Indolente Lymphome Multiples Myelom - Dr. med. Knut Wendelin Nürnberger/Erlanger Facharztkurs - NCO
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Indolente Lymphome
Multiples Myelom
Dr. med. Knut Wendelin
Nürnberger/Erlanger Facharztkurs
Hämatologie und internistische Onkologie
2.Mai 2018
Paracelsus Medizinische Privatuniversität | Nürnberg | www.pmu.ac.at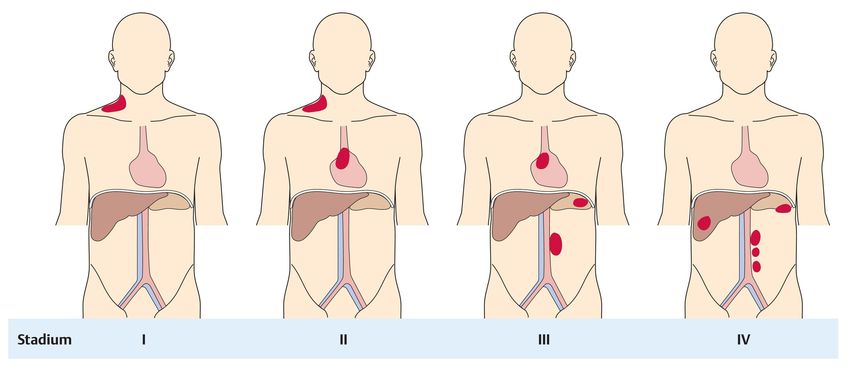
2016 Revision WHO Classification B cell lymphoma
- Chronic lymphocytic leukemia/small - Monoclonal gammopathy of undetermined
lymphocytic lymphoma significance (MGUS), IgG/A
- Monoclonal B-cell lymphocytosis - Plasma cell myeloma
- B-cell prolymphocytic leukemia - Solitary plasmacytoma of bone
- Splenic marginal zone lymphoma - Extraosseous plasmacytoma
- Hairy cell leukemia - Monoclonal immunoglobulin deposition
- Splenic B-cell lymphoma/leukemia, diseases
unclassifiable*: splenic diffuse red pulp small B- - Extranodal marginal zone lymphoma of
cell lymphoma, hairy cell leukemia-variant mucosa-associated lymphoid tissue
- Lymphoplasmacytic lymphoma, Waldenström (MALT lymphoma)
macroglobulinemia - Nodal marginal zone lymphoma
- Monoclonal gammopathy of undetermined - Pediatric nodal marginal zone lymphoma*
significance (MGUS), IgM - Follicular lymphoma
- μ heavy-chain disease - In situ follicular neoplasia
- γ heavy-chain disease - Duodenal-type follicular lymphoma
- α heavy-chain disease - Pediatric-type follicular lymphoma
- Primary cutaneous follicle center lymphoma
- Mantle cell lymphoma
- In situ mantle cell neoplasia
„Small B-cell lymphoid neoplasms“
© Paracelsus Medizinische Privatuniversität | Nürnberg
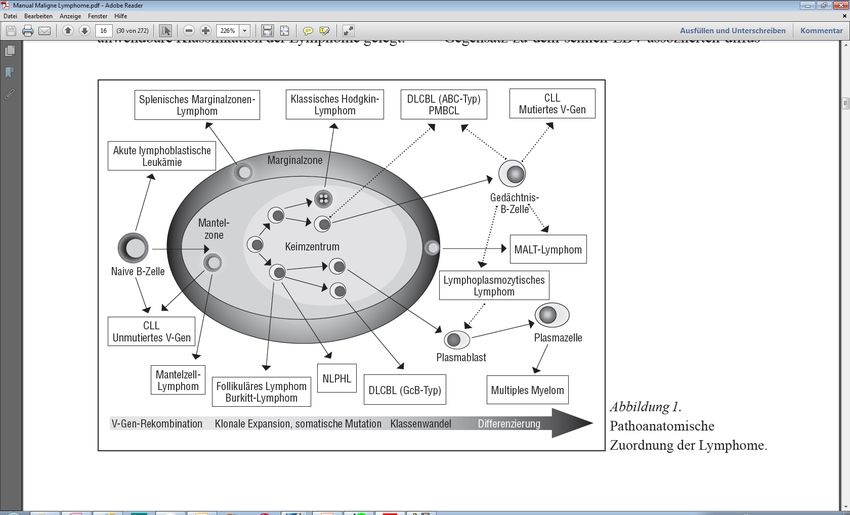
Überblick „indolente Lymphome/Multiples Myelom“ „reife/periphere B-Zell-Neoplasien“: - 1.Follikuläres Non-Hodgkin-Lymphom - 2.Nodales/splenisches Marginalzonenlymphom - extranodales Marginalzonen-/MALT-Lymphom „indolente (B-) NHL“ - 3.Mantelzell-Lymphom - Chronische lymphatische Leukämie, SLL - Haarzell-Leukämie, HCLv - 4. Lymphoplasmozytisches Lymphom „indolente (B-) NHL“ mit - 5. periphere T-Zell-Lymphome Besonderheiten - 6. Multiples Myelom © Paracelsus Medizinische Privatuniversität | Nürnberg
Indolente B-NHL
Allgemeines
Maligne Lymphome - Manual
Tumorzentrum München,
10.Auflage 2015
© Paracelsus Medizinische Privatuniversität | Nürnberg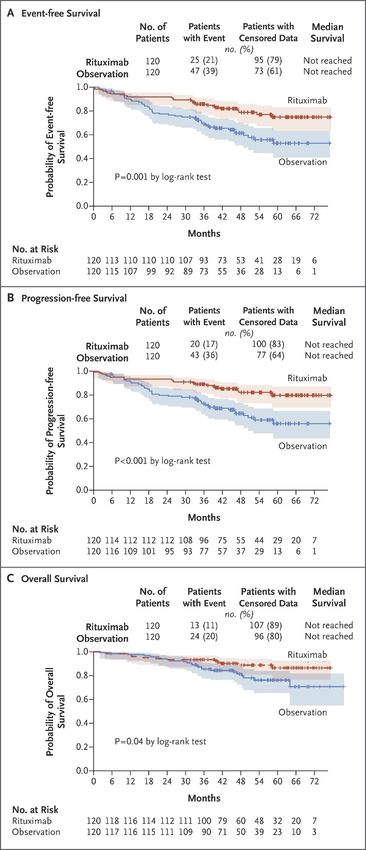
Indolente B-NHL
Allgemeines
§ Epidemiologie der Non-Hodgkin-Lymphome:
- ca. 10 Neuerkrankungen/100.000/Jahr
- ca. 50 % davon „indolent
- Männer 1,5 x häufiger betroffen
- im höheren Alter, bei HIV/AIDS oder unter Immunsuppression:
Inzidenz é
Jahresbericht 2014
Bayerisches Krebsregister
© Paracelsus Medizinische Privatuniversität | Nürnberg
Häufigkeit der verschiedenen B-NHL Quelle: WHO classification of tumours and haematopoetic and lymphoid tissues 4th edition, Lyon 2008 © Paracelsus Medizinische Privatuniversität | Nürnberg
Indolente B-NHL
Symptome
- Tastbare oder sichtbare nicht schmerzhafte („indolente“) Lymphknoten
(> 4 Wochen ->histologische Abklärung!) insbesondere zervikal, axillär,
paraaortal, inguinal, auch extranodal
- lokales Druckgefühl, paraneoplastisch : AIHA, Juckreiz uvm.
- „B-Symptome : Fieber, Nachtschweiß, Gewichtsverlust
Bei Knochenmarkinfiltration Symptome durch Störung der Blutbildung:
- Anämie: Leistungsknick, Abgeschlagenheit
- Thrombozytopenie: Blutungen (Petechien, Schleimhautblutungen)
- Abwehrschwäche durch Mangel an funktionsfähigen Leukozyten:
gehäufte oder ungewöhnliche Infektionen
-
© Paracelsus Medizinische Privatuniversität | Nürnberg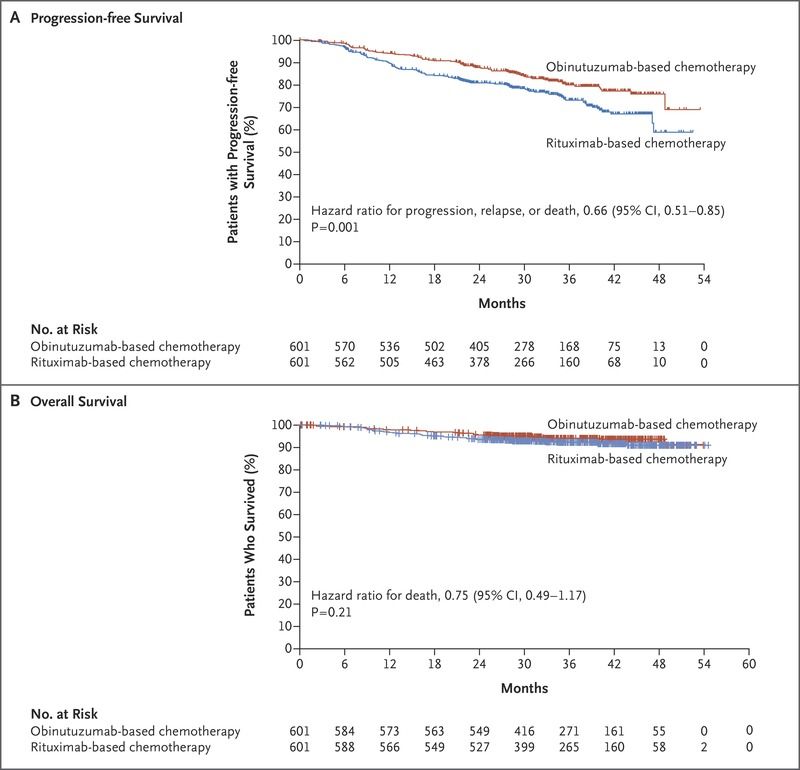
Indolente Lymphome
Diagnostik
Labordiagnostik:
-mikroskopisches Differentialblutbild, Kreatinin, Harnsäure, BZ,
LDH, GOT, GPT, AP, g-GT, Bilirubin, Quick, PTT, Fibrinogen,
BSG, Ferritin, ß2-μ-Globulin, Gesamteiweiß, Elektrophorese,
Immunglobuline, Immunfixation (Paraprotein?)
-Immunphänotypisierung (Flowzytometrie s.u.)
Bildgebung:
- Ultraschall und Computertografie von Hals, Thorax und
Abdomen, bei Symptomatik: craniale CT, evtl. auch PET-CT (kein
Standard, Überprüfung in Studien)
© Paracelsus Medizinische Privatuniversität | Nürnberg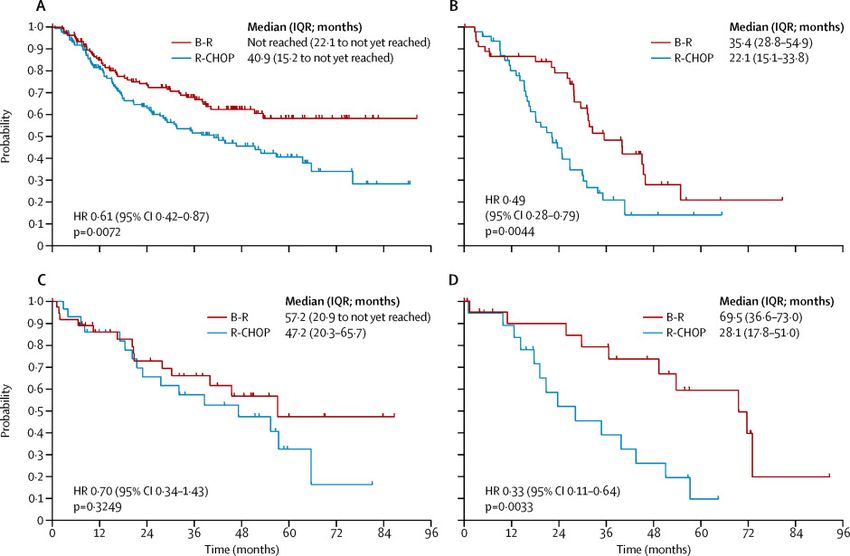
Indolente Lymphome
Diagnostik
Stadium I: Befall einer Lymphknotenstation
Stadium II: Befall von ≥ 2 Lymphknotenstationen auf einer Seite des Zwerchfells
Stadium III: Befall von Lymphknotenstationen auf beiden Seiten des Zwerchfells
Stadium IV: diffuser oder disseminierter Befall (z.B. auch KM)
A, B (-Symptome), E(-xtranodal), S(-pleen), X (bulky disease, ≥ 7,5 cm)
© Paracelsus Medizinische Privatuniversität | Nürnberg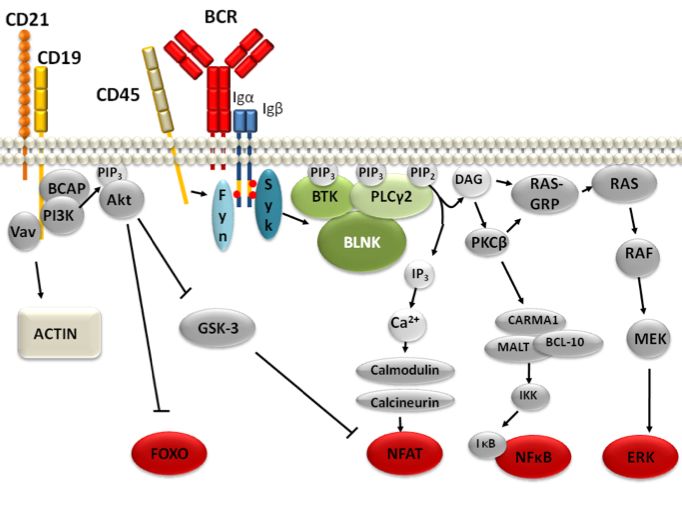
Indolente Lymphome
Diagnostik
• Lymphknotenpunktion: Zytologie, Histologie (Abtupfpräparate,
Stanzpräparate, idealerweise Exzision), Flowzytometrie
• Knochenmarkpunktion: Zytologie, Histologie, Flowzytometrie,
• Ggf. Molekularzytogenetische Untersuchung
© Paracelsus Medizinische Privatuniversität | NürnbergIndolente Lymphome
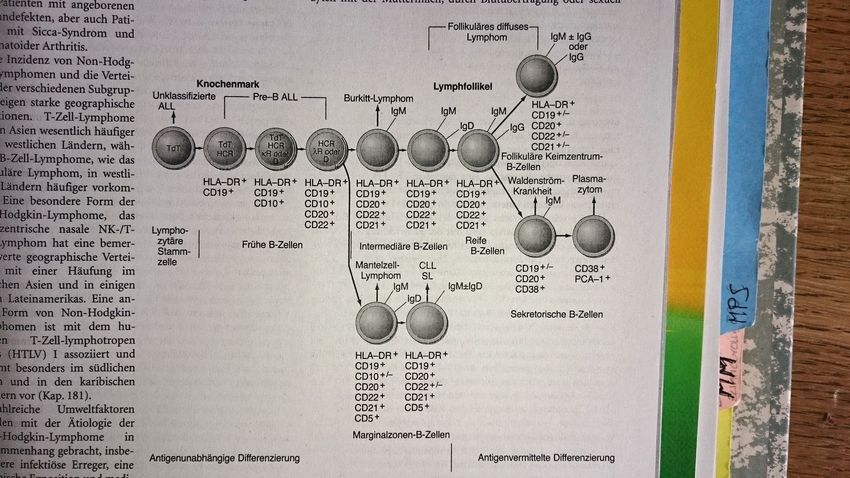
Diagnostik
Entwicklung der B-
Lymphozyten:
Veränderung,
Zugewinn und
Verlust von Ober-
flächenmarkern
Grafik: Harrisons Innere Medizin
© Paracelsus Medizinische Privatuniversität | NürnbergIndolente Lymphome
V. Bücklein, F. Lichtenegger, K. Pfannes et al. 36
Diagnostik Durchflusszytometrie
Tabelle 1. Immunzytologische Charakteristika verschiedener peripherer reifzelliger B-Zell-Lymphome.
Antigen CLL PLL SMZL HCL HCLv LPL MM PZL MALT FL MCL
CD19 + + + + + + – – + + +
CD20 +w + + + + +/– – – + + +
CD22 –/w + + + + + – – + + +
CD23 + – –/+ – – – – – – –/+ –/w
CD79b –/w + + – –/w + + +/w +
CD43 ++ + – + + –/+ – +
CD200 + – + – – v v v v –
sIgM +w ++ + ++ ++ ++ – – + + +
Leichtketten s/cy s/cy s s cy cy s s s/cy
HLA-DR + + + + + + +/– +/– + + +
CD10 – – – +/– – – – – – + –
CD5 + –/+w – – – – – – – – +
CD11c –/w –/+ +/– ++ ++ –/+w – – +w – –
FMC7 –/w + + + + –/+ –/+ + +
CD103 – – –/+ + + – – – –/+ – –
CD56 + –
CD138 + v –/+
CD38 v –/+ + ++ ++ –/+
CD28 + +
CD25 – ++ – –/w
CD123 – + – –
cy μ +
Dargestellt ist der häufigste Immunphänotyp nach [3, 4, 6] Maligne Lymphome - Manual
Tumorzentrum München,
CLL Chronische lymphatische Leukämie vom B-Zell-Typ
w=weakly, s=surface,
PLL cy=cytoplasmatic,
Prolymphozytenleukämie vom B-Zell-Typ v=variable 10.Auflage 2015
SMZL Splenisches Marginalzonenlymphom
HCL Haarzell-Leukämie
© Paracelsus Medizinische Privatuniversität | Nürnberg
HCLv Haarzell-Leukämie-VarianteScores: IPI, FLIPI, MIPI
„internationaler prognostischer Index“ IPI, je ein Punkt für:
-Alter > 60 Jahre
-Stadium III oder IV
-mehr als ein extranodaler Herd
-Schlechter AZ (ECOG ≥ 2, Karnofsky Index ≤ 60%)
-Erhöhte LDH
- niedriges Risiko (0-1 Punkte): 5-Jahres-Überlebensrate 73%
- niedrig-intermediäres Risiko (2 Punkte): 5-Jahres-Überlebensrate 51 %
- hoch-intermediäres Risiko (3 Punkte): 5-Jahres-Überlebensrate 43 %
- hohes Risiko (4-5 Punkte) 5-Jahres-Überlebensrate 26 %
FLIPI-2 (follikuläres Lymphom): MIPI (Mantelzell-Lymphom):
-Alter - Alter
-Hämoglobin - ECOG
-Größter LK (cm) - LDH
-KM-Infiltration? - WBC
-ß2-µ-Globulin PRIMA score
© Paracelsus Medizinische Privatuniversität | NürnbergIndolente Lymphome:
Therapieprinzipien – Allgemeines
- „watchful waiting“ bis zum Auftreten klinischer Symptome u.U. möglich
und auch sinnvoll (Ardeshna et al, Lancet 2003: 309 pts. Chlorambucil vs.
w&w: OS gleich, nach 10 Jahren 19% aller Patienten, 40% der >70jg. ohne
Therapie)
- durch frühzeitigen Einsatz von anti-CD20 Antikörper (Rituximab) „mono“
evtl. Vorteile (bei follikulärem Lymphom: Progression und Chemotherapie
verzögert; OS bisher nicht unterschiedlich: Ardeshna et al, Lancet oncology
2014)
- Radiatio bei lokalisiertem Befall
- Standard-Chemotherapie R-CHOP
- klinische Symptome/Befunde als Trigger für Chemotherapie-Beginn: ≥3
LK > 3cm, Bulk (7cm), Ergüsse, Zytopenien, Leuko>50/nl, LDH oder ß2-
μGl é, Splenomegalie, Organschäden...
- im Rezidiv sollte eine erneute histologische Untersuchung angestrebt
werden, um eine Transformation in ein hochmalignes Lymphom nicht zu
übersehen
© Paracelsus Medizinische Privatuniversität | Nürnberg1.Follikuläres Lymphom
- häufigster Subtyp aller neu diagnostizierten Lymphome
- Medianes Erkrankungsalter 60 – 65 Jahre, Frauen häufiger als Männer
- Risikofaktoren: Belastung mit Benzol, Pestiziden, Rauchen
- Neoplasie der B-Zellen des Keimzentrums (Zentrozyten und Zentroblasten)
- Die häufigste genetische Veränderung (in > 80% der Fälle) ist die Translokation
t(14;18)(q32;q21), welche zur Überproduktion von BCL2 führt („B-cell
lymphoma 2“, ein anti-apoptotisches Protein), d.h. die Apoptose wird inhibiert
Durchflusszytometrie positiv:
(u.a.) CD10, CD19, CD20, sIgM
pathologyoutlines.com
© Paracelsus Medizinische Privatuniversität | Nürnberg1. Follikuläres Lymphom – Grading
§ „in situ follicular lymphoma“ geringes Progressionsrisiko
§ Der Anteil an Zentroblasten in einem „high power field“ (400-fache
Vergrößerung, Histologie) bestimmt den Grad:
- Grad 1: 0 - 6
- Grad 2: 6 – 15 zählen zu den indolenten Lymphomen
- Grad 3A: > 15
- Grad 3B: > 15, keine Zentrozyten mehr nachweisbar gelten als
- Sonderformen: aggressive
- diffuses follikuläres Lymphom (keine Pseudofollikel) Lymphome
- diffuses großzelliges Lymphom mit follikulärem und werden
Lymphom so behandelt
§ FLIPI Index
§ Risikofaktor: Lymphome > 5 cm
© Paracelsus Medizinische Privatuniversität | Nürnberg1.Follikuläres Lymphom - Therapie 1 RF – Risikofaktoren (LK ≥ 5 cm) 2 AZ - Allgemeinzustand; 3 watch & wait – abwartendes Verhalten unter regelmäßiger Beobachtung 4 Induktionschemotherapie: R-Ben – Rituximab / Bendamustin oder R-CHOP – Rituximab / Cyclophosphamid / Doxorubicin / Vincristin / Prednison oder R-MCP – Rituximab / Mitoxantron / Chlorambucil / Prednison; 5 CR – komplette Remission, PR – partielle Remission Quelle: Onkopedia © Paracelsus Medizinische Privatuniversität | Nürnberg
1.Follikuläres Lymphom – Therapie
R-Benda oder R-CHOP?
Progression-free survival in histological subtypes of follicular lymphoma (A), mantle-cell lymphoma (B), marginal-zone
lymphoma (C), and Waldenstrom’s macroglobulinaemia (D)
Signifikant weniger Grad 3/4 Hämatotoxizität MJ Rummel et al. Lancet 2013; 381: 1203–10
© Paracelsus Medizinische Privatuniversität | Nürnberg1.Follikuläres Lymphom – Therapie
neue Therapien, Rezidivtherapie
§ Obinutuzumab: CD20-AK (veränderte Glykosylierung), zugelassen in
Kombination mit Chemotherapie als Induktionstherapie und bei
Rituximab-refraktärer Erkrankung (auch Rezidiv < 6 Monate):
Allerdings höhere Rate an Grad 3-5 NW:
74,6% vs. 67,8% R Marcus et al NEJM 2017
(Einsatz in der Primärtherapie)
§ 90Yttrium-Ibritumomab-Tiuxetan: Radio-
Immunkonjugat, zugelassen als Konsolidierung,
(Nutzen nach Rituximab unklar) und bei Rezidiv
oder Rituximab-refraktärer Erkrankung,
NW: Lymphozytopenie in 10%
© Paracelsus Medizinische Privatuniversität | Nürnberg1.Follikuläres Lymphom – Therapie
neue Therapien, Rezidivtherapie
§ Idelalisib: PI3Kδ-Inhibitor, zugelassen als Monotherapie im Rezidiv,
Ansprechrate 54%, NW: Neutropenie 27%, Colitis 16%
Von Altaileopard - Eigenes Werk, CC
BY-SA 3.0,
https://commons.wikimedia.org/w/ind
ex.php?curid=121932052.Marginalzonenlymphom
splenisch, nodal oder extranodal
- Insgesamt ca. 12 % aller B-NHL, Neoplasie der B – Lymphozyten der
Marginalzone der Lymphfollikel, assoziiert mit chronischen Entzündungen
(autoimmun z.B. Sjögren Syndrom oder Hashimoto Thyreoiditis, bakteriell z.B.
Helicobacter pylori)
- Je nach Ursprungsort nodal (2 %), extranodal (überwiegend im
Gastrointestinaltrakt (als „MALT“-Lymphom, 9 %) oder splenisch (auf Milz
beschränkt, 1 %), primär kutanes MZL
- (noduläre) Knochenmarkinfiltration und leukämische Ausschwemmung möglich,
beim splenischen Typ als „villöse Lymphozyten“
© Paracelsus Medizinische Privatuniversität | Nürnberg2.Marginalzonenlymphom
- Abgrenzung von anderen Lymphomen aufgrund Befallsmuster,
Oberflächenmarker (Durchflusszytometrie bzw. Immunhistochemie)
und Zellmorphologie (Mischung verschiedener Lymphozyten: kleine
atypische, größere monozytoid wirkende und blastäre Zellen wie
Zentroblasten und Immunoblasten)
Therapie je nach Entität unterschiedlich:
- Ansprechen des nodalen Typs auf Immunchemotherapie in der Regel
gut, Rezidive sind jedoch häufig, medianes Überleben ca. 5 Jahre
- Bei splenischem MZL kann eine Splenektomie zur Heilung führen
- MALT (mucosa associated lymphatic tissue) Lymphome sind häufig
assoziiert mit Helicobacter pylori – assoziierter Gastritis, im Stadium I
können sie durch eine antibiotische Eradikationsbehandlung geheilt
werden, sonst erfolgt Bestrahlung, bei aggressiveren Formen
Immunchemotherapie wie bei den anderen Lymphomen
© Paracelsus Medizinische Privatuniversität | Nürnberg3.Mantelzelllymphom - 70 % Männer, formal „indolentes“ Lymphom, jedoch meist aggressiver Verlauf - Lymphknotenbefall, häufig auch GI-Trakt (Waldeyerscher Rachenring), häufig Knochenmarkinfiltration, in 30 % leukämische Ausschwemmung, blastoide Variante möglich - Diagnosestellung am besten über Lymphknotenexstirpation - zytogenetisch fast immer Translokation t(11;14) mit Überexpression von Cyclin D1, was zur Aktivierung des Zellzyklus führt -> Überführung in „S-Phase“ © Paracelsus Medizinische Privatuniversität | Nürnberg
3.Mantelzelllymphom -MCL international prognostic index (MIPI): Alter, ECOG, LDH und Leukozytenzahl, score für 3 Risikoklassen mit 6 Jahresüberleben von 10 % bis 65 %. Proliferationsmarker Ki67 (MIB1) hat ebenfalls hohe prognostische Relevanz - zytologisch kleine Zellen mit gekerbtem Kern - histologisch angedeutet follikuläres Wachstum, häufig auch diffus mit Aufhebung der Lymphknotenstruktur - durchflusszytometrisch oder immunhisto-chemisch Nachweis einer CD5/CD19 Koexpression, CD 23 fehlt, Cyclin D1 kräftig pos. © Paracelsus Medizinische Privatuniversität | Nürnberg
3.Mantelzelllymphom Therapie § „Standard“: 6 Kurse Immunochemotherapie (R-CHOP/R-DHAP im Wechsel) gefolgt von HD-Cytarabin-haltiger Konditionierung mit autologer Stammzelltransplantation, PFS signifikant besser als Induktion mit 6 x R-CHOP: „MCL younger“ Studie des European Mantle Cell Lymphoma Network (O Hermine et al, The Lancet 2016) § Erhaltungstherapie mit Rituximab verlängert PFS (79 vs. 61 %) und OS (89 vs. 80 %) (S Le Gouill, NEJM 2017) © Paracelsus Medizinische Privatuniversität | Nürnberg
3.Mantelzelllymphom Therapie § TRIANGLE Studie rekrutiert aktuell, 3 Arme: „Standard“ +/- Ibrutinib, Standard + Ibrutinib ohne autologe SZT § Eine neuere Therapie, die bessere Resultate zeigt als 6 x R-CHOP (bisher Standard für Pat. > 65 Jahre) ist „VR-CAP“ (Bortezomib statt Vincristin): PFS 24,7 vs. 14,4 Monate (T Robak NEJM 2015) § Optionen für die Rezidivtherapie jeweils als Monotherapie zugelassen - Ibrutinib (Hemmung der Brutonkinase): ORR 67%, DOR 17,5 Monate - Lenalidomid (Immunmodulator): ORR 28 – 53%, DOR 13 – 17 Monate - Temsirolimus (mTOR Inhibitor): ORR 9,1 % © Paracelsus Medizinische Privatuniversität | Nürnberg
4.Lymphoplasmozytisches Lymphom
Makroglobulinämie - Morbus Waldenström
- Proliferation von pathologischen/monoklonalen B-Lymphozyten und
lymphoplasmozytoiden Zellen, die ein IgM produzieren – ohne „CRAB“
Kriterien, daher nicht mit Multiplem Myelom zu verwechseln
- Zellen sind typischerweise CD 19 positiv (Unterschied zu MM !)
- In 90 % „L265P“ Mutation im MYD88-Gen, in 30 % Mutation in CXCR-4
(Chemokinrezeptorgen)
- in bis zu 60 % der Patienten verstärkte Blutungsneigung, auch retinal
(Plättchenaggregation durch IgM gehemmt), Embolien/Apoplex
- Symptome durch Anämie
- Infektanfälligkeit
- Neurologische Symptome durch Hyperviskosität, z.B. Sehstörungen oder
Nervenschädigung durch IgM-Anlagerung
- Raynaud-Symptomatik („digitus mortuus“)
- Lymphadenopathie, Hepatosplenomegalie
- Im Labor (Immunfixation) Nachweis eines monoklonalen IgM - Proteins
© Paracelsus Medizinische Privatuniversität | Nürnberg4.Lymphoplasmozytisches Lymphom
Makroglobulinämie - Morbus Waldenström
- Verschiedene Stadieneinteilungen bzw. Prognose-Scores (Alter,
Symptomatik, Hb-Wert, Thrombozytopenie)
- Therapie je nach Stadium: Rituximab mono, Kombination mit
Chemotherapie
- Neu zugelassen ist Ibrutinib (Bruton-Tyrosinkinaseinhibitor) als orale,
gut verträgliche Therapie mit sehr guten Ansprechraten von 90%
innerhalb eines Monats
- Bortezomib ist ebenfalls hochwirksam, aber nicht zugelassen
Die Mutation im MYD88-Gen triggert die Interleukin-1 Receptor-Associated Kinase und die
Bruton s Tyrosinkinase. Diese beiden Kinasen aktivieren NF-κB, einen onkogenen Faktor.
Patienten, die zusätzlich die CXCR-4 Mutation aufweisen, scheinen am Besten auf Ibrutinib
anzusprechen.
© Paracelsus Medizinische Privatuniversität | NürnbergIndolente Lymphome:
Therapieprinzipien – Rezidivbehandlung
§ Grundsätzlich ist im Rezidiv eine erneut histologische Diagnostik sinnvoll,
um eine Transformation in ein hochmalignes Lymphom auszuschließen
§ Erneute Chemotherapie, gefolgt von autologer Stammzelltransplantation
ist insbesondere bei jüngeren Patienten und frühen Rezidiven eine
Option
§ In Einzelfällen kann auch eine allogene Stammzelltransplantation
erwogen werden (z.B. mehrfach rezidiviertes follikuläres Lymphom bei
jungem Patienten):
5 Jahre:
Rezidivrate 16 %
PFS 48 %
OS 51 %
(reduced Intensity allo TX bei rezidiviertem FL)
SP Robinson et al Ann Oncol. 2016
© Paracelsus Medizinische Privatuniversität | Nürnberg5. Mature T and NK neoplasms WHO 2016
- Monomorphic epitheliotropic intestinal T-cell lymphoma
- Indolent T-cell lymphoproliferative disorder of the GI tract*
- Hepatosplenic T-cell lymphoma
- T-cell prolymphocytic leukemia - Subcutaneous panniculitis-like T-cell lymphoma
- T-cell large granular lymphocytic leukemia - Mycosis fungoides
- Chronic lymphoproliferative disorder of NK cells* - Sezary syndrome
- Aggressive NK-cell leukemia - Primary cutaneous CD30+ T-cell lymphoproliferative
Systemic EBV1 T-cell lymphoma of childhood disorders:Lymphomatoid papulosis, Primary cutaneous
- Hydroa vacciniforme–like lymphoproliferative anaplastic large cell lymphoma
disorder - Primary cutaneous γδT-cell lymphoma
- Adult T-cell leukemia/lymphoma - Primary cutaneous CD8+ aggressive epidermotropic cytotoxic
- Extranodal NK-/T-cell lymphoma, nasal type T-cell lymphoma*
- Enteropathy-associated T-cell lymphoma - Primary cutaneous acral CD8+ T-cell lymphoma*
- Primary cutaneous CD4+ small/medium T-cell
lymphoproliferative disorder*
- Peripheral T-cell lymphoma, NOS
- Angioimmunoblastic T-cell lymphoma
- Follicular T-cell lymphoma*
- Nodal peripheral T-cell lymphoma with TFH phenotype*
- Anaplastic large-cell lymphoma, ALK1
- Anaplastic large-cell lymphoma, ALK2
- Breast implant–associated anaplastic large-cell lymphoma*
* = „provisional entities“
© Paracelsus Medizinische Privatuniversität | Nürnberg5. Periphere T-Zell- und NK-Zell-Lymphome
§ Lymphome aus reifen „post-thymischen T-Zellen:
- starke zytologische Variationsbreite
- Diagnostik schwierig; Klonalitätsbestimmung mittels PCR (T-Zell-
Rezeptor Gen-Rearrangement)
- häufig Nekrosen und Gefäßinvasion, häufig extranodal und
Hautbeteiligung
- >20 versch. Subtypen (auch klinische Aspekte)
- Prognose insgesamt eher schlecht: 5-Jahres OSkutane T-Zell-Lymphome
- Mycosis fungoides (70% der kutanen TCL)
TNMB Klassifikation:
T1: 10% der KÖF Flecken/Papeln/Plaques
T3: Hauttumore > 1cm
T4: Erythrodermie bzw. > 80 % der KÖF betroffen
N (LK), M (viszerale Beteiligung), B (Blut) ->
- Sezary-Syndrom: leukämischer Verlauf
- Therapie: PUVA, extrakorporale Photopherese, bei
generalisiertem Verlauf z.B. CHOP, MTX,
Kombination mit IFN.
- T1 10-Jahres OS 100%, ab T3 10-Jahres OS nur
noch 40%, viszerale Beteiligung ist prognostisch
ungünstig
© Paracelsus Medizinische Privatuniversität | Nürnberg6.Plasmazellneoplasie/Multiples Myelom
- Neoplasie monoklonaler Plasmazellen mit Produktion von pathologischem
Immunglobulin (IgG in 55%, IgA in 25%, selten IgM, IgD oder IgE) oder nur
kappa- oder lambda- Leichtketten, selten asekretorisch
- Ca. 6500 Neuerkrankungen pro Jahr in Deutschland
- Relative 5-Jahres Überlebensrate um 50%, 10-Jahre ca. 30%
• Inzidenz: 4-6/100.000/Jahr www.dgho-onkopedia.de
© Paracelsus Medizinische Privatuniversität | Nürnberg6.Multiples Myelom - Symptome § Knochenschmerzen (60%) durch Osteolysen und pathologische Frakturen § Fatigue (40%), auch anämiebedingt § Hyperkalzämie (20%) § Infektneigung (20%) mitbedingt durch AK-Mangel („Immunoparese , erniedrigte Spiegel der nicht betroffenen Immunglobuline) und unvollständige oder unphysiologische Antikörper, funktionelle Hypogammoglobulinämie § Gewichtsverlust (25%) § Schäumender Urin durch Albuminurie und Ausscheidung von Bence- Jones Proteinen (freie Leichtketten) § 25 % der Patienten sind beschwerdefrei bei Diagnosestellung © Paracelsus Medizinische Privatuniversität | Nürnberg
6.Multiples Myelom Diagnostik § Labor: Diff.BB, Elektrolyte, Nierenretentionsparameter, Gesamteiweiß und Albumin im Serum, Serumprotein-Elektrophorese mit Bestimmung des M-Gradienten, Immunfixations-Elektrophorese im Serum und Urin, Immunglobuline (IgG, IgA, IgM) im Serum, quantitativ, freie Kappa- und Lambda-Leichtketten im Serum quantitativ incl. Quotient, 24 h-Sammel- urin zur Quantifizierung der Eiweißausscheidung, LDH, Beta2-Mikro- globulin im Serum § Bildgebung: Ganzkörper CT ohne KM, ggf. auch MRT (sensitiver, aber aufwändiger), PET-CT (bisher nicht in Deutschland), Echokardiografie (Amyloidose?) § Knochenmarkpunktion: Zytologie, Histologie, Molekularzytogenetik © Paracelsus Medizinische Privatuniversität | Nürnberg
6.Multiples Myelom Diagnostik
Monoklonale
Gammazacke
M-Gradient
(auch quantifizierbar)
gesund Nachweis von monoklonalem
IgG, IgA, selten: IgD, IgE, IgM
oder nur Leichtketten kappa oder
lambda im Serum und/oder Urin
© Paracelsus Medizinische Privatuniversität | Nürnberg6.Multiples Myelom Diagnostik
Immunfixation
Anlegen von mit Antiseren getränkten Zelluloseacetatfolien, Auswaschen nicht-
präzipitierter Proteine, Anfärbung der durch Präzipitation mit monospezifischen
Antikörpern gebildeten Immunkomplexe
gesund
Als positiv gilt eine Immunfixation bei
Nachweis klar umschriebener Banden
(Pfeile). Im vorliegenden Fall existiert ein
Paraprotein vom Typ IgG Lambda
© Paracelsus Medizinische Privatuniversität | Nürnberg6.Multiples Myelom – Diagnose/Stadien § Plasmozytom = 1 solitärer Knochenherd § monoklonale Gammopathie unklarer Signifikanz (MGUS), Prävalenz bei > 50 Jährigen 3 - 4 %, Progressionsrisiko 0,5 – 1 % pro Jahr § Smoldering Multiple Myeloma: monoklonales Protein >30 g/l u./o. KM- Infiltration >10% oder Plasmozytom, Progression bis 10 % pro Jahr, bei „high risk“ (SliM Kriterien s.u.) 40 – 80 % in zwei Jahren § Asekretorisches MM: KM-Infiltration >10%, Organkomplikationen, kein Paraprotein nachweisbar § Symptomatisches MM: monoklonales Protein in Serum/Urin + Plasmazellinfiltration in KM oder Plasmozytom + Organkomplikationen („CRAB-sliM“) © Paracelsus Medizinische Privatuniversität | Nürnberg
6.Multiples Myelom – Diagnose/Stadien
International Staging System (10.750 pts.)
nach Durie und Salmon, 1975 Greipp et al, J clin oncol., 2005
Stadium Overall ISS - Kriterien Median des
Survival Stadium Overall Suvival
I Hb >10g/dl, maximal ~ 60 Monate
1 Osteolyse,
I Serum ß2- 62 Monate
monoklonales Protein
Mikroglobulin <
niedrig*
3.5mg/L
UND
II weder I noch II 25-50 Monate
Serumalbumin ≥
3.5g/dl
III Hb6.Multiples Myelom – Diagnose/Stadien
Revised international staging system (Palumbo et al, JCO 2015)
R-ISS Stadium 5-yr OS „real life OS1
I ISS I,
Standardrisiko*, 82 % 77 %
normale LDH
II Nicht I oder III 62 % 53 %
III ISS III +
Hochrisiko* oder 40 % 19 %
erhöhte LDH
*zytogenetisches Risiko:
Standard: kein Hochrisiko
Hoch: del(17p), t(4;14), t(14;16)
1E Kastritis et al, hematologica 2016
© Paracelsus Medizinische Privatuniversität | Nürnberg6. Diagnosekriterien für ein behanldungsbedürftiges
Multiples Myelom „CRAB-sliM“
1.) klonale Plasmazellen im KM > 10% oder extramedulläres MM
+ C = Hyperkalzämie (>0,25 mmol/l über normal oder >2,75)
+ R = Niereninsuffizienz GFR 2 mg/dl
+ A = Hb 2 g/dl unter normal oder 60% „sixty“
+ Leichtketten Ratio im Serum ≥ 100 „light chain“
+ > 1 fokale Läsion im MRT (> 5mm) „MRT“
SV Rajkumar, Lancet Oncology 2014
Auch andere krankheitsbedingte Symptome wie B-Symptomatik, Schmerzen, Para-
neoplastische PNP können eine Behandlungsindikation darstellen
© Paracelsus Medizinische Privatuniversität | Nürnberg6. Multiples Myelom -Therapieüberblick
Hochdosistherapie möglich? Altersgrenze 70-75 Jahre
ja
nein
Bisphosphonat über 2 Jahre
3-6 Zyklen Induktionstherapie:
VCD, VTD, VD, (VRD)1 VMP 9 Zyklen
Rd bis zum Progress
Stammzellseparation VCD 6 oder mehr Zyklen
VD 6 oder mehr Zyklen
BP
1-2 x Hochdosistherapie (HD): (VRD)1
Melphalan 140-200 mg/qm mit
autologer Stammzelltransplantation
Evtl.
Erhaltungstherapie
1Kombination nicht zugelassen
© Paracelsus Medizinische Privatuniversität | Nürnberg6.Multiples Myelom Induktionstherapie vor HD § VD Bortezomib, Dexamethason § VCD Bortezomib, Cyclophosphamid, Dexamethason § VTD Bortezomib, Thalidomid, Dexamethason § VRD1 Bortezomib, Lenalidomid, Dexamethason - Bortezomib: das Bor-Atom in Bortezomib bindet an ein Proteasom, welches essentiell für den Abbau bestimmter Proteine ist, dies führt auf verschiedenen Wegen zu einer Apoptose bevorzugt von Tumorzellen („Mülleimer verstopft“ usw.) - Thalidomid und Lenalidomid gelten als Immunmodulatoren („IMiDs“), T-Rezeptpflicht! Antiangiogenese, TNFα-Hemmung uvm... - CR Raten 20 – 40 %, Auflistung oben in (vermuteter) aufsteigender Effektivität (schlechte Vergleichbarkeit, unterschiedliche Ergebnisse aus verschiedenen Studien) - VTD mit Neurotoxizität (PNP) Grad 3-4 bei 11%, andere Regime 3-4% - bei Bortezomib Auftreten von Herpes Zoster, daher Aciclovir-Prophylaxe notwendig! - Antibiotika- und Thromboseprophylaxe in der Induktionstherapie empfohlen 1Kombination in Deutschland nicht zugelassen © Paracelsus Medizinische Privatuniversität | Nürnberg
6.Multiples Myelom Stammzellsammlung
§ Die Stammzellmobilisation erfolgt mittels G-CSF:
- nach vorgeschalteter „Mobilisations-Chemotherapie
- aus dem steady state (nur G-CSF über 4 Tage), bei Bedarf verstärkt um
eine Gabe von Plerixafor
§ Bei ansteigenden Leukozytenwerten wird eine Zählung CD34 positiver
Blutstammzellen durchgeführt, bei ausreichend hohem Wert erfolgt dann
eine Stammzellapherese
In Stickstofftanks können die
Zellen bei -140 C jahrelang auf-
bewahrt werden
© Paracelsus Medizinische Privatuniversität | Nürnberg6.Multiples Myelom Hochdosistherapie
§ Stationäre Behandlung von ca. 3 Wochen: nach Applikation von 200 bzw. 140
(für komorbide Patienten) mg/qm Melphalan erfolgt nach mindestens 24 h
Abstand die Rücktransfusion einer frisch aufgetauten ausreichenden Menge
autologer Blutstammzellen, dies führt zuverlässig zu einer Regeneration des
Blutbilds nach 8 – 12 Tagen, Mortalität 1-2%, Nebenwirkungen: protrahierte
Übelkeit, Infektionen
§ Eine Tandem-Transplantation (erneute HD innerhalb von max. 6 Monaten)
führt nach aktuellen vorläufigen Studienergebnissen zu längerem PFS (73 vs.
64 %) und OS (89 vs. 82%) (3year estimate), insbesondere Patienten im
Stadium R-ISS III scheinen zu profitieren (ASH 2017 abstract #401, Cavo et
al.). Die Frage 1 oder zwei HD-Therapien ist aber nicht abschließend geklärt.
© Paracelsus Medizinische Privatuniversität | Nürnberg6.Multiples Myelom allogene
Stammzelltransplantation
allogene Tx (meist nach 1x autolog) als kuratives Konzept –
„GvM (Graft versus Myeloma) -Effekt
Ø Bei konventioneller Konditionierung wegen hoher TRM (30-
40%) alternativen Konzepten unterlegen
Ø Bei Konditionierung in reduzierter Intensität deutlich
niedrigere TRM (10-15%) bei gutem Ansprechen (OR um
80%), Langzeitdaten (OS, Rezidiv), Problematik der
chronischen GvH-Reaktion in 50-70% der Patienten
Ø weiterhin Überprüfung in Studien sinnvoll, da Datenlage
bisher unklar
© Paracelsus Medizinische Privatuniversität | Nürnberg6. Multiples Myelom Erhaltungstherapie § Lenalidomid ist als Erhaltungstherapie nach autologer Stammzelltransplantation zugelassen und führt im Vergleich zu Placebo in mehreren Studien zu einer Verlängerung des PFS (z.B. 57 vs. 29 Monate in IFM 2005-02 Studie) und auch z.T. auch des OS (median 111 vs. 84 Monate) und sollte den Patienten daher trotz des erhöhten Risikos von sekundären Neoplasien (ca. 2/100 Patientenjahre) angeboten werden § Interferon alfa-2b, zugelassen, wird aber in den Leitlinien nicht empfohlen § Bortezomib ist als Erhaltungstherapie nicht zugelassen, zeigte aber in einer Studie nach Bortezomib-haltiger Induktion und HD-Therapie einen signifikanten Vorteil im PFS gegenüber einer Thalidomid-haltigen Therapie und konnte den negativen Effekt einer ungünstigen Zytogenetik sowie einer Niereninsuffizienz nivellieren (Goldschmidt, Leukemia 2018) © Paracelsus Medizinische Privatuniversität | Nürnberg
6.Multiples Myelom Therapie ohne Hochdosis § VMP: Bortezomib, Melphalan, Prednison, „VISTA“ Studie: - OS 56 vs. 43 Monate für MP (Miguel, JCO 2013) § Rd: Lenalidomid, Dexa, „FIRST“ Studie: Rd cont > Rd 18 > MPT - medianes OS für Rd cont: 59 Monate (Facon, Blood 2018) § MPR-R: Melphalan, Prednison, Lenalidomid - „ECOG E1A06“ OS 48 Monate (Stewart, Blood 2015) - EFS 29 Monate (Palumbo, NEJM 2012) (MPThalidomid -T gleichwertig) § VD: Bortezomib und Dexamethason, (Bortezomib s.c. PNP Grad ≥ 3: 6 vs. 16 % i.v.) § BP: Bendamustin, Prednison, PFS 15 Monate © Paracelsus Medizinische Privatuniversität | Nürnberg
6.Multiples Myelom supportive Therapie
– Bisphosphonate
– Schmerzmedikation
– Radiatio einzelner Osteolysen, Vertebroplastie (Schmerz/Stabilität)
– Transfusionen, Erythropoetin
– Antibiotika, Immunglobuline
– Amyloidose? Nierenfunktion? Hyperkalzämie?
© Paracelsus Medizinische Privatuniversität | Nürnberg6.Multiples Myelom Bisphosphonate
– ASCO 2002/Mayo Consensus 2006: bei Osteolysen Verminderung
von Skelettkomplikationen erwiesen, bei stabilem Krankheitsverlauf
nach 2 Jahren längere Intervalle (3Monate)
– Zolendronat hat günstigen Einfluss auf PFS, OS und
Skelettkomplikationen => für 2 Jahre Teil der Therapie emfpohlen,
Denosumab möglicherweise insb. bei Niereninsuffizienz besser
geeignet (Raje, The Lancet Oncology 2018)
– NW: Kieferosteonekrose in ca. 4%! Zahnärztliche Mitbetreuung!!
© Paracelsus Medizinische Privatuniversität | Nürnberg6.Multiples Myelom Rezidivtherapie § Pomalidomid („IMiD“), Dexamethason +/- Cyclophosphamid (Zulassung in Europa 2013) § Carfilzomib (Proteasominhibitor), in Kombination mit Dexamethason +/- Lenalidomid (2015) § Panobinostat, Bortezomib, Dexamethason (2015) § Ixazomib (Proteasominhibitor) oral, in Kombination mit Lenalidomid und Dexamethason (2016) § Elotuzumab (SLAMF7-Inhibitor) Lenalidomid, Dexamethason (2016) § Daratumumab, anti CD38 Antikörper auch kombiniert mit Lenalidomid oder Bortezomib und Dexamethason (2016 mono, 2017 Kombination) § Klassische Zytostatika: Adriamycin, Cyclophosphamid, Bendamustin auch in Kombination © Paracelsus Medizinische Privatuniversität | Nürnberg
Sie können auch lesen

















































