Genetische Medizin: Möglichkeiten und Herausforderungen - Medizinische Universität ...
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

02.01.2020
Genetische Medizin:
Möglichkeiten und Herausforderungen
Johannes Zschocke
Institut für Humangenetik
Medizinische Universität Innsbruck
Aufgabe der Humangenetik
Klärung und Erklärung des Zusammenhangs
zwischen klinischem Bild und erblicher Konstitution
im Einzelfall
Erbliche Klinisches
Konstitution Bild
Umwelt – Zufall – Therapie
2
1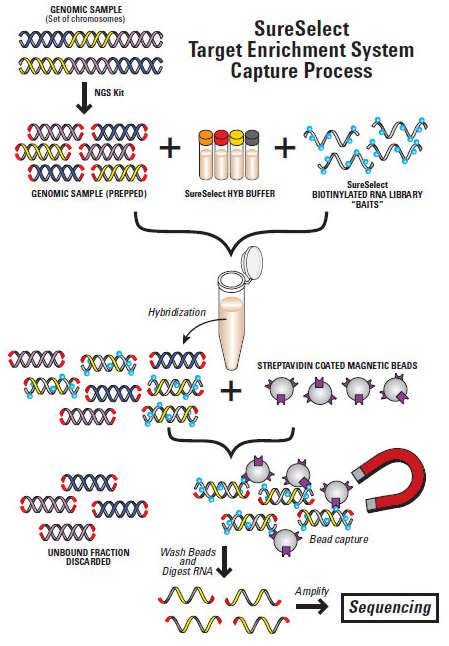
02.01.2020
Aufgabe der Humangenetik
Genotyp Phänotyp
3
Gretchenfrage der genetischen Diagnostik:
Kennen wir den Zusammenhang
zwischen Genotyp und Phänotyp?
4
2
02.01.2020
Erbgut
2 x 23 = 46 Chromosomen
3,096,649,726 Nukleotide
(„Golden Path Length“)
20,454 proteinkodierende Gene
23,940 nicht-proteinkodierende Gene
15,204 Pseudogene
6,013,113 strukturelle Varianten
665,834,144 kurze/kleine Varianten
Ensembl genome browser, www.ensembl.org
Database version 97.38 (März 2019)
5
Genetische Variabilität
• Varianten einzelner Nukleotide
– Punktmutationen,
Einzelnukleotidpolymorphismen (SNP)
• Kleine Deletionen/Insertionen
Monogene
• Variable Sequenzwiederholungen Effekte
– STRs (short tandem repeats, 2-6 nt) [u.a.] Molekulargenetik
• Größere monogene Deletionen
• Epigenetische Varianten (Methylierung)
• Kopienzahl-Varianten (CNVs)
– Deletion/Duplikation/Multiplikation Viele Gene,
– 1 kb – viele Mb Gendosis-Effekte
– Inklusive „Mikrodeletionen/-duplikationen“
• Chromosomale Varianten
Zytogenetik
6
3
02.01.2020
Genetische Variabilität
• Varianten einzelner Nukleotide
– Punktmutationen,
Einzelnukleotidpolymorphismen (SNP)
• Kleine Deletionen/Insertionen Genomweite
• Variable Sequenzwiederholungen Analysen:
– STRs (short tandem repeats, 2-6 nt) [u.a.]
• Größere monogene Deletionen Massiv-parallele
• Epigenetische Varianten (Methylierung) Sequenzierung
• Kopienzahl-Varianten (CNVs)
– Deletion/Duplikation/Multiplikation DNA-Array
– 1 kb – viele Mb
– Inklusive „Mikrodeletionen/-duplikationen“
• Chromosomale Varianten
7
Massiv-parallele Sequenzierung
“next generation” sequencing, Hochdurchsatz-Sequenzierung
parallele Durchführung vieler Sequenzierreaktionen auf einem Mikrochip
1. Vorbereitung der Probe
2. Sequenzierreaktion
3. Sortierung der Sequenzen (Alignment)
4. Identifizierung von Varianten (variant call format, vcf)
5. Funktionell-klinische Interpretation der Varianten
8
4
02.01.2020
3’ 5’
Massiv-parallele Sequenzierung
A G
DNA C
T
G
A
(0.1-1.0 µg) C
T
T
A
C C
G
G A
T
A
A
C
T
C
C
G
Fragmentierung C G
A
T
Adaptoren T C
Ggf. G
A
Anreicherung Amplifikation 5’
T
Zielsequenzen (Clusterbildung) Anlagerung
Nukleotide
Aufnahme 1 2 3 4 5 6 7 8 9
Fluoreszenz- T G C T A C G A T …
bilder Sequenz
9
Anreicherung von Zielsequenzen
• Zum Beispiel:
– Ausgewählte Gene (Panel)
• nur die klinisch relevanten Gene
• Kostengünstig
• maximale Coverage
– Alle kodierenden Gene (Exom)
• alle kodierenden Exone im Genom
• begrenzte Coverage
– Transkriptom
• Alle RNA-Transkripte
• Warum?
– Kosten
– Abdeckung (Coverage)
auch Del/Dup-Analyse
– Weniger Analyseaufwand
– Vermeidung von Nebenbefunden
10
5
02.01.2020
Massiv-parallele Sequenzierung: Datenverarbeitung
1. Zuordnung zur Referenzsequenz (Alignment)
2. Erstellung Varianten-Liste (variant call format, vcf)
– Automatische Sequenzanalyse
– Deletionsanalyse
– „Manuelle“ Inspektion der Rohdaten
3. Funktionell-klinische Interpretation
– Häufigkeiten (Allelfrequenz), Auswahl möglicher/wahrscheinlicher
– Beschreibung in Datenbanken/Fachliteratur Gene bzw. Varianten
– Erwarteter biologischer Effekt (Protein, Spleißen u.a.)
– evolutionäre Konservierung
– Klinische Daten, Auftreten/Segregation in der Familie
4. Klassifizierung C1-C5
– Sicher/wahrscheinlich pathogen/benigne
– Varianten unbekannter Signifikanz (VUS)
11 Plon et al., Human Mutation 2008
Sequenzierung eines Gens – relevante Fragen
• Was bedeuten die nachgewiesenen Varianten?
– Sicher/wahrscheinlich krankheitsrelevant oder irrelevant?
– Hypomorph/Risikofaktor? Mit welcher Wahrscheinlichkeit?
– Variante mit unbekannter Signifikanz (VUS) – Unklassifizierte Variante (UV)
• Seltene genetische Variante, mögliche aber nicht sichere Krankheitsbedeutung
• Kann nicht für prädiktive Diagnostik verwendet werden
• Welche Mutationen werden NICHT nachgewiesen?
– Nicht im vcf Datensatz
– Schlechte Abdeckung, schlechte Qualität
– Ungewöhnliche Mutationen
– Außerhalb der Zielsequenz
– Mosaikbefund
Nicht erfasste Varianten meist im unteren einstelligen Prozentbereich
12
6
02.01.2020
Europäischer Ringversuch PKU-Mutationsanalyse (seit 2004)
Jahr Labs Länder Genotypisierungsfehler Interpretations- Volle
Fehler/Mängel Punkte
2011 27 16 6% 3 Labs je 1 Genotyp, 1 Lab beide, 42 % 32/76 Befunde 3 Labs
1 Lab Genotyp-Fehler
2012/ 23 16 1,4 % 1 Lab 1 Mutation 61 % 42/69 Befunde 1 Lab
2013
2014 25 (27) 17 1,3 % 1 Lab 1 Mutation, 15 Labs Exon-Deletion* 59 % 44/74 Befunde 1 Lab
10 x falsche Risikoberechnung
2015 26 (27) 14 5% 1 Lab 2 Mutation (falsche Methode) 24 % 18/76 Befunde 14 Labs
1 Lab nicht-existente Deletion
2016 25 16 Keine 1 Lab alle Mutationen (falsche Methode) 45 % 34/75 Befunde 9 Labs
2017 23 (24) 13 Keine 1 Lab alle Mutationen (falsche Methode) 64 % 46/72 Befunde 1 Lab
2018 24 (26) 13 Keine Keine 66 % 48/72 Befunde 1 Lab
2019 27 (28) 15 3,7% 2 Labs je 1 Spleißmutation; 78% 63/81 Befunde 3 Labs
9 Labs Exon-Deletion*
Überträgeranalyse 2007: 5/11 Labors falsche klinische Bewertung, 2 Labors PD für non-disease
Überträgeranalyse 2012: 5/22 Labors gefährliche Fehlinterpretation, 3 Labors PD für non-disease
13 * nicht als Genotypisierungsfehler gewertet; 2019: 2x keine MLPA-Empfehlung
Sequenzierung vieler Gene?
• Welche Gene…
… sind für die Fragestellung wichtig?
… könnten für die Fragestellung wichtig sein?
… könnten für die Fragestellung neue Erkenntnisse liefern?
… liefern hilfreiche Nebenbefunde?
… führen in die Irre?
… sind Zeit- und Geldverschwendung?
… sollten nicht untersucht werden?
14
7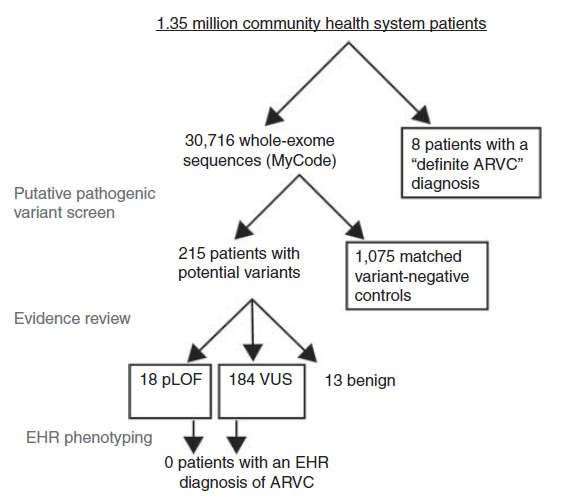
02.01.2020
15
CMAMMA: A Non-Disease
16
8
02.01.2020
Histidinämie ist keine Krankheit
…und wurde doch Jahrzehnte im Neugeborenenscreening erfasst
17 Brosco et al. (2010) Pediatrics 125:417-19
18
9
02.01.2020
Neue Sequenzierverfahren
Welche Gene Welche Gene
sequenziert? ausgewertet?
• Technische Strategien: Kosten/Abdeckung
– Anreicherung von Zielgenen
– Anreicherung aller kodierenden Gene (Exom)
– Genom
• Analytische Strategien: Interpretation
– Bekannte/bestimmte Gene ↔ „alle“ Gene
– Bekannte Mutationen ↔ alle Varianten
Auch bei einer Exomanalyse werden (zunächst)(nur) bestimmte Zielgene
aufgrund der spezifischen Indikation ausgewertet
19
Miller-Syndrom
• Unklares Fehlbildungssyndrom
– Postaxiale Strahldefekte Hände/Füße, faziale Auffälligkeiten
– Normale Intelligenz (ggf. Hörstörung)
• Exom-Sequenzierung in 3 Familien
• Bestätigung in 3 weiteren Familien
DHOD
DNAH5
• Ursache: Mutationen im DHOD-Gen
• Atyp. Atemwegssymptome in 1 Familie – Zusätzlich DNAH5-Mutationen
– Primäre ciliäre Dyskinesie, ebenfalls autosomal rezessiv
20
1002.01.2020
Brust- und Eierstockkrebs: Hauptgene BRCA1 und BRCA2
BRCA1 BRCA2 Allgemein
Brustkrebs der Frau 50-80 % 40-80 % 12 %
Deutsche Verbundstudie 60-85 % 40-85 %
Prospektive Studien* 40-75% 30-70%
2. Brustkrebs innerhalb 10 Jahren 21 % 11 % 2 % (in 5 Jahren)
Eierstockkrebs 40-60 % 16-27 % 1-2 %
Deutsche Verbundstudie 45 % 15-20 %
Prospektive Studien* 7-35%
Brustkrebs beim Mann 1-2 % 5-10 % 0,1 %
Prostatakarzinom 8,5 % (bis 65 J.) 15-20% 6 % (bis 69 J.)
Pankreas 1-3 % 2-7 % 0,5 %
BRCA2-Mutation: Erhöhtes Hautkrebsrisiko (Melanome). Generell: möglicherweise andere Tumorrisiken.
Autosomal dominanter Erbgang
Kind mit 50% Wahrscheinlichkeit ebenfalls Mutationsträger/in (unabhängig vom Geschlecht)
21 *Mavaddad et al, 2013, doi:10.1093/jnci/djt095; Evans et al, 2014, doi:10.1136/jmedgenet-2014-102336
Erblicher Brust- und Eierstockkrebs: Früherkennung und Prävention
Frau aus Hochrisikofamilie bzw. Mutationsträgerin
• Ab 25. Lebensjahr bzw. ab 5 Jahre vor dem
frühesten Erkrankungsalter in der Familie
– regelmäßige Selbstuntersuchungen der Brust
– alle 6 Monate ärztliche Brustuntersuchung inkl. Ultraschall
(>7,5 MHz)
– Jährliches MRT der Brust (bis 50 Jahre/Involution der Drüse)
– Jährliche gynäkologische Untersuchung
inkl. transvaginaler Sonographie der Ovarien
• Ab dem 35. Lebensjahr zusätzlich:
– Jährliche Mammographie
• Operative Prophylaxe
– bilaterale Mastektomie
– bilaterale Salpingo-Oophorektomie
22
1102.01.2020
Brust- und Eierstockkrebs: Weitere Gene
Risikoerhöhung entweder für Brustkrebs oder für Eierstockkrebs
↑ Lebenszeitrisiko für Brustkrebs ↑ Lebenszeitrisiko für Eierstockkrebs
PALB2 35 % BRIP1 6%
ATM 38 % (Mutation c.7271T>G: 69 %) RAD51C 5-6-fach
CHEK2 28-37 % RAD51D 5-6-fach
NBN 3-fach (Mutation c.657_661del5)
Spezifische Krankheitsbilder Gen(e) Krebsrisiko
• Li-Fraumeni-Syndrom TP53 Brustkrebs: >50 %
• Peutz-Jeghers-Syndrom STK11 Brustkrebs: 45 %
• Cowden-Syndrom PTEN Brustkrebs: 25-50 %
• Hereditäres diffuses Magenkrebssyndrom CDH1 Brustkrebs: 39-52 %
• Neurofibromatose Typ 1 NF1 Brustkrebs prämenopausal: 2-3-fach
• Lynch-Syndrom MLH1, MSH2, MSH6, PMS2 Eierstockkrebs: bis 24 %
23 Daly et al., J Natl Compr Canc Netw 2017;15:9–20;
Brust- und Eierstockkrebs: andere genetische Faktoren
Verändertes Krebsrisiko durch
eine Vielzahl häufiger genetischer Varianten
polygene Risikoscores
24
1202.01.2020
Erblicher Brust- und Eierstockkrebs:
Krebsrisiko abhängig von der Familienanamnese
Brustkrebs 70 J.
(ED 40 J.)
Kein
Brustkrebs
20 J. 20 J. 50 J.
25
Erblicher Brust- und Eierstockkrebs:
Krebsrisiko abhängig von der Familienanamnese
Mutter hat ATM-Mutation. Tochter trägt
Mutation nicht: Wie hoch ist ihr Krebsrisiko?
a) Ca. 20 %
Brustkrebsrisiko >18 %
b) Ca.
trotz 18%
Ausschluss der maternalen
c) Ca. 15 %
ATM-Mutation
d) Ca. 12 %
e) Ca. 10 %
26
1302.01.2020
Massiv-parallele Sequenzierung als Screeningtest
The ACMG recommends that
Long-QT-Syndrom:
laboratories performing
Ausgelöst durch Anstrengung, clinical
Emotion, Schlaf
sequencing
Risiko plötzlicherseek and(bis
Herztod report
zum mutations
40. LJ.): < 4 %
of Tod
Plötzlicher theohne
specified classes
vorherige or types
Symptome: < 0,4-0,6 %
Hilft
in thedie[56]
genetische
genes Information?
listed here.
27
Arrhythmogene rechtsventrikuläre Kardiomyopathie:
„Nebenbefunde“ der genetischen Diagnostik
• >30.000 Exome
• 215 Personen mit „möglicher“ Diagnose
– Keine Diagnose
– EKG nicht anders als bei Kontrollen
– Keine sonstigen Auffälligkeiten
• 18 Personen mit „sicher“ pathogener Mutationen
– Keine Diagnose
– Keine Symptome
– Keine signifikanten EKG-Auffälligkeiten
Was sagt uns der Screeningbefund?
28 Haggerty et al., Genet Med 2017;19:1245-1252
1402.01.2020
Genetische Diskriminierung bei Mukoviszidose (CF)
11jähriger Bub mit CFTR-Mutationen (im Neugeborenenalter nachgewiesen) ohne klinische CF
wurde auf Betreiben der Eltern von zwei Kindern mit CF von der Schule verwiesen (Angst vor
Infektion). Außergerichtlicher Vergleich (2012).
29
Genetisches Screening in der Bevölkerung wird kommen
• Präsymptomatische Diagnostik mit Therapierelevanz
(z.B. Neugeborenenscreening)
• Krankheiten mit präventiven bzw. therapeutischen Optionen
• Genetische Risikofaktoren?
• Carrier-Screening von Paaren mit Kinderwunsch?
• Pharmakogenetik
• …
Große Herausforderung
Potenziell großer Nutzen – mögliche Nachteile/Schäden
Einbindung in ärztliche Versorgungsstrukturen
Information/Diskurs der Allgemeinbevölkerung
30
1502.01.2020
Genetische Medizin
• Das vollständige Lesen des genetischen Textes ist kein technisches
(und eigentlich auch kein finanzielles) Problem mehr.
• Klärung der genetischen Ursachen von monogenen und zunehmend auch
multifaktoriellen Krankheiten.
• Entwicklung neuer Therapieansätze aufgrund von molekularen Erkenntnissen
auch für einzelne PatientInnen.
• Einführung genetischer Diagnostik in die Routineversorgung.
Aber weiterhin große Herausforderungen…
• Nachweis komplexer Veränderungen, Interpretation genetischer Varianten
• Aufklärung, Einverständnis, genetische Beratung
• Wie bringen wir das genetische Wissen zu PatientInnen und ÄrztInnen?
31
16Sie können auch lesen

















































