STRUKTURELLE UND FUNKTIONELLE UNTERSUCHUNG DES MYELINPROTEINS 36K AUS DEM ZNS DER REGENBOGENFORELLE (ONCORHYNCHUS MYKISS) - repOSitorium
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
STRUKTURELLE UND FUNKTIONELLE UNTERSUCHUNG
DES MYELINPROTEINS 36K AUS DEM ZNS DER
REGENBOGENFORELLE (ONCORHYNCHUS MYKISS)
DISSERTATION
zur Erlangung des akademischen Grades
„Doktor der Naturwissenschaften (Dr. rer. nat.)“
des Fachbereiches Biologie/Chemie
der Universität Osnabrück
vorgelegt von
Wolfgang Moll
November 2004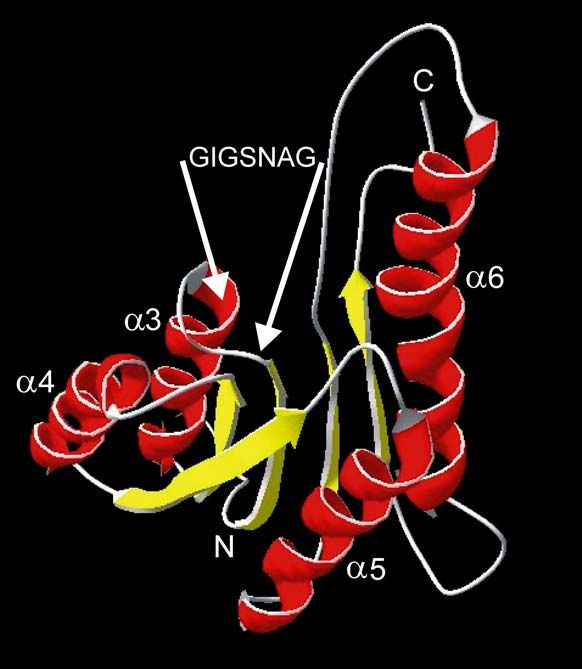
INHALTSVERZEICHNIS
INHALTSVERZEICHNIS
1. EINLEITUNG 1
2. MATERIAL UND METHODEN
2.1 Material
2.2.1 Bakterienstämme 4
2.2.2 Regenbogenforellen (Oncorhynchus mykiss) 4
2.2 Methoden
2.2.1 Computergestützte Aminosäure-Sequenzanalysen 4
2.2.2 Animpfen einer Bakterien-Über-Nacht-Kultur (ÜK) 5
2.2.3 Heterologe Expression mit E. coli/pET-System (Fa. Novagen) 5
2.2.4 Affinitätschromatographische Aufreinigung mit Ni2+-chelatierender
Sepharose 5
2.2.5 SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) (nach Laemmli) 6
2.2.6 Proteinanfärbung mit Coomassie-Lösung 7
2.2.7 Proteinanfärbung durch Silberfärbung 7
2.2.8 Proteintransfer auf Nitrozellulose- oder Polyvinylidenfluorid- (PVDF-)
Membran (Western-Blot) 8
2.2.9 Immunologischer Nachweis von Proteinen auf Nitrozellulose- oder
Polyvinylidenfluorid- (PVDF-) Membran 9
2.2.10 Fern-UV-Circulardichroismus-Spektroskopie (CD-Spektroskopie) von
rekombinantem 36K in Lösung 9
2.2.11 Tryptophan-Fluoreszenz-Studien von rekombinantem 36K in Lösung 10
2.2.12 Elektronenspin-Resonanz-Spektroskopie (ESR-Spektroskopie) von
rekombinantem 36K in Lösung 10
2.2.13 Röntgenkleinwinkelstreuung von rekombinantem 36K in Lösung 11
2.2.14 Assoziation von rekombinantem 36K an phospholipidbeschichtete
Silica- Kügelchen („TRANSIL“; Fa. Nimbus, Leipzig) 11
2.2.15 Kontrollierter proteolytischer Verdau von rekombinantem 36K mit
Trypsin (Fa. Sigma-Aldrich, München) 12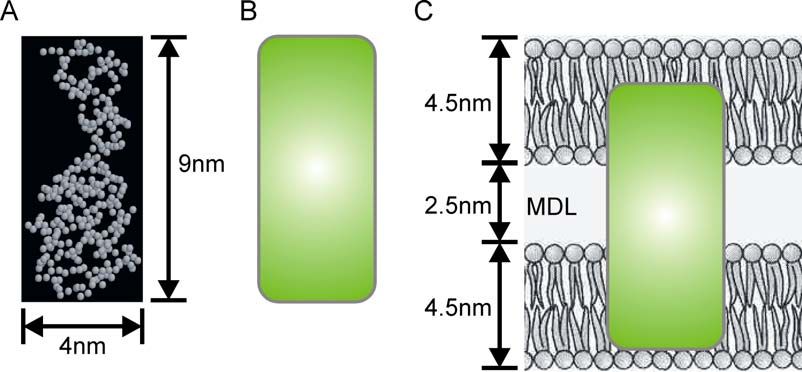
INHALTSVERZEICHNIS
2.2.16 Chemische Spaltung von rekombinantem 36K mit 2-Nitro-5-
Thiocyanobenzonsäure (NTCB) 12
2.2.17 Random-Fluoreszenz-Resonanz-Energie-Transfer-(Random-FRET-)
Untersuchungen von membranassoziiertem rekombinanten 36K 13
2.2.18 Isolieren von Myelin aus dem ZNS der Regenbogenforelle
(Oncorhynchus mykiss) 14
2.2.19 Herstellen einer 5%igen (w/v) Digitonin-Stammlösung und Myelinprotein-
Extraktion mit Digitonin 14
2.2.20 Protein-Konzentrationsbestimmung mit Amidoschwarz-Tüpfel-Test 15
2.2.21 Messung einer Dehydrogenase-Aktivität im isolierten ZNS-Myelin der
Regenbogenforelle (Oncorhynchus mykiss) 15
2.2.22 Herstellung von Phosphatidylcholin (PC)/Phosphatidylethanolamin
(PE)-Liposomen 16
2.2.23 Aggregationsassay von Phosphatidylcholin (PC)/Phosphatidyl-
ethanolamin (PE)-Liposomen mit rekombinantem 36K 16
2.2.24 Blue-Native Gradienten-Polyacrylamidgel-Elektrophorese (BN-PAGE)
(1. Dimension) 17
2.2.25 SDS-PAGE der durch BN-PAGE aufgetrennten Myelinprotein-Komplexe
(2. Dimension) 18
2.2.26 Separation der Myelinprotein-Komplexe mit ProFound-Co-Immuno-
präzipitation-Kit (Perbio Sciences, Bonn) 18
3. ERGEBNISSE
3.1 Computerunterstützte Analyse der 36K-Aminosäuresequenz
3.1.1 Homologie-Alignment der Aminosäure-Sequenzen von 36K und
Proteinen aus der Reduktase/Epimerase/Oxidoreduktase Superfamilie
(RED) 20
3.1.2 Sekundärstruktur-Vorhersage des 36K-Proteins mit dem JPred-Internet-
Server 22
3.1.3 3-dimensionales Modell des N-terminalen Teils von 36K 24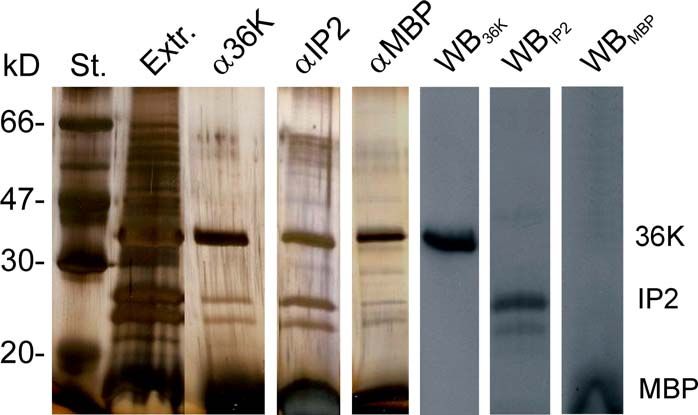
INHALTSVERZEICHNIS
3.2 Untersuchung der Sekundärstrukturzusammensetzung des
rekombinanten 36K-Proteins mittels Fern-UV-Circulardichroismus-
Spektroskopie (CD-Spektroskopie) 25
3.3 Röntgenkleinwinkelstreuung von rekombinantem 36K in Lösung 26
3.4 Untersuchungen zur Funktion von 36K
3.4.1 Messung einer Dehydrogenase-Aktivität im isolierten Myelin 27
3.4.2 Aggregationsassay von Phosphatidylcholin (PC)/Phosphatidyl-
ethanolamin (PE)-Liposomen mit rekombinantem 36K 29
3.5 Einfluss von Nukleotiden auf die Konformation des rekombinanten
36K-Proteins
3.5.1 Tryptophan-Fluoreszenz-Spektren von rekombinantem 36K ± Nukleotide 31
3.5.2 Fern-UV-Circulardichroismus-Spektroskopie von rekombinantem 32
36K ± Nukleotide
3.5.3 Elektronenspin-Resonanz-Spektroskopie (ESR-Spektroskopie) von
rekombinantem 36K ± Nukleotide 33
3.6 Untersuchungen zur Penetration des 36K-Proteins in einen
Phospholipid-Bilayer
3.6.1 Kontrollierter proteolytischer Verdau von membrangebundenem 36K 34
3.6.2 Einfluss von Nukleotiden auf Trypsinisierung von membrangebundenem
36K 38
3.6.3 Spaltung von rekombinantem 36K mit 2-Nitro-5-Thiocyanobenzonsäure
(NTCB) in Lösung 40
3.6.4 Spaltung von membrangebundenem rekombinanten 36K mit
2-Nitro-5-Thiocyanobenzonsäure (NTCB) 42
3.6.5 Random-Fluoreszenz-Resonanz-Energie-Transfer- (Random-FRET-)
Untersuchung zwischen rekombinantem 36K-QSY und 7-Nitrobenz-2-
oxa-1,3-Diazol-4-yl-Phosphatidylcholin (NBD-PC) 42
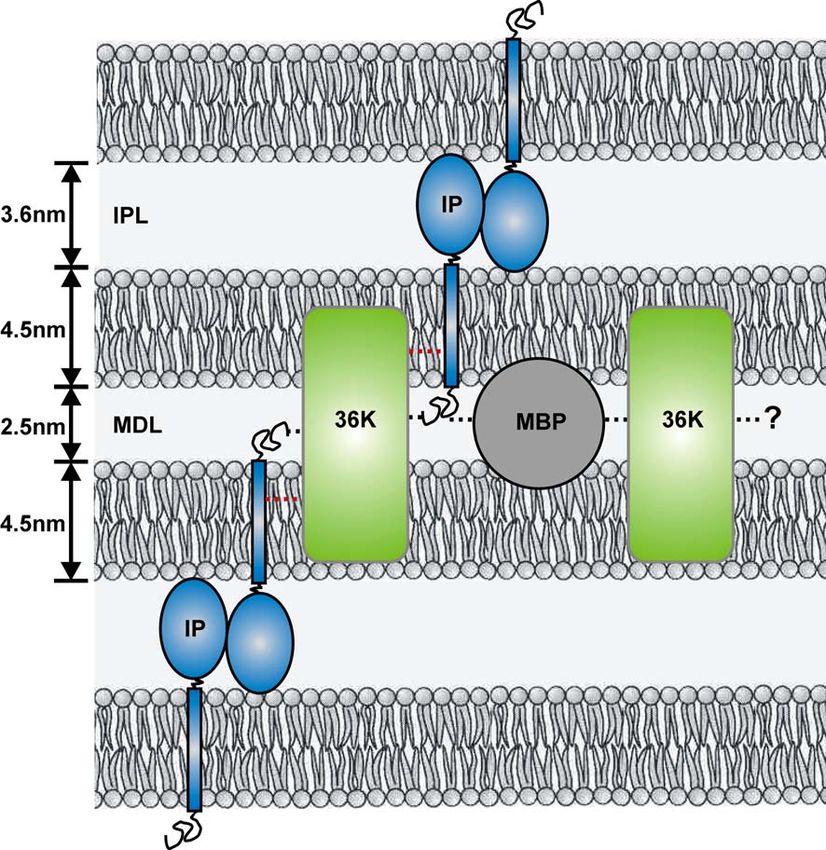
3.6.6 Hypothetisches Modell der Lage von 36K in der „Major Dense Line“ des
ZNS-Myelins der Regenbogenforelle (Oncorhynchus mykiss) 46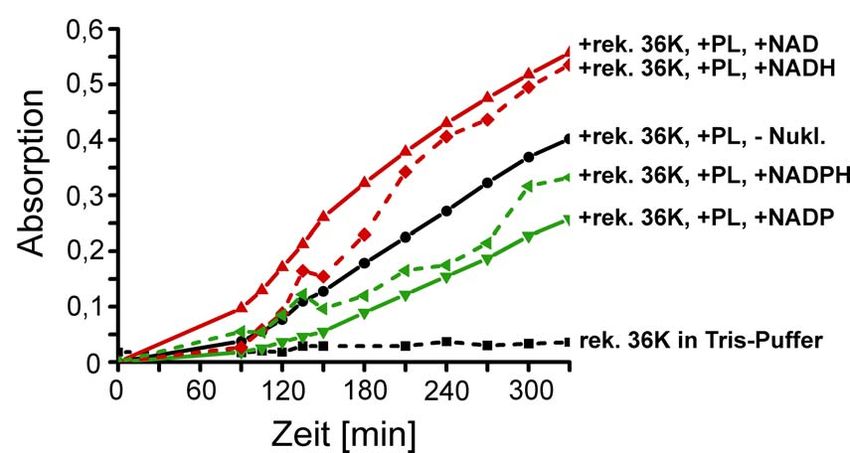
INHALTSVERZEICHNIS
3.7 Interaktionen zwischen 36K und anderen Myelinproteinen aus dem
ZNS der Regenbogenforelle (Oncorhynchus mykiss)
3.7.1 Myelinprotein-Extraktionen aus dem Fisch-ZNS mit dem nicht-ionischen
Detergenz Digitonin (Fa. Merck, Schwalbach/Ts.) 47
3.7.2 Blue-Native Polyacrylamidgel-Elektrophorese (BN-PAGE) der
Myelinprotein-Extraktion aus dem ZNS der Forelle 48
3.7.3 Co-Immunopräzipitation von Myelinprotein-Komplexen aus dem ZNS-
Myelin der Forelle 52
3.8 Hypothetisches Modell der Lage und der Interaktionen von 36K mit
Proteinen in der „Major Dense Line“ des ZNS-Myelins der
Regenbogenforelle (Oncorhynchus mykiss) 53
4. DISKUSSION 55
5. LITERATUR 63
6. ANHANG
6.1 36K-Aminosäure-Sequnenz mit Sekundärstruktur-Vorhersage
(nach 3.1.2) und Trypsinisierungsstellen nach PeptidCutter
(Expasy-Internet-Server) 69
6.2 Klonierungsschema der 36KcDNA in die MCS („multiple cloning site“)
des pET23a(+)-Vektors (Fa. Novagen, Schwalbach/Ts.)
(nach Diplom-Arbeit W. Moll, 2001) 70
6.3 Abkürzungen 71
6.4 Publikationen 72
DANKSAGUNG 73
ERKLÄRUNG ÜBER DIE EIGENSTÄNDIGKEIT DER ERBRACHTEN
WISSENSCHAFTLICHEN LEISTUNG 74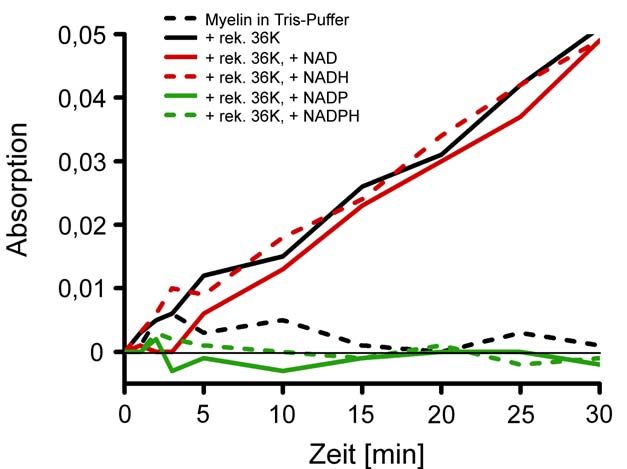
INHALTSVERZEICHNIS ERKLÄRUNG ÜBER ETWAIGE FRÜHERE PROMOTIONSVERSUCHE 75 CURRICULUM VITAE 76
1. EINLEITUNG
1. EINLEITUNG
Im Laufe der Evolution wurde es für das Überleben der Tiere immer wichtiger,
aufgenommene Sinnesreize schnell zu verarbeiten und entsprechend reagieren zu
können. Die Reize werden dabei von den Sinnesorganen registriert und als
elektrische Erregung (Aktionspotentiale) über weit verzweigte Nervenbahnen
weitergeleitet. Dabei entwickelten sich zwei Wege, um eine hohe Geschwindigkeit
der Impulsfortleitung zu erreichen. Der eine Weg ist die Vergrößerung des
Axondurchmessers, was in eine Verminderung des inneren Längswiderstandes
resultiert. Dieses ist z. B. beim Riesenaxon der Tintenfische verwirklicht. Bei den
Vertebraten wird diese Möglichkeit durch die zunehmende Anzahl von Nervenzellen
bzw. deren Ausläufern (Axone) und den daraus resultierenden Platzmangel limitiert.
Um transmembrane Leckströme zu minimieren und die Membrankapazität zu
verringern, werden deren Axone mit einer isolierenden Schicht, dem Myelin, das zu
ca. 70% aus Lipiden besteht
(Morell, 1977), umhüllt (Ritchie,
1995). Diese Myelinhülle wird von
spezialisierten Gliazellen, den
Schwannschen Zellen im
peripheren Nervensystem (PNS)
und den Oligodendrozyten im
zentralen Nervensystem (ZNS),
gebildet. Bei der Myelinisisierung
wickeln sich deren abgeflachte
Membranfortsätze mehrfach Abb. 1: A. Schematische Darstellung eines
konzentrisch um die Axone und myelinisierten Axons (aus Roger Eckert:
„Tierphysiologie“). B. Elektronenmikroskopische
bilden nach Verdichtung die Aufnahme der multilamellaren Struktur des Myelins
aus einem peripheren Nerv der Maus
typische kompakte, multi- (Vergrößerung 87200fach, aus J. B. Metzler „Linder
Biologie“)
lammelare Struktur des Myelins.
-1-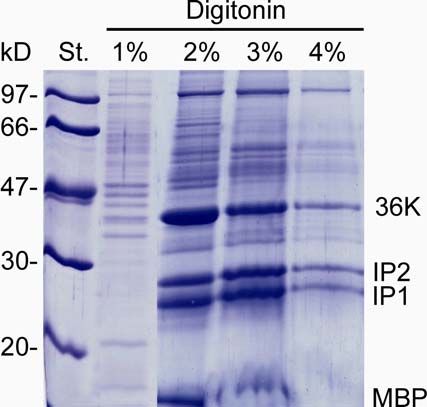
1. EINLEITUNG
In regelmäßigen Abständen (ca. alle 100 µm) ist die Myelinscheide durch die so
genannten „Ranviersche Schnürringe“ unterbrochen. Eine Erregung breitet sich
ausschließlich über diese freiliegenden Axonbereiche aus
(„Saltatorische Erregungsleitung“). Innerhalb des kompakten Myelins unterscheidet
man zwei unterschiedliche Bereiche: Die aus der extrazellulären Apposition gebildete
„Intraperiod Line“ und die aus der cytosolischen Apposition gebildete „Major Dense
Line“. Innerhalb dieser Bereiche befindet sich eine limitierte Anzahl von Proteinen,
die aufgrund ihrer adhäsiven Eigenschaften zur Stabilisierung der übereinander
liegenden Membranlamellen beitragen. Dabei gibt es deutliche Unterschiede in der
Proteinzusammensetzung des ZNS-Myelins phylogenetisch älterer Vertebraten wie
den Fischen und jener phylogenetisch jüngerer Vertebraten (Abbildung 2)
(Jeserich, 1983; Waehneldt & Jeserich,
1984). Das Proteolipid-Protein (PLP), für
das eine adhäsive Funktion auf der
extrazellulären Seite angenommen wird
(Lees & Brostoff, 1984), ist bei den
Fischen durch 2 Isoformen von
P0-ähnlichen Glykoproteinen ersetzt
(Intermediär Protein 1 & 2; Stratmann &
Jeserich, 1995; Lanwert & Jeserich,
2001). Ein weiteres Protein, das nach
seinem Molekulargewicht als 36K
bezeichnet wird (Jeserich, 1983), sucht Abb. 2: Coomassie-gefärbtes SDS-
PAGE-Gel von Myelinproteinen aus dem
man bei höheren Vertebraten vergebens. ZNS der Ratte und der Forelle. CNP: 2´, 3´
Dieses neue, myelinspezifische Protein ist Cyclisches Nukleotid 3´-Phospho-
diesterase, PLP: Proteolipid Protein, (L/S)
nicht glykolisiert (Jeserich & Waehneldt, BP: (large/small) Basisches Protein,
IP: Intermediär Protein.
1987) und ausschließlich in der Aus Neuron-Glia Interrelations during
phylogony: I. Phylogeny and Ontogeny of
cytosolischen Apposition („Major Dense Glial Cells, A. Vernadaktis & B. Roots, Eds.
Humana Press Inc., Totowa, Nj
Line“) des ZNS der Fische zu finden
(Jeserich & Waehneldt, 1986b; Jeserich &
Rauen, 1990). Da rekombinantes 36K mit Phospholipid-Bilayern interagiert
(Diplom-Arbeit W. Moll, 2001) scheint dieses Protein mit der Myelinmembran
assoziiert zu sein.
-2-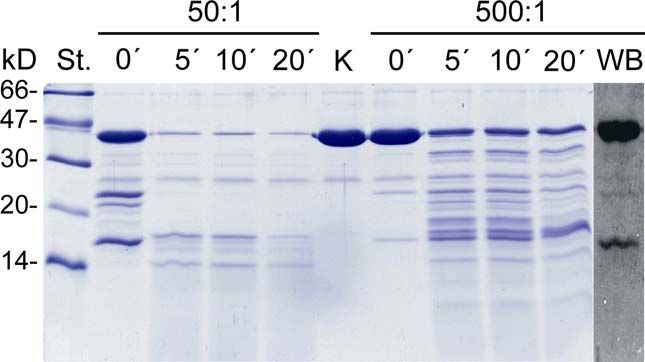
1. EINLEITUNG
Immunologische Untersuchungen zeigten, dass 36K mit keinem der polyklonalen
Antikörper gegen eines der bekannten Myelinproteine reagierte. Ebenso zeigten
erste Datenbank-Analysen überraschenderweise keine Homologien zu den
Myelinproteinen, sondern zu den NAD(P)(H)-abhängigen Oxidoreduktasen
(Dissertation Strelau, 1997). Im Rahmen der vorliegenden Arbeit wurde versucht
über einen Dehydrogenase-Assay sowohl eine entsprechende Aktivität als auch ein
mögliches Substrat zu identifizieren. Alternativ wurde für das membranassoziierte
36K, das neben IP1 & 2 und Basischen Myelinprotein (MBP) eines der
Hauptmyelinproteine im ZNS der Fische darstellt (vgl. Abbildung 2), eine Funktion als
Strukturprotein innerhalb der „Major Dense Line“ vermutet. In diesem
Zusammenhang ist die von Blaurock et al. (1986) festgestellte strukturelle Dynamik
der Myelinlamellen im Fisch-ZNS zu erwähnen. Blaurocks Experimente zeigten
erstmals, dass es unter bestimmten physiologischen Bedingungen zu einer
Veränderung der Abstände innerhalb der Myelinlamellen und damit zu einer
Auflockerung des scheinbar starren Myelins kam. In wie weit 36K dabei eine Rolle
spielen könnte, wurde sowohl mit Hilfe von Liposomen als auch mit isoliertem
ZNS-Myelin untersucht. Aufgrund der Sequenzähnlichkeiten von 36K zu den
NAD(P)(H)-abhängigen Oxidoreduktasen wurde dabei auch die Beeinflussbarkeit
durch Nukleotide geprüft. Dieser Einfluss könnte auf Konformationsänderungen
basieren, die wiederum mit biophysikalischen Methoden analysiert wurden. Die
beschriebene Myelindynamik könnte auch das Resultat eines koordinierten
Zusammenspiels einiger, eventuell komplexierter Myelinproteine sein. Im Rahmen
dieser Dissertation wurde deshalb erstmals versucht, Protein-Komplexe aus dem
ZNS-Myelin nativ aufzutrennen und deren Komponenten zu identifizieren.
-3-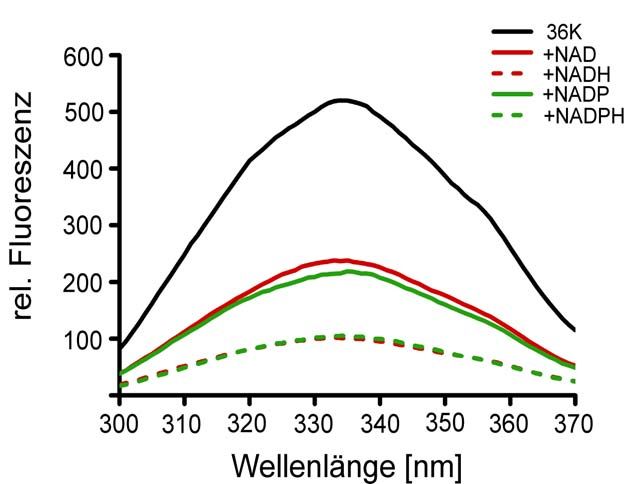
2. MATERIAL UND METHODEN
2. Material und Methoden
2.1 Material
Soweit nicht anders vermerkt, wurden alle Chemikalien von der Fa. Roth (Karlsruhe)
in p. A.-Qualität bezogen.
2.1.1 Bakterienstämme
STAMM GENOM
E. coli BL21(DE3)/pLysS E. coli B F- dcm ompT hsdS(rB-mB- galλ(DE3[pLysS Camr]
2.1.2 Regenbogenforellen (Oncorhynchus mykiss)
Vom Forellenzuchtbetrieb Lindhorst-Emme (Stukenbrock) wurden 3-4 cm lange
Jungfische bezogen und in den hauseigenen Aquarien bei ca. 8-10°C gehalten.
2.2 Methoden
2.2.1 Computerunterstützte Aminosäure-Sequenzanalysen
Die aus der 36K-cDNA resultierende Aminosäure-Sequenz (Diplom-Arbeit W. Moll,
2001) wurde mit Hilfe des JPred-Internet-Servers (University of Dundee,
Dundee, UK) auf vorhandene Sekundärstruktur-Motive untersucht (Cuff et al., 1998).
Die Protein-Datenbank-Recherche wurde mit dem BLAST-Internet-Server
(Altschul et al., 1990; Madden et al., 1996) durchgeführt und Sequenzähnlichkeiten
zwischen 36K und homologen Proteinen mit MultAlin, Version 5.4.1, (Corpet, 1988)
analysiert. Das resultierende Alignment wurde mit dem Computerprogramm
GeneDoc, Version 2.6.002, bearbeitet.
Mit dem SWISS-MODEL-Server (Guex und Peitsch, 1997) und dem
SwissPdb-Viewer, Version 3.7, wurde eine mögliche Tertiärstruktur der N-terminalen
Hälfte von 36K (L39 – V186) modelliert.
-4-2. MATERIAL UND METHODEN
2.2.2 Animpfen einer Bakterien-Über-Nacht-Kultur (ÜK)
Zu Beginn dieser Arbeit lag die für das 36K-Protein codierende cDNA im
prokaryotischen Expressionsvektor pET23a(+) (Fa. Novagen, Schwalbach/Ts.) in
E. coli als –80°C-Dauerkultur vor (Diplom-Arbeit W. Moll, 2001). Um das Protein
heterolog zu exprimieren, wurden pro 500 ml Expressionsansatz 5 ml ÜK angelegt.
In sterile Kulturröhrchen wurde mit entsprechenden Antibiotika versetztes
LB-Medium (1% (w/v) Bacto-Trypton, 0.5% (w/v) Hefe-Extrakt; Fa. Otto Nordwald,
Hamburg; 1% (w/v) NaCl) gegeben und Bakterien aus der Dauerkultur überimpft. Die
Inkubation erfolgte über Nacht bei 37°C im Rüttler (180 rpm).
2.2.3 Heterologe Expression mit E. coli/pET-System (Fa. Novagen)
Die heterologe Expression in E. coli/pET-System erfolgte im Wesentlichen nach
Protokoll des Herstellers (Fa. Novagen, Schwalbach/Ts.).
Der pET23a(+)-Vektor trägt unter der Kontrolle des induzierbaren
Bakteriophagenpromotors T7 eine Nukleotidsequenz, die für 6 aufeinanderfolgende
Histidine codiert. Durch Klonierung des Zielproteins unter Beachtung des
Leserahmens in den distalen Polylinker entsteht bei Expression eine c-terminale
„His6-Tag“-Zielprotein-Fusion, mit der dieses Fusionsprotein durch Ni2+-chelatierende
Sepharose (Affinitätschromatographie) aufgereinigt werden kann.
Zur heterologen Expression wurden 500 ml LBamp/cam-Medium (Endkonz.: 100 µg/ml
Ampicillin; 34 µg/ml Chloramphenicol) mit einer ÜK aus 2.2.2 angeimpft und bei 37°C
im Schikanekolben inkubiert (180 rpm). Bei einer OD600 von 0.7 bis 0.8 wurde mit
IPTG (Endkonz. 100 mM) die Translation induziert und für weitere 2 h bei 16°C
inkubiert. Nach dieser Zeit wurden die Zellen pelletiert (15 Minuten, 5000xg, 4°C), in
Lyse-Puffer (20 mM Tris/HCl pH 8, 500 mM NaCl) resuspendiert und ggf. bis zur
weiteren Verarbeitung bei –80°C gelagert (Moll et al., 2003).
2.2.4 Affinitätschromatographie mit Ni2+-chelatierender Sepharose
Die Affinitätschromatographie mit Ni2+-chelatierender Sepharose wurde unter nativen
Bedingungen durchgeführt. Sie erfolgte nach einem modifizierten Protokoll des
Herstellers (His-Bind Kit, Fa. Novagen, Schwalbach/Ts.).
-5-2. MATERIAL UND METHODEN
Die resuspendierten Bakterien wurden bis zur Zelllyse sonifiziert und zur Pelletierung
der unlöslichen Bestandteile und Zelltrümmer für 30 min bei 16000xg und 4°C
zentrifugiert. Der filtrierte (0.42 µm-Filter) Überstand wurde auf eine mit Nickelsulfat
(NiSO4) geladene (Charge-Buffer mit 50 mM NiSO4) und mit Binding-Buffer
(20 mM Tris/HCl, pH 8.0, 500 mM NaCl, 50 mM Imidazol) equilibrierte His-Bind
Sepharose-Säule (Fa. Novagen, Schwalbach/Ts.) gegeben. Das anschließende
Säulenwaschen erfolgte mit 10fachem Säulenbettvolumen Binding-Buffer und
13fachem Säulenbettvolumen Wash-Buffer (Lysepuffer mit 100 mM Imidazol). Das
Zielprotein wurde mit 3fachem Bettvolumen Elution-Buffer (Lyse-Puffer mit
150 mM Imidazol) von der Säule gelöst. In das Auffanggefäß vorgegebenes EDTA
(Endkonzentration 2 mM) komplexierte ausgewaschene Nickelionen. Vor der Dialyse
(Dialyseschläuche Fa. Serva, Heidelberg) gegen 100fach Volumen PBS-
(50 mM Na-/K-Phosphat, pH 7.4, 150 mM NaCl) oder Tris-Puffer (30 mM Tris/HCl,
pH 7.4, 150 mM NaCl) und vor jedem Experiment wurde die Proteinlösung zur
Entfernung von 36K-Aggregaten und sonstigen Partikeln ultrazentrifugiert
(30 Minuten, 200000xg, 4°C).
Die elektrophoretische Analyse durch SDS-PAGE (2.2.5) zeigte nach Anfärbung der
Proteine (2.2.6 oder 2.2.7) in der Elutionsfraktion eine einzige Bande mit dem
erwarteten Molekulargewicht. Im Western Blot (2.2.8 & 2.2.9) reagierte die Bande
eindeutig mit polyklonalen Antikörpern gegen das native Protein (Jeserich und
Waehneldt, 1986b; Moll et al., 2003). Die Protein-Konzentrationsbestimmung über
die Absorption von aromatischen Aminosäure-Resten bei 280 nm (Warburg-Formel;
Layne, 1957) zeigte, dass rekombinantes 36K-Protein im Milligramm-Maßstab
aufgereinigt werden konnte.
2.2.5 SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) (nach Laemmli)
Zur analytischen Proteingelelektrophorese nach Laemmli (1970) wurde zunächst das
Trenngel mit entsprechender Acrylamid-Endkonzentration (12 bis 15% (v/v) aus
37.5:1 Acrylamid/Bisacrylamid-Fertiglösung), 1/1000 Volumen TEMED,
1/400 Volumen Ammoniumpersulfatlösung (10% (w/v)), 0.375 M Tris/HCl pH 8.8,
1% (w/v) SDS im Hoefer Mighty Small Gelcaster (Amersham Biosciences, Freiburg)
gegossen und mit Isopropanol überschichtet.
-6-2. MATERIAL UND METHODEN
Nach Polymerisation wurde das Isopropanol abgegossen, kurz mit Wasser gespült,
das 5%ige Sammelgel (wie Trenngel mit 0.375 M Tris/HCl, pH 6.8) eingefüllt und ein
Teflonkamm eingeführt. Die Proteinproben wurden vor dem Auftragen mit
Laemmlipuffer (0.125 mM Tris/HCl pH 6.8, 2% (w/v) SDS, 6% (w/v) Glycerin,
10% (v/v) ß-Mercaptoethanol), 0.4 mM Bromphenolblau) versetzt und für
5 min aufgekocht. Die Elektrophorese erfolgte bei 60 Volt im Sammelgel und
180 Volt im Trenngel. Nach Auslaufen der Bromphenolblaufront aus dem Trenngel
wurden die Proteinbanden nach 2.2.6, 2.2.7 oder 2.2.8 sichtbar gemacht.
2.2.6 Proteinanfärbung mit Coomassie-Lösung
Zum Anfärben der Proteine wurde das SDS-PAGE-Gel aus 2.2.5 in Coomassie-
Lösung (0.1% (w/v) Brillant blue R250 in 500 ml Methanol, 75 ml Essigsäure, 425 ml
Aqua dest.) gegeben und mindestens 1 Stunde geschwenkt. Durch Aufkochen in
Aqua dest. (30 Min.) oder Schwenken in Entfärberlösung (7.5% (v/v) Essigsäure,
7.5% (v/v) Methanol, 5% (v/v) Glycerin) konnte überschüssiger Farbstoff entfernt
werden. Das Gel wurde nach Stabilisierung in Geltrocknungslösung
(22% (v/v) Ethanol, 1% (v/v) Methanol, 1% (v/v) Isopropanol, 2% (v/v) Glycerin) in
Zellophanfolie (Fa. Deti, Meckesheim) eingeschweißt und über Nacht getrocknet.
2.2.7 Proteinanfärbung durch Silberfärbung
Für die Silberfärbung wurde das SDS-PAGE-Gel zunächst für mindestens
15 Minuten in Fixierer (45% (v/v) Methanol, 10% (v/v) Essigsäure, 45% (v/v) Aqua
dest.) geschwenkt. Nach zweiminütiger Inkubation in „Farmers Reducer“
(0.03 M K3[Fe(CN)6], 0.032 M Na2S2O3) wurde das Gel mit Aqua dest. gewaschen
bis der gelbe Hintergrund verschwunden war. Die Anfärbung der Proteine erfolgte im
0.1% (v/v)-Silbernitrat-Bad (30 min.). Nach je zweimaligem Waschen mit A. dest.
(30 sec.) und 2.5% (v/v) Na2CO3-Lösung (30 sec.) folgte die Entwicklung in
Formaldehyd-Lösung (0.4% (v/v) in 2.5% (v/v) Na2CO3-Lösung) bis die
Proteinbanden zu sehen waren. Die Reaktion wurde mit 10% (v/v) Essigsäure
gestoppt. Das Gel wurde nach Stabilisierung in Geltrocknungslösung (22% (v/v)
Ethanol, 1% (v/v) Methanol, 1% (v/v) Isopropanol, 2% (v/v) Glycerin) in
Zellophanfolie (Fa. Deti, Meckesheim) eingeschweißt und über Nacht getrocknet.
-7-2. MATERIAL UND METHODEN
Zur Reduzierung des Hintergrundes bzw. zum Entfärben von silbergefärbten
SDS-PAGE-Gelen wurde die Lösung A (15% (w/v) Na2S2O3, 1.2% (w/v)
Thioharnstoff in Aqua dest.) und Lösung B (5% (w/v) K3[Fe(CN)6]) in Verhältnis 1:1
gemischt. Je nach Grad der Abschwächung wurde die Misch-Lösung mit 2 (stark)
oder 4 (mäßig) Volumenteilen Aqua dest. versetzt und das Gel bis zur gewünschten
Entfärbung darin geschwenkt. Anschließend wurde das Gel unter häufigem
Wasserwechsel in Aqua dest. gewässert bis der gelbliche Hintergrund vollständig
verschwunden war.
2.2.8 Proteintransfer auf Nitrozellulose- oder Polyvinylidenfluorid- (PVDF-)
Membran (Western-Blot)
Es wurden drei Lagen Blotpapier (Fa. Whatman, Bruchsal) in Größe des zu
blottenden Gels in Transferpuffer (25 mM Tris/HCl, pH 8.0, 20 mM Glycin,
20% (v/v) Methanol) getränkt und auf die Anode der Semi-Dry-Blotting Apparatur
(Fa. Phase, Lübeck) gelegt. Darüber wurde ein gleich großes mit Transferpuffer
getränktes Stück Nitrozellulose- (Fa. Schleicher & Schuell, Dassel) oder
Polyvinylidenfluorid- (PVDF-) Membran (Fa. Amersham Biosciences, Freiburg)
luftblasenfrei aufgelegt. Das darüber gelegte SDS-PAGE-Gel wurde vorher kurz in
Transferpuffer equilibriert und mit weiteren drei Lagen getränktem Blotpapier
blasenfrei bedeckt. Die Kathode der Blotkammer wurde auf das Sandwich gelegt und
mit ca. 1 kg beschwert. Der Protein-Transfer erfolgte bei 150 mA pro SDS-PAGE-Gel
(max. 25 V) für 1.5 h. Die auf die Membran transferierten Proteine wurden durch
Schwenken in Ponceau-S-Lösung (0.1% (w/v) Ponceau-S, 5% (v/v) Essigsäure)
reversibel angefärbt und überschüssige Ponceau-S-Lösung mit Aqua. dest. entfernt.
Der Größenstandard wurde vor der Protein-Entfärbung mit TBST (10 mM Tris/HCl
pH 8.0; 150 mM NaCl, 0.05% (v/v) Tween-20) und anschließendem immuno-
logischen Protein-Nachweis (nach 2.2.9) mit dem Skalpell abgetrennt.
-8-2. MATERIAL UND METHODEN
2.2.9 Immunologischer Nachweis von Proteinen auf Nitrozellulose- oder
Polyvinylidenfluorid- (PVDF-) Membran
Um unspezifische Antikörperbindung zu vermeiden, wurde die proteintragende
Nitrozellulose- bzw. Polyvinylidenfluorid- (PVDF-) Membran aus 2.2.8 für 30 min in
TBST/Gelatine (10 mM Tris/HCl pH 8.0, 150 mM NaCl, 0.05% (v/v) Tween-20,
0.3% (w/v) Gelatine) geschwenkt. Nach 1 bis 2 Stunden Inkubation mit
entsprechenden Antikörpern (verdünnt in TSBT/Gelatine) und 6x5 minütigem
Waschen der Membran mit TBST/Gelatine wurde der sekundäre
Meerrettich-Peroxidase gekoppelte Antikörper (ebenfalls verdünnt in TBST/Gelatine)
für 1 Stunde dazugegeben. Überschüssige Antikörper wurden durch abschließendes
Waschen der Membran in TBS (10 mM Tris/HCl pH 8.0 150 mM NaCl) entfernt. Die
Peroxidase gekoppelten Antikörper wurden mit Hilfe von Chemoluminiszenz auf
ECL-Film (Fa. Amersham Pharmacia, Freiburg) sichtbar gemacht.
2.2.10 Fern-UV-Circulardichroismus-Spektroskopie (CD-Spektroskopie) von
rekombinantem 36K in Lösung
Die Fern-UV-Circulardichroismus-Spektroskopie (CD-Spektroskopie) wurde mit
einem Jasco J-600 Spektropolarimeter bei 20°C durchgeführt. Dazu wurden bei einer
Integrationszeit von 50 nm/s drei Spektren von je 0.12 mg/ml (3 µM) rekombinantem
36K (2.2.2 - 2.2.4) in PBS (50 mM Na/K-Phosphat, 150 mM NaCl, pH 7.4) über einen
Wellenlängen-Bereich von 185 bis 260 nm gemittelt. Die path length der Küvette
(Hellma, Müllheim) betrug 1 mm. Der Anteil von α-Helices, β-Faltblättern, β-Turns
und ungeordneter Strukturen („random coil“) wurde mit dem Computer-Programm
CDNN, Version 2.1 kalkuliert. Dabei wurde das erhaltene CD-Spektrum mit denen
von Proteinen mit bekannter Sekundärstruktur verglichen (Böhm et al., 1992). Der
Einfluss von Nukleotiden auf die Sekundärstruktur-Verteilung wurde durch Zugabe
von 0.6 mM NAD, NADP oder deren reduzierter Formen untersucht.
-9-2. MATERIAL UND METHODEN
2.2.11 Tryptophan-Fluoreszenz-Studien von rekombinantem 36K in Lösung
In der Primärstruktur von 36K (3.1.2) befinden sich 6 Tryptophane. Deren
Fluoreszenz nach Erregung bei 280 nm wurde mit einem SLM-Aminco
8100-Spektrometer (Fa. Polytec, Waldbronn) bei 4°C und einer Spaltbreite von 8 mm
gemessen. Dazu wurde 2 µM rekombinantes 36K in PBS pH 7.4
(50 mM Na/K-Phosphat, 150 mM NaCl) (2.2.2 - 2.2.4) in eine Quarzküvette
(Fa. Hellma, Müllheim) gegeben und Fluoreszenz-Spektren (300 bis 370 nm)
aufgenommen. Hinweise auf Konformationsänderungen sollten aufgrund von
Fluoreszenzlöschung nach Zugabe von 0.2 bzw. 0.4 mM Nukleotide erhalten
werden.
Nach Kd=[Nukl.]*(∆Fmax-∆F)*(∆F)-1 (Lin et al., 1997) wurden die jeweiligen
apparenten Dissoziationskonstanten berechnet, wobei für Fmax die größte
gemessene Fluoreszenzreduktion bei 0.4 mM NADH angenommen wurde.
2.2.12 Elektronenspin-Resonanz-Spektroskopie (ESR-Spektroskopie)
rekombinantem 36K in Lösung
Die ESR-Spektroskopie wurde in Zusammenarbeit mit Prof. Dr. H.-J. Steinhoff
(AG Experimentelle Physik, Universität Osnabrück) durchgeführt. Zum Einfügen des
Nitroxid-Spinlabels (1-oxyl-2,2,5,5-tetramethylpyrrolin-3-methyl)-Methanthiosulfonat
(MTS, Fa. Toronto Research Chemicals, Kanada) über die Sulfhydrylgruppen der
beiden einzigen im Protein vorhandenen Cystein-Reste wurde die sonifizierte
Bakteriensuspension aus 2.2.3 filtriert (0.45 µm) und auf eine nach 2.2.4 vorbereitete
His-Bind Sepharose-Säule (Fa. Novagen, Schwalbach/Ts.) gegeben. Nach Waschen
mit 10 bzw. 8fachem Säulenbettvolumen Binding-Buffer (20 mM Tris/HCl, pH 8.0,
500 mM NaCl, 50 mM Imidazol) und Wash-Buffer (20 mM Tris/HCl, pH 8.0,
500 mM NaCl, 100 mM Imidazol) wurde 0.15 mM (~10fach molare
Proteinkonzentration auf Säule) MTS aus einer 10 mM Stock-Lösung (in DMSO) in
6 ml Wash-Buffer zu dem gebundenen rekombinanten 36K gegeben und die
Sepharose über Nacht bei 4°C geschwenkt. Nach Waschen mit weiteren
7 Bettvolumen Wash-Buffer wurde MTS-gelabeltes 36K (36K-MTS) wie unter 2.2.4
beschrieben von der Säule eluiert und gegen 100fach PBS pH 7.4
(50 mM Na/K-Phosphat, 150 mM NaCl) dialysiert.
-10-2. MATERIAL UND METHODEN
Für die ESR-Spektroskopie wurde das Protein mit Centriprep-Säulen (Fa. Millipore,
Schwalbach/Ts.) nach Vorschrift des Herstellers auf mindestens 10 µM (0.4 mg/ml)
konzentriert und vor jeder Messung ultrazentrifugiert (30 Minuten, 200000xg, 4°C).
Das ESR-Spektrum wurde von Prof. Dr. H.-J. Steinhoff aufgenommen.
2.2.13 Röntgenkleinwinkelstreuung von rekombinantem 36K in Lösung
Die Röntgenkleinwinkeldaten von rekombinantem 36K in Lösung (2.2.2 - 2.2.4)
wurden von PD Dr. G. Grüber (Inst. f. Biophysik, Universität des Saarlandes,
Homburg/Saar) aufgenommen und mit Hilfe des Programms DAMMIN ausgewertet.
2.2.14 Assoziation von rekombinantem 36K an phospholipidbeschichtete
Silica-Kügelchen („TRANSIL“; Fa. Nimbus, Leipzig)
Die mit einem Phosphatidylcholin- (PC-) Bilayer beschichteten Transil-Kügelchen
wurden von Firma Nimbus (Leipzig) synthetisiert. Vor der Bindung von
rekombinantem 36K an die PC-Membran, wurden 2 µl der 50%igen
Transil-Suspension (111 µM PC) zunächst zweimal in 200 µl PBS pH 7.4
(50 mM Na/K-Phosphat, 150 mM NaCl) aufgenommen und bei Raumtemperatur (RT)
5 Minuten mit 3000xg zentrifugiert.
Zu den gewaschenen Kügelchen wurde 16 µM rekombinantes 36K (2.2.2 - 2.2.4)
gegeben (200 µl) und 1 Stunde bei RT unter Schütteln (500 rpm) inkubiert.
Anschließend wurden die Kügelchen pelletiert (5 Minuten, 3000xg, RT) und zum
Entfernen von nicht membranassoziiertem Protein zweimal mit PBS gewaschen. Für
die SDS-PAGE wurden die Membranen mit 2% SDS aufgelöst (15 Minuten, 500 rpm,
RT) und die Elektrophorese mit anschließender Proteinfärbung wie unter 2.2.5 und
2.2.6 beschrieben durchgeführt.
-11-2. MATERIAL UND METHODEN
2.2.15 Kontrollierter proteolytischer Verdau von rekombinantem 36K mit
Trypsin (Fa. Sigma-Aldrich, München)
Der kontrollierte proteolytische Verdau von rekombinantem 36K (2.2.2 - 2.2.4) wurde
statt bei (für Typsin) optimalen 37°C bei 30°C durchgeführt. Unter definierten 36K- zu
Trypsin-Verhältnissen (50:1, 500:1) wurde das Protein zwischen 0 und 20 Minuten
unter leichtem Schütteln (450 rpm) in PBS pH 7.4 (50 mM Na/K-Phosphat,
150 mM NaCl) trypsinisiert und die Reaktion nach entsprechender Zeit mit
8 mM Pefabloc (Fa. Serva, Heidelberg) gestoppt. Zur Identifizierung von
Trypsinisierungsstellen, die durch eine Membranassoziation geschützt sind, wurde
rekombinantes 36K vor dem proteolytischen Verdau an einen PC-Bilayer
(„TRANSIL“) gebunden (2.2.14) und nach Entfernen von nicht-assoziiertem Protein
einer Trypsinisierung ausgesetzt. Die entstandenen Fragmente wurden über
SDS-PAGE (2.2.5) aufgetrennt und mit Coomassie-Färbung (2.2.6) sichtbar
gemacht.
2.2.16 Chemische Spaltung von rekombinantem 36K mit 2-Nitro-5-
Thiocyanobenzonsäure (NTCB)
NTCB (Fa. Sigma-Aldrich, München) spaltet Proteine chemisch an freien
Thiol-Resten (Jacobson et al., 1973; Bähler et al., 1989). Um zu prüfen, ob die 2 in
der Primärstruktur von 36K vorhandenen Cystein-Reste als Thiole vorliegen, wurde
5 mM NTCB (aus 40 mM NTCB in 50 mM Na/K-Phosphat pH 7.6) zu
13 µM (0.5 mg/ml) rekombinantem 36K (2.2.2 - 2.2.4) in PBS pH 7.4
(50 mM Na/K-Phosphat, 150 mM NaCl) gegeben und im Dunkeln für bis zu
48 Stunden bei 37°C inkubiert. Durch Zugabe von Laemmlipuffer (0.125 mM Tris/HCl
pH 6.8, 2% (w/v) SDS, 6% (w/v) Glycerin, 10% (v/v) ß-Mercaptoethanol,
0.4 mM Bromphenolblau) wurde die Reaktion durch das vorhandene
ß-Mercaptoethanol gestoppt und die Reaktionslösung für die SDS-PAGE (2.2.5)
vorbereitet.
Für die Spaltung von membranassoziiertem 36K wurde das Protein zunächst an
einen PC-Bilayer („TRANSIL“) gebunden (2.2.14) und nach Entfernen von
nicht-assoziiertem Protein der NTCB-Spaltung ausgesetzt. Die Spaltprodukte wurden
nach der SDS-PAGE (2.2.5) mit Coomassie-Färbung sichtbar gemacht (2.2.6).
-12-2. MATERIAL UND METHODEN
2.2.17 Random-Fluoreszenz-Resonanz-Energie-Transfer (Random-FRET)-
Untersuchungen von membranassoziiertem rekombinanten 36K
Für die Fluoreszenz-Resonanz-Energie-Transfer (FRET)-Untersuchungen wurden
von der Fa. Nimbus (Leipzig) Silica-Kügelchen hergestellt, die mit einem Bilayer aus
Phosphatidylcholin (PC) und 1 mol% 7-Nitrobenz-2-oxa-1,3-Diazol-4-yl-Gruppe-PC
(NBD-PC) beschichtet waren. Dabei war das als Fluoreszenz-Donor fungierende
NBD an das Ende je einer Fettsäurekette fusioniert. Wie bei der Einfügung des
Nitroxid-Spinlabels für die ESR-Spektroskopie (2.2.12), wurde als
Fluoreszenz-Akzeptor QSY® 7 Maleimid (Fa. MoBiTec, Göttingen) im Laufe der
affinitätschromatographischen Aufreinigung über die 2 im Protein vorhandenen
Cystein-Reste in das rekombinante 36K (2.2.2 - 2.2.4) gebracht. Dazu wurden
0.25 mM QSY® 7 Maleimid (~ 10fach molare Proteinkonzentration auf Säule) aus
einer 10 mM Stocklösung (in DMSO) zu 6 ml Wash-Buffer (20 mM Tris/HCl pH 8.0,
500 mM NaCl, 100 mM Imidazol) gegeben und über Nacht bei 4°C mit dem an die
NiSO4-Sepharose gebundenen rekombinanten 36K geschwenkt. Nach Waschen mit
weiteren 7 Bettvolumen Wash-Buffer wurde QSY-markiertes 36K (36K-QSY) wie
unter 2.2.4 beschrieben von der Säule eluiert, gegen 100fach PBS pH 7.4
(50 mM Na/K-Phosphat, 150 mM NaCl) dialysiert und vor jeder Messung
ultrazentrifugiert (30 Minuten, 200000xg, 4°C).
Zur Bestimmung des spektralen Überlappungsintegrals (Jda=Σ(Fd*εa*λ4)/ΣFd;
Fd: Donor-Fluoreszenz, εa: molarer Extinktionskoeffizient vom Akzeptor [M-1*cm-1], λ:
Wellenlänge [nm]) und dem daraus resultierenden Förster-Abstand R0
-5 2 -4 1/6
(R0=[(8.79*10 )*(κ *n *Qd*Jda)] Å; Wu, P. & Brand, L., 1994) wurde mit Hilfe eines
Shimadzu UV-2501 PC Zweistrahlphotomerters (Fa. Shimadzu Deutschland,
Duisburg) ein Absorptionsspektrum vom Akzeptor (36K-QSY) sowie mit einem
SLM-Aminco 8100-Spektrometer (Fa. Polytec, Waldbronn) ein Emissionsspektrum
vom Donor (NBD-PC) in PBS pH 7.4 (50 mM Na/K-Phosphat, 150 mM NaCl)
aufgenommen. Für die Berechnung von R0 wurde für den
Dipol-Dipol-Orientierungsfaktor (κ2) 2/3 (Wu, P. & Brand, L., 1994), für den
Brechungsindex des Mediums (n) 1.4 und für die Quantenausbeute von NBD in
Membranen in Abwesenheit des Akzeptors (Qd) 0.32 (Chattopadhyay, A., 1990)
angenommen.
-13-2. MATERIAL UND METHODEN
Um die Energietransfer-Effizienz (Fluoreszenz-Reduktion des NDB durch
ausreichende Annäherung des proteingebundenen Akzeptors; Wu, P. & Brand, L.,
1994; Szöllősi et al., 2002) zwischen den Chromophoren zu bestimmen, wurde
rekombinantes 36K (2.2.2 - 2.2.4) bzw. 36K-QSY wie unter 2.2.14 beschrieben mit
dem PC/NBD-PC-Bilayer assoziiert, nicht gebundene Proteine ausgewaschen und
die NBD-Fluoreszenz-Spektren nach Erregung bei 467 nm aufgenommen. Mit Hilfe
des Computerprogramms Mathcad 2001 Professional (MathSoft, München) wurden
minimal mögliche Abstände zwischen Donor und Akzeptor bei verschiedenen
vertikalen Abständen mit der von Fung und Stryer ermittelten (1978)
Integral-Gleichung kalkuliert. Aufgrund des großen Durchmessers der
Silica-Kügelchen konnte dabei von einem planaren Bilayer ausgegangen werden.
2.2.18 Isolieren von Myelin aus ZNS der Regenbogenforelle (Oncorhynchus
mykiss)
Zum Isolieren von Myelin aus dem ZNS der Regenbogenforelle wurden
800 mg Gesamthirn von 3 bis 8 cm langen Forellen in 10 ml 0.2 M eiskalter
Sucrose-Lösung homogenisiert und je 5 ml auf auf ein 0.6 M Sucrose-Bett (7 ml)
geschichtet. Nach der Zentrifugation in einem Ausschwingrotor (1 Stunde, 100000xg,
4°C) wurde das weiße Myelin in der Grenzschicht zwischen den Sucrose-Lösungen
entnommen, in 10 ml Eiswasser transferiert und in einem Festwinkelrotor erneut
zentrifugiert (10 Minuten, 8500xg, 4°C). Das pelletierte Myelin wurde noch 3 mal in
Eiswasser resuspendiert, zentrifugiert und schließlich bis zur Weiterverarbeitung bei
–80°C eingefroren.
2.2.19 Herstellen einer 5%igen (w/v) Digitonin-Stammlösung und
Myelinprotein-Extraktion mit Digitonin
Eine 5% (maximale Löslichkeit) entsprechende Menge des Detergenzes Digitonin
(Merck, Schwalbach/Ts.) wurde in 98°C heißem Aqua dest. aufgenommen,
15 Minuten gekocht und anschließend bei 4°C mindestens 30 Minuten abkühlen
lassen. Aus dieser Stammlösung wurden die bei der im Folgenden beschriebenen
Myelinprotein-Extraktion verwendeten Digitonin-Puffer hergestellt.
-14-2. MATERIAL UND METHODEN
Das nach 2.2.18 isolierte ZNS-Myelin wurde zunächst in je 100 µl 0.5% (w/v) und
1% (w/v) Digitonin (in 20 mM Tris/HCl pH 7.4, 0.1 mM EDTA, 50 mM NaCl,
10% (v/v) Glycerin, 1 mM PMSF) resuspendiert, für 10 Minuten bei Raumtemperatur
(RT) inkubiert und anschließend zentrifugiert (25000xg, 15 Minuten, 4°C). Zur
Extraktion der Myelinproteine wurde das resultierende (Myelin-)Pellet zweimal in je
100 µl 4% (w/v) Digitonin-Puffer aufgenommen, 10 Minuten bei RT inkubiert und die
nicht solubilisierten Membranen bei 25000xg, 15 Minuten, 4°C pelletiert. Der
Überstand mit den gelösten Proteinen wurde abgenommen und die
Protein-Konzentration mit einem Amidoschwarz-Tüpfel-Test (2.2.20) bestimmt.
2.2.20 Protein-Konzentrationsbestimmung mit Amidoschwarz-Tüpfel-Test
Zur Bestimmung der Protein-Konzentration wurde 1 µl Proteinlösung auf eine
Nitroacetat-Membran gegeben und für 2 Minuten in Amidoschwarzlösung
(0.5% (w/v) Amidoschwarz in 90% (v/v) Methanol, 10% (v/v) Essigsäure) gebadet.
Überschüssiger Farbstoff wurde im Methanol/Essigsäure-Bad (9:1) entfernt und die
Proteinkonzentration mit Hilfe einer BSA-Eichreihe bestimmt.
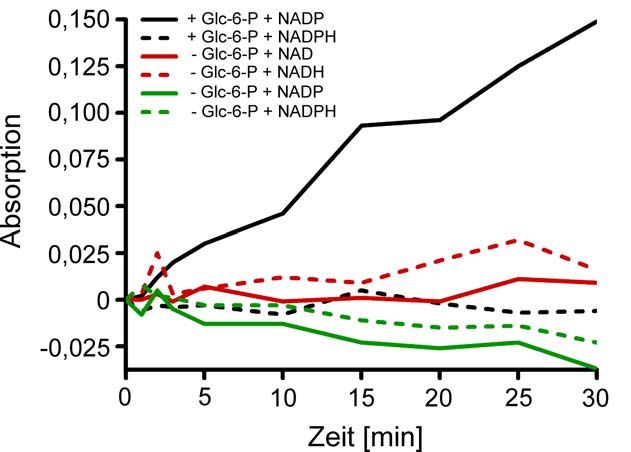
2.2.21 Messung einer Dehydrogenase-Aktivität im isolierten ZNS-Myelin der
Regenbogenforelle (Oncorhynchus mykiss)
Nach Cammer et al. (1982) wurde die Glucose-6-Phosphat-Dehydrogenase-Aktivität
im isolierten Myelin bestimmt. Dabei wird das bei der Umwandlung von
Glucose-6-Phosphat zu 6-Phosphoglucono-δ-lacton aus der Reduktion von NADP
entstehende NADPH durch die Absorptionszunahme bei 320 nm nachgewiesen.
Es wurden 3 µl nach 2.2.18 isoliertes Fisch-ZNS-Myelin (resuspendiert in
200 µl 30 mM Tris/HCl, pH 7.4) in Reaktionspuffer (30 mM Tris/HCl pH 7.4,
6.25 mM MgCl2, 1% (v/v) Triton X-100, 1 mM Glucose-6-Phosphat) gegeben und die
Reaktion nach 20 Minuten Vor-Inkubation (Absorptionsabnahme durch Triton X-100)
mit 0.1 mM NADP (zur Kontrolle NADPH) gestartet. Die Zunahme der Absorption bei
320 nm (Biophotometer Fa. Eppendorf, Hamburg) wurde über die Zeit beobachtet
und die Aktivität nach (A320/t)*(Vtotal/6.22)*(1/VMyelin) [µmol/min/mg] berechnet.
-15-2. MATERIAL UND METHODEN
Zur Untersuchung einer Dehydrogenase-Aktivität von myelininternem 36K wurde der
beschriebene Ansatz ohne Glucose-6-Phosphat im Reaktionspuffer durchgeführt.
Dazu wurde eine mögliche Reaktion mit Myelinkomponenten als Substrat mit
0.1 mM Nukleotiden gestartet.
Zur Verstärkung der potentiellen Dehydrogenase-Aktivität wurden nach Vor-
Inkubation in 30 mM Tris/HCl pH 7.4, 6.25 mM MgCl2, 1% (v/v) Triton X-100 und
0.1 mM Nukleotide als Reaktionsstart 80 µg/ml rekombinantes 36K (2.2.2 - 2.2.4)
zugegeben und die Absorption bei 320 nm über die Zeit verfolgt.
2.2.22 Herstellung von Phosphatidylcholin/Phosphatidylethanolamin-
Liposomen
Phosphatidylcholin (PC) und Phosphatidylethanolamin (PE) stellen die
Hauptphospholipide im ZNS-Myelin der Forellen dar. Zur Herstellung von großen,
mulitlamellaren Vesikeln (large multilamellar vesicles, LMV) wurden
75% (w/v) Phosphatidylcholin und 25% (w/v) Phosphatidylethanolamin (in
Chloroform; Fa. Larodan, Schweden) zusammengegeben und das Chloroform unter
einem Stickstoff-Strahl verblasen. Der entstandene Lipidfilm wurde mit mind.
74°C (Tm von PE) heißem Tris-Puffer (30 mM Tris/HCl pH 7.4, 150 mM NaCl) für
1 Stunde unter starkem Rühren rehydriert und die Liposomen-Suspension (~1.3 mM)
bis zur weiteren Verwendung bei 4°C gelagert.
2.2.23 Aggregationsassay von Phosphatidylcholin (PC)/Phosphatidyl-
ethanolamin/(PE)-Liposomen mit rekombinantem 36K
Nach de la Fuente und Parra (1995), Lee und Pollard (1997) und Stieglitz et al.
(2001) wurden 0.1 mM PC/PE-Liposomen aus 2.2.22 mit 0.4 mg/ml rekombinantem
36K in Tris-Puffer (30 mM Tris/HCl pH 7.4, 150 mM NaCl) (2.2.2 - 2.2.4) versetzt und
die Absorption bei 320 nm (Biophotometer Fa. Eppendorf, Hamburg) über die Zeit
beobachtet. Um den Einfluss von Nukleotiden zu untersuchen, wurde entweder
0.1 mM NAD, NADP oder deren reduzierte Formen zu dem Ansatz gegeben.
-16-2. MATERIAL UND METHODEN
2.2.24 Blue-Native Gradienten-Polyacrylamidgel-Elektrophorese (BN-PAGE)
(1. Dimension)
Nach Schägger & von Jagow (1991), Schägger et al. (1994) und Brookes et al.
(2002) wurden die nach 2.2.18 aus der Myelin-Membran extrahierten Proteine bzw.
Protein-Komplexe über ein natives Gradienten-Polyacrylamidgel elektrophoretisch
aufgetrennt. Dazu wurde mit Hilfe einer Hoefer Mighty Small Gradientengelcaster
(Amersham Biosciences, Freiburg), in dem eine ~1 cm Isopropanol-Schicht
vorgegeben wurde, ein 6-16%iges Polyacrylamid-Trenngel (6 bzw.
16% Acrylamid/Bisacrylamid aus 24:1-Stocklösung, 500 mM Aminocapronsäure,
50 mM Bis-Tris pH 7.0, 10% (w/v) Glycerin, 1/400 Volumen
Ammoniumpersulfatlösung (10% (w/v)) und 1/4000 Volumen TEMED gegossen.
Nach der Polymerisation wurde der Alkohol entfernt, vorsichtig mit Wasser gespült
und nach Auffüllen mit 4%iger Sammelgel-Lösung (4% Acrylamid/Bisacrylamid aus
24:1-Stocklösung, 500 mM Aminocapronsäure, 50 mM Bis-Tris pH 7.0,
10% (w/v) Glycerin, 1/125 Volumen Ammoniumpersulfatlösung (10% (w/v)) und
1/1250 Volumen TEMED) gegossen ein Teflonkamm eingefügt. Die Proteinproben
wurden auf 0.8 mg/ml verdünnt, mit BN-Probenpuffer (0.25% (w/v) Brillant Blue G250
in 25 mM Aminocapronsäure) versetzt und kurz vor dem Auftragen zentrifugiert
(2 Minuten, 10000xg, 4°C). Die nötige negative Ladung wurde durch den Farbstoff
Brillant Blue G250 an die Proteine gebracht. Aufgrund der daraus resultierenden
elektrischen Abstoßung wurde außerdem die Aggregation der Proteine im Gel
verhindert. Um dieses während der gesamten Elektrophorese-Dauer zu
gewährleisten, wurde zum Kathoden-Puffer (15 mM Bis-Tris pH 7.0, 50 mM Tricine)
0.02% (w/v) Brilliant Blue G250 geben, das nach der Hälfte der Elektrophorese auf
0.01% (w/v) reduziert werden konnte. Zu der Anode wurde ein Puffer aus
50 mM Bis-Tris pH 7.0 gegeben.
Die Elektrophorese wurde bei 4°C mit 40 Volt gestartet und nach Eintritt der
Farbstoff-Front in das Sammelgel auf 110 Volt erhöht. Nachdem die Front den
Anoden-Puffer erreicht hatte, wurde das Gel zur Proteinanfärbung in einem
Coomassie-Bad (Brilliant Blue R250) (2.2.6) geschwenkt.
-17-2. MATERIAL UND METHODEN
2.2.25 SDS-PAGE der durch BN-PAGE aufgetrennten Myelinprotein-Komplexe
(2. Dimension)
Zur Analyse der Protein-Zusammensetzung der durch BN-PAGE (1. Dimension)
(2.2.24) aufgetrennten Komplexe wurde entweder die gesamte Spur oder die
Komplexe mit einem scharfen Skalpell aus dem Gel geschnitten. Diese wurden auf
die Aluminium-Platte der Elektrophorese-Einrichtung gelegt und zum Einbringen der
nötigen negativen Ladung an die Protein bzw. zum Hydrolysieren von
Disulfid-Brücken für 2 Stunden mit 1% (w/v) SDS/1% (v/v) β-Mercaptoethanol
überschichtet. Im Anschluss wurde wie unter 2.2.5 beschrieben ein Trenngel und ein
dünnes Sammelgel bis ~0.5 cm unter die Gel-Fragmente gegossen. Diese wurden in
0.8% Agarose eingebettet und die Elektrophorese unter den üblichen Bedingungen
durchgeführt. Durch Silberfärbung wurden die aufgetrennten Proteine schließlich
sichtbar gemacht (2.2.7).
2.2.26 Separation der Myelin-Proteinkomplexe mit ProFound-Co-
Immunopräzipitation Kit (Perbio Science, Bonn)
Mit Hilfe der entsprechenden polyklonalen Antikörper wurden die mit 36K, IP2 und
MBP assoziierten Proteine mit dem ProFound-Co-Immunopräzipitation Kit
(Perbio Science, Bonn) separiert. Dabei wurde im Wesentlichen nach Vorschrift des
Herstellers verfahren. Die Seren wurden mit 400 µl „Coupling Buffer“ 1/80 verdünnt,
zu 100 µl Matrix (aus 200 µl Suspension) gegeben und nach Zugabe von
1 µl 5 M Natrium-Cyanoborohydrid für 5 Stunden inkubiert. Zur längeren
Aufbewahrung der Antikörper-tragenden Matrix bei 4°C in „Coupling Buffer“ wurde
Natriumazid (Endkonzentration 0.02% (w/v)) zu der Suspension (400 µl) gegeben.
Für die Co-Immunopräzipitation wurden die Antikörper-Matrix-Suspensionen zum
entfernen des Lagerungspuffers zentrifugiert (1 Minute, 4000xg, RT) und mit der auf
400 µl verdünnten Protein-Extraktion (2.2.18) resuspendiert. Nach 3-stündiger
Inkubation bei RT in einem Überkopftaumler wurde der Ansatz erneut zentrifugiert
und nicht gebundene Proteine durch viermaliges Waschen in 300 µl gefolgt von
einmaligem Waschen in 100 µl Extraktionspuffer (4% Digitonin (w/v) in
20 mM Tris/HCl pH 7.4, 0.1 mM EDTA, 50 mM NaCl, 10% (v/v) Glycerin,
1 mM PMSF) entfernt.
-18-2. MATERIAL UND METHODEN
Die mit den Antikörpern assoziierten Proteine bzw. Protein-Komplexe wurden durch
dreimalige Inkubation mit „Elution-Buffer“ (5 Minuten, 750 rpm, RT) abgelöst und
durch Zentrifugation (1 Minute, 4000xg, RT) separiert. Die Proteinlösung wurde mit
Laemmli-Puffer (0.125 mM Tris/HCl pH 6.8, 2% (w/v) SDS, 6% (w/v) Glycerin,
10% (v/v) ß-Mercaptoethanol, 0.4 mM Bromphenolblau) versetzt und einer
SDS-PAGE (2.2.5) unterzogen. Mit Hilfe einer Silberfärbung (2.2.7) wurden die
auftrennten Proteine sichtbar gemacht und die Gele anschließend zur Aufbewahrung
in Zellophanfolie eingeschweißt.
-19-3. ERGEBNISSE
3. ERGEBNISSE
3.1 COMPUTERUNTERSTÜTZTE ANALYSE DER 36K-AMINOSÄURE-
SEQUENZ
Zu Beginn dieser Arbeit lag die vollständige, exprimierbare cDNA des Myelinproteins
36K als Bakteriendauerkultur vor (Diplom-Arbeit W. Moll, 2001). Die daraus
resultierende Sequenz von 325 Aminosäuren wurde für die folgenden
computergestützten Sequenz-Analysen verwendet.
3.1.1 Homologie-Alignment der Aminosäure-Sequenzen von 36K und
Proteinen aus der Reduktase/Epimerase/Oxidoreduktase Superfamilie
(RED)
Datenbank-Analysen mit dem BLAST-Internet-Server (Altschul et al., 1990; Madden
et al., 1996) der 36K-Aminosäure-Sequenz (2.2.1) zeigten keine signifikanten
Homologien zu allen bisher bekannten Myelinproteinen. Stattdessen gab es deutliche
Sequenzähnlichkeiten zu Proteinen aus der Reduktase/Epimerase/Oxidoreduktase
Superfamilie (RED) (Labesse et al., 1994; Kallberg et al., 2002). Abbildung 1 zeigt
ein Homologie-Alignment (2.2.1) der Aminosäure-Sequenzen von 36K und einem
humanen Protein unbekannter Funktion (FLJ13639), Retinol Dehydrogenasen von
Mensch (RDH 11) und Maus (UBE-1b) sowie der bereits kristallisierten
Carbonyl Reduktase vom Aal (Tanaka et al., 1996) und der
20-β-Hydroxysteriod-Dehydrogenase vom Zebrafisch (Gosh et al., 1991). Besonders
im N-terminalen Bereich, in dem sich auch das für die REDs charakteristische
Nukleotid-Bindemotif (GXXXGXG) befindet, stimmen die Aminosäuren abgesehen
vom FLJ13639 signifikant überein (Moll et al., 2003). Trotz abnehmender
Übereinstimmungen im Kern- und C-terminalen Bereich scheinen typische
Sequenzmotive der REDs im Bereich der Substrat-Bindtasche (YXXXK, PGXXXT)
zwischen 36K, FLJ13639, UBE-1b und RDH11 konserviert zu sein. Diese Motive
sind bei der Carbonyl Reduktase und der 20-β-Hydroxysteriod-Dehydrogenase durch
das Alignment in Richtung C-Terminus verschoben, können aber vermutlich trotzdem
als konserviert betrachtet werden. Ein Hinweis auf eine potentielles Substrat von 36K
konnte daraus nicht erhalten werden.
-20-3. ERGEBNISSE
Aufgrund dieser signifikanten Motif-Übereinstimmungen könnte 36K in die
Reduktase/Epimerase/Oxidoreduktase Superfamilie eingeordnet werden. Nach den
Kriterien von Kallberg et al. (2002) wäre dieses Myelinprotein eine „klassische
Kurzkettige Dehydrogenase/Reduktase (SDR)“.
Abb. 1: Computerunterstützter Aminosäuresequenz-Vergleich (1-Buchstaben-Code) von 36K mit
FLJ13639 (Protein unbekannter Funktion vom Menschen; Q9H8H1), UBE-1b (Maus Retinol
Dehydrogenase; Q9R1R8), RDH11 (Retinol Dehydrogenase vom Menschen; Q8TC12), CarbRed
(Carbonyl Reduktase vom Aal; Q90X71) und Steroid-DH (20 β-Hydroxysteroid-Dehydrogenase vom
Zebrafisch; Q9DF44). Swissprot-TrEMBL-Dateinummern sind kursiv angegeben; Aminosäure-
Nummerierung beginnend vom N-Terminus; identische/ähnliche Aminosäuren sind schwarz markiert.
-21-3. ERGEBNISSE
3.1.2 Sekundärstruktur-Vorhersage des 36K-Proteins mit dem JPred-Internet-
Server
Mit Hilfe des JPred-Internet-Servers (2.2.1), der die Vorhersagen von
6 verschiedenen Algorithmen verarbeitet (Cuff et al., 1998) wurde eine
Sekundärstruktur-Analyse des 36K-Proteins durchgeführt. Abbildung 2 zeigt die
36K-cDNA-Sequenz (Moll et al., 2003) und die daraus abgeleitete Primärstruktur aus
325 Aminosäuren mit der vorhergesagten Sekundärstruktur.
1 M G N S T M S L Y R N S A
1 ATG GGT AAT TCA ACG ATG TCT CTT TAC CGC AAC TCC GCG
14 W F L K G M T E F T R S A
40 TGG TTT CTG AAA GGA ATG ACT GAA TTC ACC AGG AGT GCG
27 F L S A A K H F V E K D L
79 TTC CTG TCT GCA GCC AAG CAC TTT GTG GAA AAG GAC CTG
40 E V S M A G R V F M I T G
118 GAA GTG TCC ATG GCA GGC AGG GTC TTC ATG ATC ACT GGA
53 A N S G I G R A T A M A I
157 GCC AAC AGT GGC ATA GGG AGA GCT ACA GCC ATG GCC ATC
66 A K R G G T V H M V C R N
196 GCC AAG AGA GGT GGT ACA GTC CAC ATG GTG TGC AGG AAC
79 K D K A E E A R A D I V K
235 AAG GAT AAA GCA GAG GAG GCC AGG GCT GAC ATT GTC AAG
92 E S G N K E I Y V H I L D
274 GAG TCA GGA AAT AAA GAA ATA TAT GTC CAC ATT CTG GAC
105 L S E T R K V W E F A E A
313 CTA TCA GAG ACA CGG AAG GTT TGG GAA TTT GCA GAG GCC
118 F K R K Y K V L N L L I N
352 TTC AAG AGG AAG TAC AAA GTC TTG AAT TTA CTG ATA AAT
131 N A G C I M S E R D V N A
391 AAT GCA GGC TGC ATC ATG AGT GAG AGG GAT GTG AAC GCT
144 E G L E K S F A S N V M G
430 GAG GGG CTG GAG AAG AGC TTT GCC AGT AAC GTC ATG GGT
157 V Y I L T R G L I P L L E
469 GTG TAT ATC CTG ACC AGG GGA CTG ATT CCC TTA CTG GAG
170 K S A E P R V I T V S S G
508 AAG AGT GCA GAG CCC AGA GTG ATC ACA GTT TCA TCT GGA
183 G M L V Q K L R T G N L Q
547 GGG ATG CTG GTC CAG AAG CTG AGG ACA GGG AAC CTG CAA
-22-3. ERGEBNISSE
196 T E I G R Y D G T M V Y A
586 ACA GAA ATA GGC CGC TAT GAC GGG ACC ATG GTC TAC GCA
209 Q H K R Q Q V V M T E Q W
625 CAG CAC AAG AGG CAA CAG GTG GTG ATG ACA GAG CAG TGG
222 A Q T H S N I H F S V M H
664 GCC CAA ACC CAT AGC AAC ATC CAC TTC TCA GTG ATG CAT
235 P G W V D T P A V A N A M
703 CCT GGC TGG GTG GAC ACA CCA GCG GTG GCC AAT GCC ATG
248 P D F H Q S M K D S L R T
742 CCT GAC TTC CAC CAG TCT ATG AAG GAC AGC CTG AGG ACC
261 P E Q G A D T V V W L A I
781 CCG GAG CAG GGG GCT GAC ACC GTG GTG TGG TTG GCC ATC
274 S E A A A T K P S G S F F
820 TCA GAA GCC GCC GCC ACC AAG CCC AGC GGA AGC TTC TTC
287 Q D R R M V S A H L P L A
859 CAG GAT CGG AGG ATG GTG TCG GCC CAC CTG CCC CTG GCC
300 W T H S S Q L E Q Q K F M
898 TGG ACC CAC AGT TCC CAG TTG GAG CAG CAG AAG TTC ATG
313 S V M E D L A K T F Q P H
937 AGT GTG ATG GAG GAT CTG GCC AAG ACC TTC CAG CCC CAC
326 *
976 TGA
Abb.2: cDNA-Nukleotidsequenz und daraus abgeleitete Aminosäuresequenz von 36K
mit Sekundärstruktur-Vorhersage nach JPret. ROT: α-Helices, GELB: β-Faltblätter.
Demnach besteht das Protein aus alternierenden α-Helices (10) und β-Faltblättern
(7) (βαβ-Struktur). Diese sogenannte „Rossmann fold“ (Rossmann et al., 1974) ist
charakteristisch für Proteine aus der Reduktase/Epimerase/Oxidoreduktase
Superfamilie (RED). Wie bei den Proteinen aus dieser Familie ist das
Nukleotid-Bindemotif (GXXXGXG) zwischen dem ersten β-Faltblatt und der
darauffolgenden α-Helix lokalisiert (Moll et al., 2003).
-23-3. ERGEBNISSE
3.1.3 3-dimensionales Modell des N-terminalen Teils von 36K
Aus der in Abbildung 2 dargestellten 36K-Aminosäuresequenz konnte mit Hilfe des
SWISS-MODELL-Servers (Guex und Peitsch, 1997) und des SWISS-pdb-Viewer 3.7
ein 3-dimensionales Modell des N-terminalen Teils von 36K erzeugt werden.
Abbildung 3 zeigt den modellierten Teil des Proteins zwischen L39 und V186, der
nach 3.1.1 starke Homologien zu Proteinen aus der RED-Superfamilie aufweist. Das
dargestellte Molekül hat eine sehr geordnete Struktur mit β-Faltblättern im Zentrum
und α-Helices in der Peripherie. Das eingezeichnete Nukleotid-Bindemotif liegt in
einer „Tasche“ zwischen einem β-Faltblatt und der darauffolgenden α-Helix. Das
Modell bestätigt für den N-terminalen Teil von 36K die Sekundärstruktur-Vorhersage
aus 3.1.2 und zeigt die für REDs charakteristische βαβ-Struktur („Rossmann fold“)
(Rossmann et al., 1974).
Abb. 3: Struktur-Modell des N-terminalen Teils von 36K (L39 bis V186)
modelliert mit SWISS-pdb-Viewer 3.7 zeigt βαβ-Struktur (Rossmann fold).
ROT: α-Helices und GELB: β-Faltblätter. Putatives Nukleotid-Bindmotif
GXXXGXG ist angezeigt. α-Helix-Nummerierungen entsprechend
Sekundärstruktur-Vorhersage aus 3.1.2
-24-3. ERGEBNISSE
3.2 UNTERSUCHUNG DER SEKUNDÄRSTRUKTURZUSAMMENSETZUNG
DES REKOMBINANTEN 36K-PROTEINS MITTELS FERN-UV-CIRCULAR-
DICHROISMUS-SPEKTROSKOPIE (CD-SPEKTROSKOPIE)
Zur Analyse der Sekundärstrukturzusammensetzung von 36K in Lösung wurde eine
Fern-UV-Circulardichroismus-Spektroskopie (CD-Spektroskopie) durchgeführt. Dabei
ergibt die Differenz der Absorptionen von links- und rechtscircular polarisiertem Licht
in Bezug zur Wellenlänge ein CD-Spektrum, dass Voraussagen zur
Sekundärstrukturzusammensetzung erlaubt. Dazu wurde mit 0.12 mg/ml (3 µM)
rekombinantem 36K in PBS pH 7.4 (2.2.2 - 2.2.4) wie unter 2.2.10 beschrieben das
in Abbildung 4A dargestellte gemittelte Spektrum aufgenommen. Dieses zeigt ein
Minimum bei 219 nm und ein Maximum bei 195 nm, was auf eine größtenteils
α-helicale Struktur des Proteins hindeutet (Yang et al., 1986).
A B
Abb. 4: Sekundärstruktur-Zusammensetzung von rek. 36K nach CD-Spektroskopie.
A: Fern-UV-CD-Spektrum in 50 mM PBS (150 mM NaCl) pH 7.4. B: Prozentuale Verteilung der
Sekundärstrukturen kalkuliert nach Böhm et al. (1992)
Die Auswertung dieses UV-Spektrums mit Hilfe des Computerprogramms CDNN
(Böhm et al., 1992) ergab für das rekombinante 36K in Lösung die in Abbildung 4B
dargestellte Sekundärstrukturzusammensetzung von 35% ± 2% α-Helix,
12% ± 2% β-Faltblatt, 17% ± 2% β-Turn und 36% ± 2% ungeordnete Strukturen
(„random coil“). Dies steht in gutem Einklang mit den computergestützten
Vorhersagen aus 3.1.2 und 3.1.3, nach denen das Protein einen hohen Anteil
α-helicaler Strukturen aufweist.
-25-3. ERGEBNISSE
3.3 RÖNTGENKLEINWINKELSTREUUNG VON REKOMBINANTEM 36K IN
LÖSUNG
Die umhüllende Struktur des 36K-Proteins wurde in Kooperation mit
PD Dr. G. Grüber (Institut für Biophysik, Universität des Saarlandes, Homburg/Saar)
über Röntgenkleinwinkelstreuung von rekombinantem 36K in Lösung (2.2.2 - 2.2.4)
untersucht. Mit Hilfe des Programms DAMMIN konnte aus den gewonnenen
Streudaten die Quartärstruktur des Moleküls bestimmt werden. In Lösung hat das
36K-Protein demnach eine Länge von ~9 nm und setzt sich aus 2 Domänen mit den
Dimensionen von ~4 x 3 nm und ~5 x 4 nm zusammen (Abbildung 5). Mit BSA als
Referenzmolekül konnte eine apparente Molekularmasse von 35 ± 2 kD bestimmt
werden. In Zukunft soll rekombinantes 36K ebenfalls in Kooperation mit
PD Dr. G. Grüber kristallisiert werden, um über Röntgenstreuungsstudien eine
atomare Struktur des Moleküls zu erhalten.
Abb. 5: Die umhüllende Struktur des 36K-Proteins
abgeleitet aus nach Röntgenkleinwinkeldaten; Studie
wurde von PD Dr. G. Grüber (Inst. f. Biophysik,
Universität des Saarlandes, Homburg/Saar)
durchgeführt
-26-Sie können auch lesen

















































