Klinische Chemie und Laboratoriumsdiagnostik - Vorlesung: Arteriosklerose
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Klinische Chemie und Laboratoriumsdiagnostik
Vorlesung: Arteriosklerose
Prof. Dr. med. Jerzy-Roch Nofer
Centrum für Laboratoriumsmedizin
– Zentrallaboratorium –
Universitätsklinikum Münster
Albert-Schweitzer-Campus 1
D-48149 Münster
Tel.: 0251 83-47228
Fax: 0251 83-47225
nofer@uni-muenster.de
www.klichi.uni-muenster.de
QR Code / Link dieser Vorlesung:
www.klichi.uni-muenster.de/folien2
Sommersemester 2019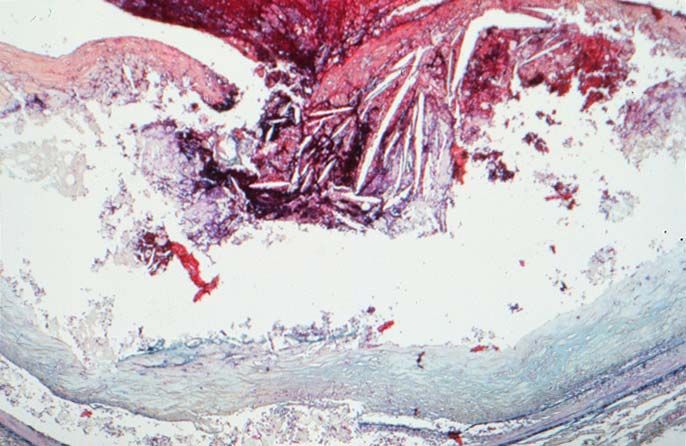
Arteriosklerose und Atherosklerose
Arteriosklerose (Lobstein 1833)
- charakterisiert die durch Alterung und hämodynamisch bedingten
Veränderungen der Arterienwand, an erster Stelle die Kalzifizierung.
Lobstein JE. Classe seconde. Maladies des arteres. In : Traites d'Anatomie
Pathologique. Tome second: Conetant l'anatomie patologique speciale. Levrault
Paris 1833.
Atherosklerose (Marchand 1904)
- bezeichnet die die Arteriosklerose begleitenden Intimaverfettungen von
Arterienwand
Marchand F. Über Arteriosklerose (Athero-Sklerose). Verh. dzsch. Congr. inn.
Med. 1904;21:23-59
Anitschkow-Hypothese
Cholesterinakkumulation als Ursache der Arteriosklerose
Cholesterinreiche Diät führt
zur Akkumulation des
Cholesterins in der
Arterienwand und schließlich
zur Arteriosklerose
Anitschkow N, Chalatow S. „Über
experimentelle Cholesterinsteatose und
ihrer Bedeutung für die Entstehung
einiger pathologischer Prozesse. Zentrbl.
Allg. Pathol. Pathol. Anat. 1913;24:1-9
Nikolai N. Anitschkov (1885 – 1964)
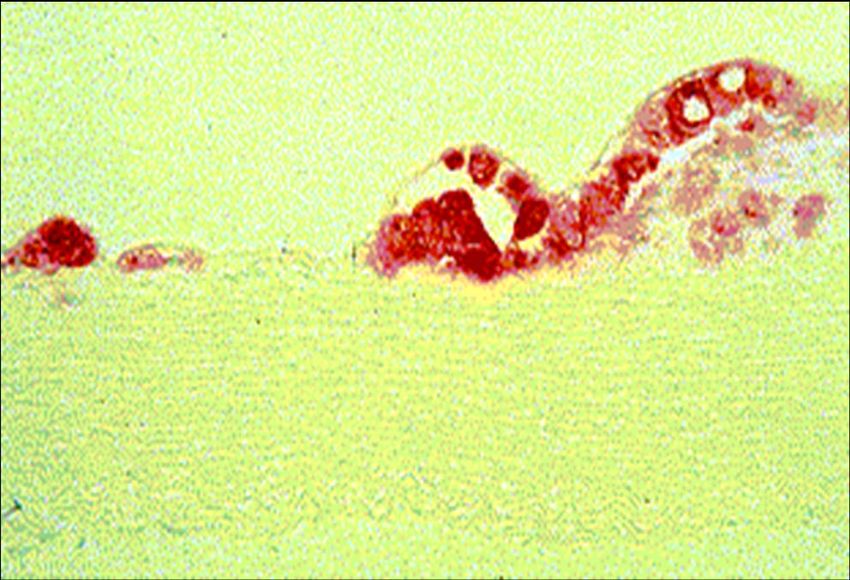
Schaumzelle
Cholesterin-Akkumulation in Makrophagen in vivo and in vivo
Lichtmikroskopie Elektronenmikroskopie
In vitro
In vivo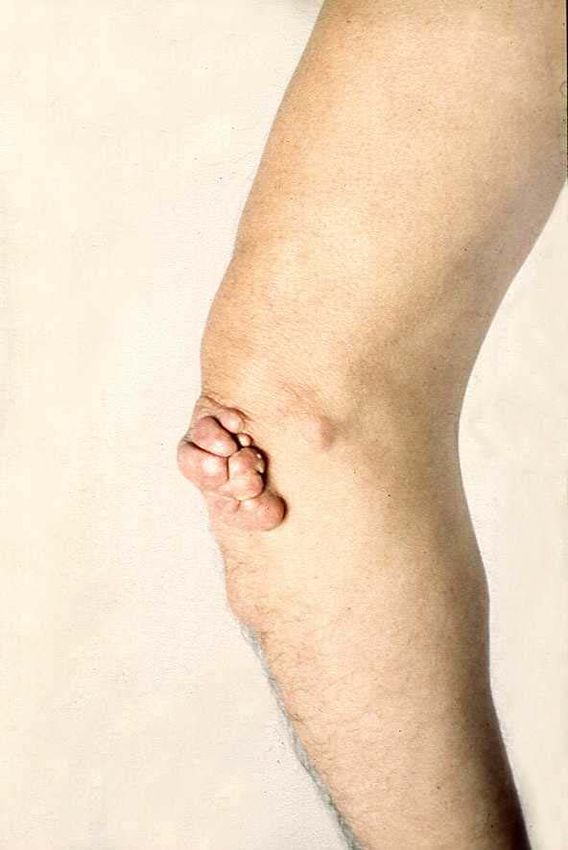
Arteriosklerose - Pathologie
Nomenklatur Wachs- Beginn
Histologie Progressionstufen Klinik
tum
Typ I Läsion – „initial“
Isolierte einzelne Schaumzellen
ab erster
Typ II Läsion – „fatty streak“ Dekade
Intrazelluläre Akkumulation ohne
von Cholesterin durch Symptome
Lipidakku-
Typ III Läsion – „intermediär“ mulierung
Typ II + einzelne extrazelluläre
Cholesterinansammlungen
ab dritter
Typ IV Läsion – „Atherom“ Dekade
Typ II + extrazellulärer Lipidkern
(Cholesterinkrystalle )
Typ V Läsion – „Fibroatherom“ durch
extrazellulärer Lipidkern + Kollagen
fibrotische Veränderungen und mit oder
(Akkumulation von Kollagen, glatte ohne
Einwanderung und Proliferation Muskel- ab vierter Symptome
von glatten Muskelzellen) zellen Dekade
Typ V Läsion – „kompliziert“ durch
Ruptur des Atheroms Thrombose
Hämatom , Thrombus Hämor-
rhagie
Rupturierte
Plaque
Thrombus
Fibrose Kappe
Lipidkern
Rupturierte Plaque
Koronare Thrombose
und
Myokardinfarkt
Koronare
Thrombose
Myokard-
infarkt
Pathogenese der Arteriosklerose
Hauptfragen der letzten 100 Jahre
• Welche Mechanismen liegen der Akkumulation des
Cholesterins in Makrophagen zugrunde?
• Woher stammen die Makrophagen der Arterienwand?
Pathogenese der Arteriosklerose
Hauptfragen der letzten 100 Jahre
•Welche Mechanismen liegen der Akkumulation des
Cholesterins in Makrophagen zugrunde?
•Woher stammen die Makrophagen der Arterienwand?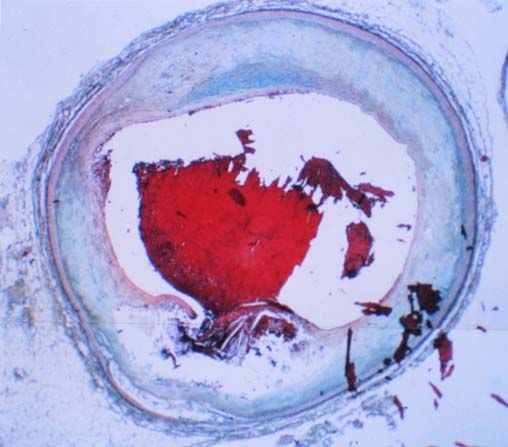
Struktur der Lipoproteine
Oberfläche-Lipide
Freies Cholesterin
Phospholipide
Proteine
(Apoproteine, Enzyme,
sonstige)
Kern-Lipide
Cholesterinester
TriglyzerideDistribution Zusammen- Molekulare
von setzung (%) Masse (Da)
Apoproteinen
Dichte (g/L)
Klassifikation von LipoproteinenFamiliäre Hypercholsterinämie
Familiäre Hypercholesterinämie
• Gesamt Cholesterin 700 – 1200 mg/dL
• erhöhtes LDL-Cholesterin
• Hautxanthome
• Sehnenxanthome
• Arcus lipoides
• Premature Arteriosklerose: Angina pectoris
im frühen Kindesalter, Aortenstenose
Mike S. Brown Joe L. GoldsteinKlinische Zeichen in FH
Koronares Risiko bei
familiärer Hypercholsterinämie (FH)
FH Patienten erleiden früher und häufiger
kardiovaskuläre Erkrankungen.
1,0
Überleben ohne Herz‐Kreislauferkrankung
Differenz zu nicht‐betroffenen Verwandten
0,8
LDL‐Cholesterol (mg/dL)
Nicht‐betroffene Verwandte
0,6 (n=1087)
N543H/2393del9 LDL‐R‐Mutation
0,4 (n=124)
alle anderen FH‐Mutationen
(n=484)
0,2
Andere
Varianten
0
0 20 40 60 80
Alter (Jahre)Familiäre Hypercholsterinämie (FH)
Geschätzte Diagnoseraten in verschiedenen Länder
Deutschland – ca. 10 %Familiäre Hypercholsterinämie (FH)
Diagnose
LDL‐Cholesterin > 190 mg/dl (4,9 mmol/l)
Kinder unter 16 Jahren: LDL‐Cholesterin > 155 mg/dl (4,0 mmol/l)
+
Positive Familienanamnese Beim Index‐Patienten
Familienangehörige ersten Grades mit LDL‐C > 190 Nachweis von tendinösen Xanthomen
mg/dl (4,9 mmol/l) oder vorzeitiger KHK (Frauen < 60 oder oder Arcus corneae < 45 Jahre
Jahre, Männer < 55 Jahre) oder mit Xanthomen
Klinische Diagnose: familiäre Hypercholesterinämie
Genetische Diagnostik
Sequenzierung LDL‐R, PCSK9
G. Klose et al., Deutsches Ärzteblatt 111, 523 (2014).Struktur des LDL-Rezeptors
DOMÄNE
Ligandenbindung EGF-Homologie O-gykosylierte Transmembran
(292 Aminosäure) (400 Aminosäure) (58 Aminosäure) (22 Aminosäure)
Zytoplasmatisch
(50 Aminosäure)
LDL LDL Triglyzeride
LDL
LDL LDLFunktion des LDL-Rezeptors
Coated pit
LDL-Rezeptor LDL
Golgi
E-plasmatisches
Reticulum
Endosom
Cholesterin
ApoB Synthese
Cholesterin
Ester Cholesterin ↓↓↓
Lysosom LDL-Rez ↓↓↓Metabolismus des LDL-Cholesterins
Normal Familiäre Hypercholesterinämie
Aufnahme Aufnahme Aufnahme Aufnahme
durch Leber (80%) außer Leber (20%) durch Leber (20%) außer Leber (80%)
LDL-R-abhängig LDL-R-unabhängig LDL-R-abhängig LDL-R-unabhängig LDL-R-abhängig LDL-R-unabhängig LDL-R-abhängig LDL-R-unabhängig
(80%) (20%) (66%) (34%) (0%) (100%) (0%) (100%)Cholesterinaufnahme in Makrophagen
Rolle von Scavenger-Receptoren
Klasse A
I
II
Cholesterinester III
MARCO
Freies Klasse B
Cholesterin
SR-BI/II
CD36
LDL
modifiziertes CD68 (Makrosialin)
LDL
Schaumzelle SR-CI
LOX -I
Kollagen-ähnlich Muzin-ähnlich
Cystein-reich
C-terminal Immunogen
Fettsäure-azyliert LektinModifikation von LDL
+ +
+
Modifikation von LDL
+
Natives LDL +
• Oxidation
+ +
+ + • Glykosylierung
+ +
• Phospholipase A2
Modifikation
• Sphingomyelinase
-
+ • Homocystein
+
-
- • Carbon Disulfid
Modifiziertes +
-
LDL
+
-
-
-LDL-Cholesterin und KHK-Risiko
Koronare Herzkrankheit Ischämische Schlaganfall
3 - nicht adjustiert 3 - nicht adjustiert
- adjustiert - adjustiert
Relatives Risiko
Relatives Risiko
2 2
1 1
110 150 190 230 110 150 190 230
LDL-Spiegel im Plasma (mg/dL)
Metaanalyse : 68 Studien bei 302.430 Probanden; Danesh et al., JAMA 2009;302:1993LDL-Cholesterin: „The Lower the Better“
30 Placebo Statin 4S
Herzinfarkte (%)
25
4S
20
LIPID
15 LIPID
CARE CARE
10 HPS
HPS
TNT (10 mg of atorvastatin)
5 TNT (80 mg of atorvastatin)
0
70 90 110 130 150 170 190 210
LDL Cholesterin (mg/dl)Meta-Analyse: 14 Statin-Studien bei 90.056 Personen Die Absenkung des LDL-C mit Statinen um 1mmol/l (39mg/dl) führt zu einer Reduktion • der Gesamtmortalität um 12%, • der koronaren Mortalität um 19%, • Koronarer Ereignisse (nicht tödlicher Herzinfarkt oder Koronartod) um 23%, • der Inzidenzrate des Schlaganfalls um 17%. Baigent C, Keech A, Kearney PM, et al., Lancet 2005; 366:1267-1278
Proprotein convertase subtilisin/kexin Typ 9 (PCSK9)
Bedeutung für LDL-Metabolismus
PCSK9
„gain-of-function“
Mutations
• Gesamt-Cholesterin
> 500 mg/dL
• Triglyzeride
< 200 mg/dL
• Anwesenheit von
Xanthomas
• Erhöhtes
Koronares Risiko
• Familiäre Hyper-
cholesterinämiePCSK9 „loss-of-function“ Mutationen senken LDL-C-Spiegel und schützen von KHK
PCSK9 Inhibitoren für die Behandlung von
Hypercholesterinämie: FOURIER Trial
Standardtherapie
Standardtherapie
Kumulative Inzidenz (%)
Standardtherapie
LDL (mg/dL)
Evolucumab
Evolucumab
Evolucumab
Wochen Monate nach Randomisierung
26.564 Patienten randomisiert 2:1 zu Standardtherapie (Statine oder Statine + Ezetimib)
oder Evolucumab (humanisiertes Antikörper gegen PCSK9) und beobachtet über 36 Monate
Sabatine et al. N Engl J Med 2017;376:1713Transport von Lipiden im Blut
Diät-Lipide
C
Cholesterin (C)
C
Triglyzeride (TG)
LDL
VLDL C HDL
C C C
VLDL
C Chylo-
Remnant C
micron
Remnants
Fettgewebe
TG
Chylo- TG Makrophagen
FFS
mikronen
Periphere
Gewebe
MuskelLipolyse von triglyzeridreichen Lipoproteinen
Endothel Aufnahme von Rem-
nants in der Leber
Oberfläche-Remnants (HDL-Vorstufen)
CM LDLR
VLDL
TG
TG
TG CMR
IDL
GPI- LPL LPL Proteo-
HBP glykane ApoE-Rezeptore
Endothel (LRP1, VLDL-R)
Myozyten
Adipo- Fettsäure
zyten (+ DAG, MAG)Pro-atherogene Wirkung von Triglyzeriden
Rolle von ApoB-haltigen Remnantpartikeln
Lipolytische
Remnanthypothese
Toxin-Hypothese
ApoB-haltige Remnantpartikel
VLDL- Proteo- Produkte
-Rezeptor GPIHBP-1 glykane Lipolyse
Endothel
Entzündung
Schaum- Adhäsion von Leukozyten
Zelle Aktivierung der GerinnungVLDL-Remnants sind pro-atherogen
Tag 0 Aufnahme von VLDL- Nicht-nüchterndes
Remnants in Makrophagen Lipidprofil
Kontrollzellen
µg esterified cholesterol/mg cell protein
VLDL Remnants
Tag 2 LDL
Tag 5Copenhagen Heart Study und Copenhagen General Population Study
Koronare und Gesamtmortalität in Abhängigkeit von Remnant-Cholesterin und LDL-Cholesterin
Koronare Sterblichkeit
Relatives Risiko
Relatives Risiko
Remnant-Cholesterin (mmol/l) LDL-Cholesterin (mmol/l)
Gesamtsterblichkeit
Relatives Risiko
Relatives Risiko
Remnant-Cholesterin (mmol/l) LDL-Cholesterin (mmol/l)
Varbo A et al. Clin Chem. 2015;61:533–543Alle Studienbeteiligte
Senkung von
Log OR für kardiovaskuläre Ereignisse
OR für kardiovaskuläre Ereignisse
54% Risikoreduktion bei Senkung
von TG-Spiegel um 1 mmol/L
Triglyzeriden im Plasma
reduziert
kardiovaskuläres Risiko
Log OR für kardiovaskuläre Ereignisse
Studienbeteiligte mit TG > 2 mmol/L
OR für kardiovaskuläre Ereignisse
43% Risikoreduktion bei Senkung
von TG-Spiegel um 1 mmol/L
Metaanalyse von 6 Studien mit
Fibraten und Niacin
Nordestgaard und Vrabo,
Senkung von Triglyzeriden (mmol/L) Lancet 2014;384:626Pathogenese der Arteriosklerose
Hauptfragen der letzten 100 Jahre
•Welche Mechanismen liegen der Akkumulation des
Cholesterins in Makrophagen zugrunde
• Woher stammen die Makrophagen der Arterienwand?LDL, Monozyten und Arteriosklerose
Zirkulierende Monozyten
Natives LDL
Endothelzellen
Makrophagen
Glatte Muskel- Makrophagen
zellen
Minimal
Oxidiertes LDL
Oxidiertes LDL
Oxidiertes LDL
SchaumzelleAktivierung des Endothels
Hyperlipidemia Rauchen
Hypertonie Homocystein
oxLDL Diabetes
Endotheliale
oxidativerDysfunktion
Stress
Transkriptions-
faktoren NF-B
Endothelzelle
proinflammatorische
Gene MCP-1 VCAM-1, ICAM-1
Monozyten-Aktivierung/InfiltrationChemotaxis von Monozyten und Arteriosklerose
Chemotaktische Faktoren Monozyten-Migration
Wild-Typ Maus
• Monocyte chemoattractant protein-1 (MCP-1)
• Interleukin 8 (IL-8)
• Andere Chemokine
• Komplement (C3a, C5a)
MCP-1-defiziente Maus
• Hydroperoxide (HPETE)
• Angiotensin-IIAktivierung von Makrophagen und die Entzündung Ox LDL
CD4+ T-Zellen begünstigen die Entwicklung
der Arteriosklerose in Tiermodellen (Maus)
IFN im Plasma (pg/mL) Lesiongröße (µm2/Sektion)
ApoE-/- Maus 150000
CD4 T-Zellen in Läsionen
entwickelt Arteriosklerose
75000
scid Maus
Ø T-Zellen Ø B-Zellen
0
150
apoE-defizient
scid Maus
75
+ adoptiver Transfer
von T-Zellen
0
ApoE-/-/scid ApoE-/-/scid ApoE-/-
apoE-/-/ scid + +
apoE-/-/scid
+ CD4 T-Zellen CD4 T-ZellenDiagnostische Marker: CRP und Fibrinogen
bei Arteriosklerose
Einfluß auf Mortalität bei instabiler KHK:
20 20
Wahrscheinlichkeit von Tod (%)
Wahrscheinlichkeit von Tod (%)
C-reaktives Protein >10 mg/l
(n=309) Fibrinogen 4.0 g/l
(n=303)
p=0.69
p=0.001
10 C-reaktives Protein 2-10 mg/l 10 Fibrinogen 3.4-3.9 g/l
(n=294) (n=300)
p=0.29 p=0.004
C-reaktives ProteinCRP und Koronare Herzkrankheit
CANTOS Study
LDL-Cholesterin ~ 82 mg/dL NIEDRIG
hsCRP 0.2 mg/dL ERHÖHT
CRP-Konzentration Herzinfarkt oder Herztod
% Senkung
% Inzidenz
Monate Jahren
• Randomisierte, doppelblinde Studie von Canakinumab (anti-IL1beta)
• Drei Dosen (50 mg, 150 mg, and 300 mg), subkutan, vierteljährlich
• 10,061 Patienten mit Myokardinfarkt und CRP > 0.2 mg/dL
Ridker et al. N Engl J Med 2017;377:1119‐31Risikofaktoren der Arteriosklerose
Klassische Neue
1. Alter nein 1. CRP ?
2. LDL-Cholesterin ja 2. SAA ?
3. Rauchen ja 3. Fibrinogen ?
4. HDL-Cholesterin ja 4. IL-6 nein
5. Bluthochdruck ja 5. IL-1beta nein
6. Diabetes mellitus ja 6. Myeloperoxidase nein
7. Familiäre Belastung nein 7. PLA2 nein
8. Triglyzeride ja 8. Fetuin nein
9. Harnsäure ja
10. NT-proBNP nein
11. PAPP-A nein
12. CD40/CD40L nein
13. und vieles mehr ???Sie können auch lesen

















































