiologie hat die Gruppe
um Detlef Weigel am Max-Planck-Institut für Entwicklungsbiologie das 1001 Genomes Projekt
(http://1001genomes.org) für diese Art ins Leben gerufen. Dieses wiederum sehr ehrgeizige Unterneh-
men ist dadurch möglich geworden, dass die Kosten für die komplette Entschlüsselung von Geno-
men in den letzten Jahren drastisch gefallen sind. Während die Sequenzierung des ersten A. thaliana
Genoms noch fast 100 Millionen US Dollar kostete, benötigt man mit der neuesten Technologie nur
noch etwa ein Zehntausendstel dieses Betrags! Fairerweise muss jedoch zugestanden werden, dass
die neuen Methoden nicht immer genauso akkurat sind wie die alten und viel teureren sind. Ebenso
profitiert man bei der Interpretation der Sequenzen von neuen Stämmen immer noch erheblich von der
ersten, sehr hochwertigen Genomsequenz. Diese dient nämlich oft als Vorlage, um die zum Teil nur
sehr kurzen Sequenzfragmente, die mit der neuen Technologie gewonnen werden, zusammenzufügen.
Auf der anderen Seite ist es mit den neuen Methoden aber auch gelungen, die Mehrzahl der knapp
über tausend Fehler in der Referenzsequenz zu korrigieren [8].
Die Arbeitsgruppe um Detlef Weigel hat bereits ihre Analyse des kompletten Erbguts von zwei A. tha-
liana Wildstämmen veröffentlicht, noch vor der Publikation entsprechender Befunde aus menschlichen
Genomen [8]. Beide A. thaliana-Stämme wiesen Hunderttausende von kleinen Unterschieden auf, bei
denen einzelne Basenpaare ausgetauscht waren oder einige wenige Basenpaare fehlten beziehungswei-
se zusätzlich vorhanden waren. Größere Unterschiede, die sich auf bis zu mehrere Hundert Basenpaare
erstreckten, konnten ebenfalls nachgewiesen werden (Abb. 1 und 2). Die Brauchbarkeit des metho-
dischen Ansatzes ist also erwiesen, wobei man noch berücksichtigen muss, dass die neuen Techniken
fast monatlich verbessert werden, sodass immer größere Abschnitte des Erbguts auf einmal abgelesen
werden können.
Abb. 1: Auffinden von Sequenzen, die nicht im Erbgut des Referenzstamms von Arabidopsis thaliana vorhanden
sind. Die einzelnen kurzen Sequenzschnipsel, die mit den neuen Methoden gelesen werden können, und wie sie
miteinander überlappen, ist dargestellt. Unterschiede in einzelnen Basenpaaren sind farbig unterlegt. Sequen-
zschnipsel, die nicht direkt einer Abfolge von Basenpaaren im Referenzgenom entsprechen, sind grün gekenn-
zeichnet (nach [8]).
Urheber: Max-Planck-Institut für Entwicklungsbiologie/Weigel
© 2008 Max-Planck-Gesellschaft www.mpg.deTätigkeitsbericht 2008 Weigel, Detlef | Die 1001 Genome der Ackerschmalwand
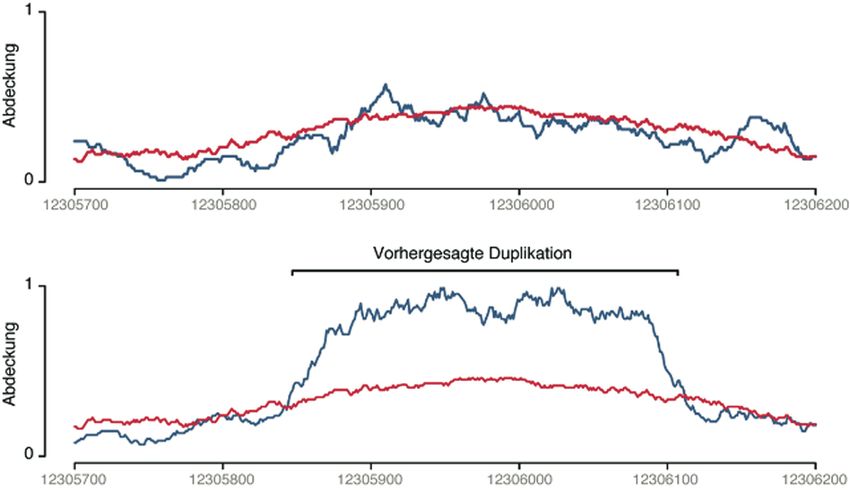
Abb. 2: Nachweis duplizierter Sequenzabschnitte in Arabidopsis thaliana. Oben: Vergleich der erwarteten (rot)
und beobachteten (blau) Anzahl an Sequenzfragmenten bei Untersuchung des Referenzstamms. Unten: Vergleich
für einen anderen Stamm im selbigen Abschnitt. Da etwa doppelt so viele Sequenzfragmente gefunden wurden,
wurde eine Duplikation vorhergesagt, die durch unabhängige Methoden bestätigt werden konnte (nach [8]).
Zahlen verweisen auf die Koordinaten des Referenzgenoms.
Urheber: Max-Planck-Institut für Entwicklungsbiologie/Weigel
In den nächsten zwei Jahren plant die Gruppe um Detlef Weigel, in enger Zusammenarbeit mit Kol-
legen aus dem In- und Ausland die Anzahl der A. thaliana-Stämme mit vollständig sequenziertem
Genom auf 1001 zu erhöhen. Die Samen dieser Sorten, die aus weit auseinander liegenden Gegenden
kommen und deshalb an sehr diversen Standorten überleben können, werden Wissenschaftlern aus
aller Welt frei zur Verfügung stehen. Die Wissenschaftler können die Stämme dann auf die ihnen
besonders am Herzen liegenden Eigenschaften überprüfen, vom Blühbeginn bis zur Fähigkeit, auf
Böden zu wachsen, die mit Salz verseucht sind. Mit dieser Information in der Hand, können sie dann
das Genom nach Genvarianten absuchen, die nur in Sorten vorkommen, die sich durch ganz bestimmte
Eigenschaften auszeichnen.
Einerseits wird die Kenntnis von Genen, deren Präsenz unter bestimmten Umweltbedingungen vorteil-
haft ist, unmittelbare praktische Konsequenzen haben, da man dann die Genome von Kulturpflanzen-
sorten nach entsprechenden Varianten sichten kann. Zum anderen werden mit diesem Projekt viele der
Methoden und Techniken entwickelt, um die Information zu finden, die sich im Erbgut der unzähligen
Reis-, Mais- oder Kartoffelstämme verbirgt. Dem 1001 Genomes Projekt für A. thaliana werden ohne
Zweifel bald ähnliche Projekte für unsere Kultur- und Forstpflanzen folgen. Man schätzt, dass der Er-
trag allein beim Reis bis zum Jahr 2025 um etwa ein Viertel ansteigen muss, um die immer noch dra-
matisch wachsende Erdbevölkerung zu ernähren [9], und zwar bei begrenzter Anbaufläche und unter
zunehmender Konkurrenz um wertvolles Agrarland durch andere Verwendungen von Kulturpflanzen,
wie beispielsweise für Biokraftstoffe, aber vor allem auch für die Fütterung von Tieren, die von immer
mehr Menschen gegessen werden wollen. Das 1001 Genomes Projekt wird ein wichtiger Schritt zur
Lösung dieser gewaltigen Aufgabe darstellen.
www.mpg.de © 2008 Max-Planck-GesellschaftWeigel, Detlef | Die 1001 Genome der Ackerschmalwand Tätigkeitsbericht 2008
Literaturhinweise
[1] T. F. Sharbel, B. Haubold, T. Mitchell-Olds:
Genetic isolation by distance in Arabidopsis thaliana: Biogeography and postglacial colonization
of Europe.
Molecular Ecology 9, 2109-2118 (2000).
[2] M. H. Hoffmann:
Biogeography of Arabidopsis thaliana (L.) Heynh. (Brassicaceae).
Journal of Biogeography 29, 125-134 (2002).
[3] K. J. Schmid, O. Torjek, R. Meyer, H. Schmuths, M. H. Hoffmann, T. Altmann:
Evidence for a large-scale population structure of Arabidopsis thaliana from genome-wide single
nucleotide polymorphism markers.
Theoretical and Applied Genetics 112, 1104-1114 (2006).
[4] M. Nordborg, T. T. Hu, Y. Ishino, J. Jhaveri, C. Toomajian, H. Zheng, E. Bakker, P. Calabrese,
J. Gladstone, R. Goyal, M. Jakobsson, S. Kim, Y. Morozov, B. Padhukasahasram, V. Plagnol,
N. A. Rosenberg, C. Shah, J. D. Wall, J. Wang, K. Zhao, T. Kalbfleisch, V. Schulz, M. Kreitman,
J. Bergelson:
The pattern of polymorphism in Arabidopsis thaliana.
PLoS Biology 3, e196 (2005).
[5] O. François, M. G. Blum, M. Jakobsson, N. A. Rosenberg:
Demographic history of European populations of Arabidopsis thaliana.
PLoS Genetics 4, e1000075 (2008).
[6] R. M. Clark, G. Schweikert, C. Toomajian, S. Ossowski, G. Zeller, P. Shinn, N. Warthmann,
T. T. Hu, G. Fu, D. A. Hinds, H. Chen, K. A. Frazer, D. H. Huson, B. Schölkopf, M. Nordborg,
G. Rätsch, J. R. Ecker, D. Weigel:
Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana.
Science 317, 338-342 (2007).
[7] G. Zeller, R. M. Clark, K. Schneeberger, A. Bohlen, D. Weigel, G. Rätsch:
Detecting polymorphic regions in the Arabidopsis thaliana genome with resequencing microarrays.
Genome Research 18, 918-929 (2008).
[8] S. Ossowski, K. Schneeberger, R. M. Clark, C. Lanz, N. Warthmann, D. Weigel:
Sequencing of natural strains of Arabidopsis thaliana with short reads.
Genome Research 18, 2024-2033 (2008).
[9] International Rice Research Institute
Bringing hope, improving lives: strategic plan, 2007-2015.
Manila, Philippines (2006), IRRI Press.
© 2008 Max-Planck-Gesellschaft www.mpg.deSie können auch lesen