Effekt des Hitzeschockproteins HspB5/alpha-B-Crystallin auf den basalen Dendritenbaum hippocampaler Pyramidenzellen der Maus
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

Universität Ulm
Institut für Anatomie und Zellbiologie
Institutsleiter: Prof. Dr. med. Tobias M. Böckers
Effekt des Hitzeschockproteins
HspB5/alpha-B-Crystallin auf den basalen
Dendritenbaum hippocampaler Pyramidenzellen der
Maus
Dissertation zur Erlangung des Doktorgrades der Medizin der
Medizinischen Fakultät der Universität Ulm
Samuel Birk
geboren in Reutlingen, Deutschland
2020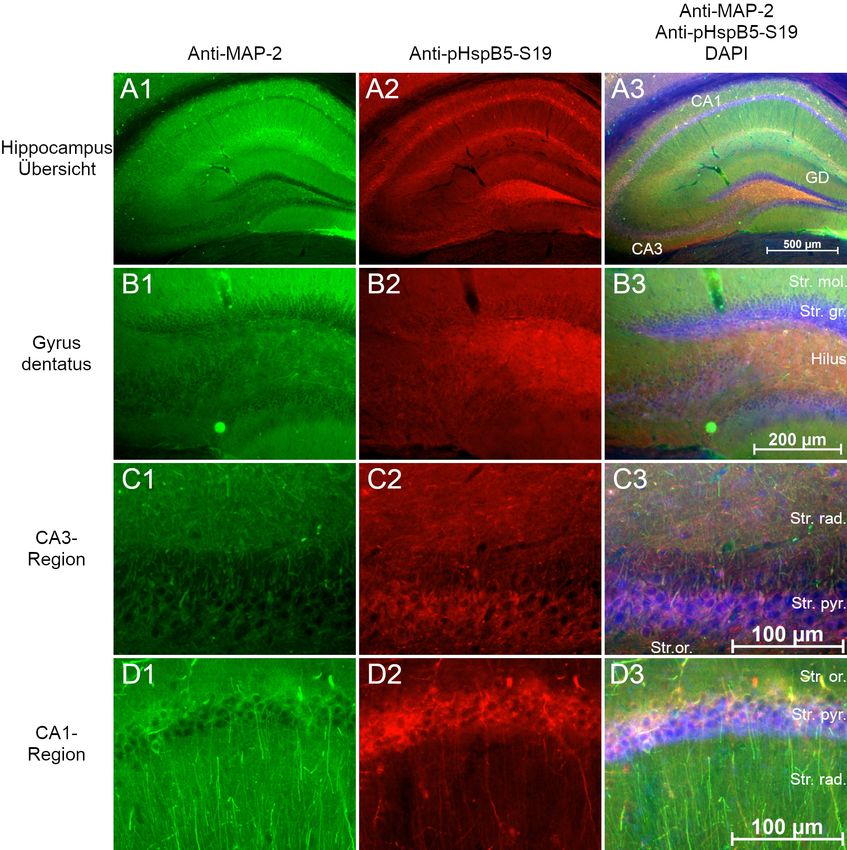
Amtierender Dekan: Prof. Dr. Thomas Wirth 1. Berichterstatter: Prof. Dr. Nikola Golenhofen 2. Berichterstatter: Prof. Dr. Gilbert Weidinger Tag der Promotion: 16.04.2021
Die Widmung wurde aus Gründen des Datenschutzes entfernt.
Teile dieser Arbeit wurden veröffentlicht in: B. BarteltKirbach, C. Wiegreffe, S. Birk, T. Baur, M. Moron, S. Britsch, N. Golen- hofen, HspB5/Bcrystallin phosphorylation at S45 and S59 is essential for protection of the dendritic tree of rat hippocampal neurons, Journal of Neurochemistry, 2020; 00:1-15, https://doi.org/10.1111/jnc.15247 © 2020 The Authors Open acces article published under the terms of the Crea- tive Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND 4.0), https://creativecommons.org/licenses/by-nc-nd/4.0/
Inhaltsverzeichnis
Abkürzungsverzeichnis vii
1 Einleitung 1
1.1 Aufbau des Nervengewebes und des Hippocampus . . . . . . . . . . 1
1.1.1 Aufbau des Nervengewebes . . . . . . . . . . . . . . . . . . . 1
1.1.2 Der Hippocampus . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2 Hitzeschockproteine und Zellstress . . . . . . . . . . . . . . . . . . . . 3
1.2.1 Die Gruppe der kleinen Hitzeschockproteine, HspBs . . . . . . 4
1.3 HspB5 / alpha-B-Crystallin . . . . . . . . . . . . . . . . . . . . . . . . . 5
1.3.1 Struktur von HspB5 / alpha-B-Crystallin . . . . . . . . . . . . . 5
1.3.2 Phosphorylierung von HspB5 . . . . . . . . . . . . . . . . . . . 5
1.4 HspB5 im Gehirn . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
1.4.1 HspB5 bei neurodegenerativen Erkrankungen . . . . . . . . . 6
1.4.2 HspB5 in Neuronen . . . . . . . . . . . . . . . . . . . . . . . . 7
1.5 Fragestellung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
2 Material und Methoden 10
2.1 Material . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
2.1.1 Allgemeine Reagenzien . . . . . . . . . . . . . . . . . . . . . . 10
2.1.2 Antikörper . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
Primärantikörper . . . . . . . . . . . . . . . . . . . . . . . . . . 11
Sekundärantikörper . . . . . . . . . . . . . . . . . . . . . . . . 11
2.1.3 Verbrauchsmaterial . . . . . . . . . . . . . . . . . . . . . . . . 12
2.1.4 Geräte . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
2.1.5 Kit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.1.6 Plasmide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
2.1.7 Tiertötungen und Tierversuche . . . . . . . . . . . . . . . . . . 15
2.1.8 Software . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.2 Molekularbiologische Methoden . . . . . . . . . . . . . . . . . . . . . . 16
2.2.1 Plasmidpräparation . . . . . . . . . . . . . . . . . . . . . . . . 16
2.3 Lokalisation von HspB5 in Gehirnen der Maus . . . . . . . . . . . . . 17
2.4 Transfektion von hippocampalen Neuronen mittels Elektroporation . . 18
2.4.1 Beschichtung der Zellkultureinsätze . . . . . . . . . . . . . . . 19
2.4.2 Herstellen der Ultra LowMeltingPoint-Agarose . . . . . . . . . . 20
2.4.3 Ansetzen des Injektions-Plasmids . . . . . . . . . . . . . . . . 20
2.4.4 Ausziehen und Schleifen der Injektionspipetten . . . . . . . . . 21
2.4.5 Transfektion und Elektroporation . . . . . . . . . . . . . . . . . 21
2.4.6 Anfertigen von Schnittkulturen des Gehirns . . . . . . . . . . . 23
v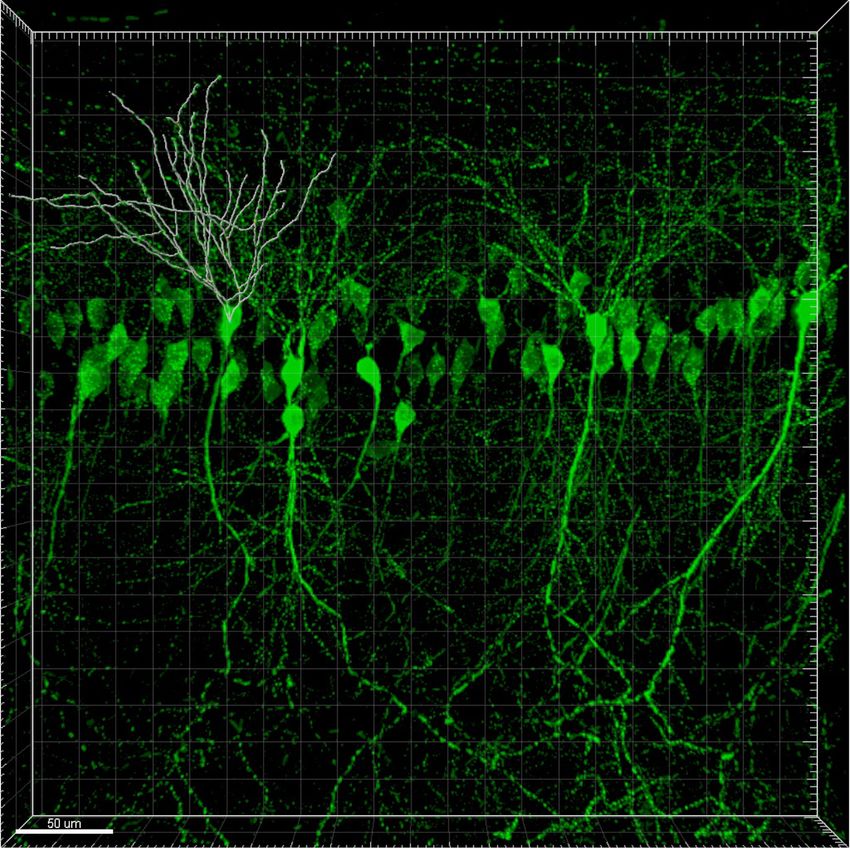
Inhaltsverzeichnis
2.5 Überexpression von HspB5 mittels in utero Elektroporation . . . . . . 26
2.5.1 Immunmarkierung von Gehirnschnitten . . . . . . . . . . . . . 28
2.5.2 Konfokale Mikroskopie . . . . . . . . . . . . . . . . . . . . . . . 31
2.5.3 Dreidimensionale Rekonstruktion des Dendritenbaums mit Ima-
ris . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
2.5.4 Statistische Auswertung der Daten . . . . . . . . . . . . . . . . 33
3 Ergebnisse 34
3.1 Phosphorylierungsabhängige Lokalisation von HspB5 im Gehirn der
Maus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
3.2 Überexpression von HspB5 im Hippocampus der Maus . . . . . . . . 41
3.2.1 Intraventrikuläre Plasmidinjektion und Elektroporation des Hip-
pocampus mit anschließender Anfertigung von Schnittkulturen 41
3.2.2 Überexpression von HspB5 mittels in utero Elektroporation in
vivo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
3.2.3 Nachweis der erfolgreichen Überexpression von HspB5 und
der nichtphosphorylierbaren Mutante HspB5-AAA . . . . . . . 46
3.2.4 Analyse des basalen Dendritenbaums von CA1-Pyramidenzellen
nach Überexpression von HspB5 und von HspB5-AAA . . . . . 47
4 Diskussion 54
4.1 Methodik . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
4.1.1 Überexpression von Proteinen im Hippocampus in kultivierten
Gehirnschnitten . . . . . . . . . . . . . . . . . . . . . . . . . . 54
4.1.2 Überexpression von Proteinen im Hippocampus in vivo . . . . 55
4.2 Phosphorylierungsabhängige Lokalisation von HspB5 im Gehirn . . . 57
4.3 Effekt von HspB5 auf den Dendritenbaum hippocampaler Pyramiden-
zellen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
4.3.1 Mögliche molekulare Angriffspunkte von HspB5 in Dendriten . 59
4.4 Mögliche Bedeutung von HspB5 bei neurodegenerativen Erkrankungen 62
5 Zusammenfassung 64
Literaturverzeichnis 66
Abbildungsverzeichnis 75
Tabellenverzeichnis 77
Danksagung 78
Lebenslauf 79
vi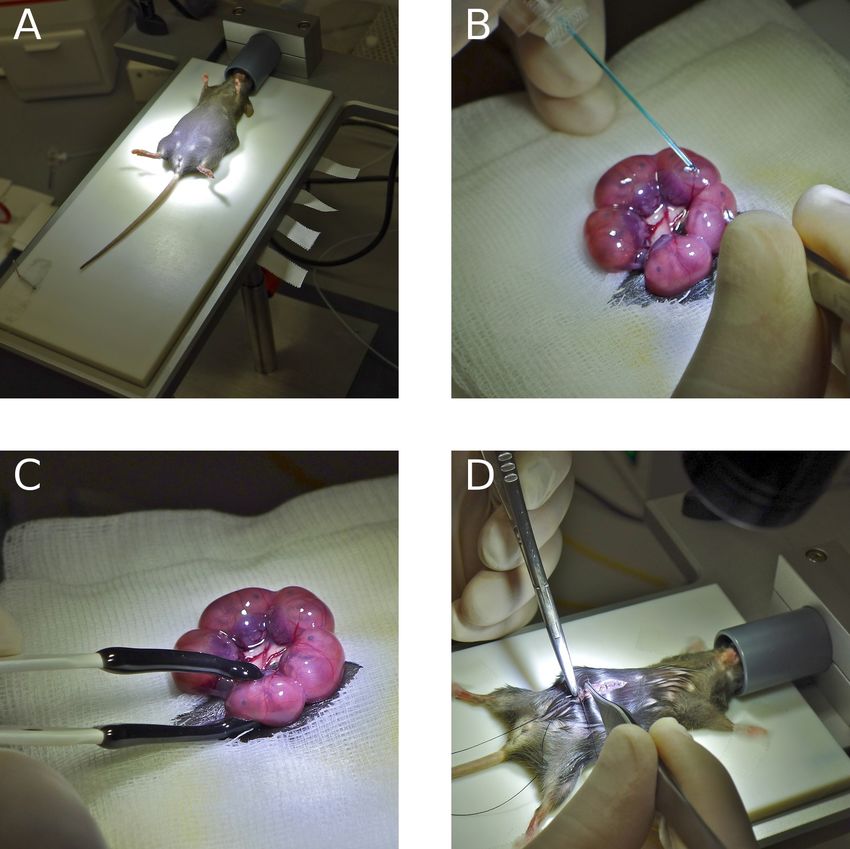
Abkürzungsverzeichnis
A Einheit: Ampere, Größe: Stromstärke, Größensymbol: I
Akt Proteinkinase B
bspw. beispielsweise
bzw. beziehungsweise
BCL11B B-Zell-Lymphom / Leukämie 11B
BDNF brain-derived neurotrophic factor
BSA Bovines Serum Albumin
◦
C Einheit: Grad Celsius, Größe: Temperatur
CA Cornu ammonis
CaCl2 Calciumchlorid
CaMKII Calcium/Calmodulin-dependent protein kinase II
CO2 Kohlenstoffdioxid
CREST Calcium responsive transactivator
CREB adenosine 3’, 5’ - monophosphate (cAMP) response
element-binding protein
DAPI 4’,6-Diamidin-2-phenylindol
d. h. das heißt
E Embryonal
EYFP enhanced yellow fluorescent protein
FA-Lösung Formaldehyd-Lösung
g Schwerebeschleunigung
G Gauge
GFP Green fluorescent protein
GD Gyrus dentatus
HBSS complete-Puffer Complete Hank’s Balanced Salt Solution Puffer
HEPES 2-(4-(2-Hydroxyethyl)-1-piperazinyl)-ethansulfonsäure
Hsp heat shock protein - Hitzeschockprotein
vii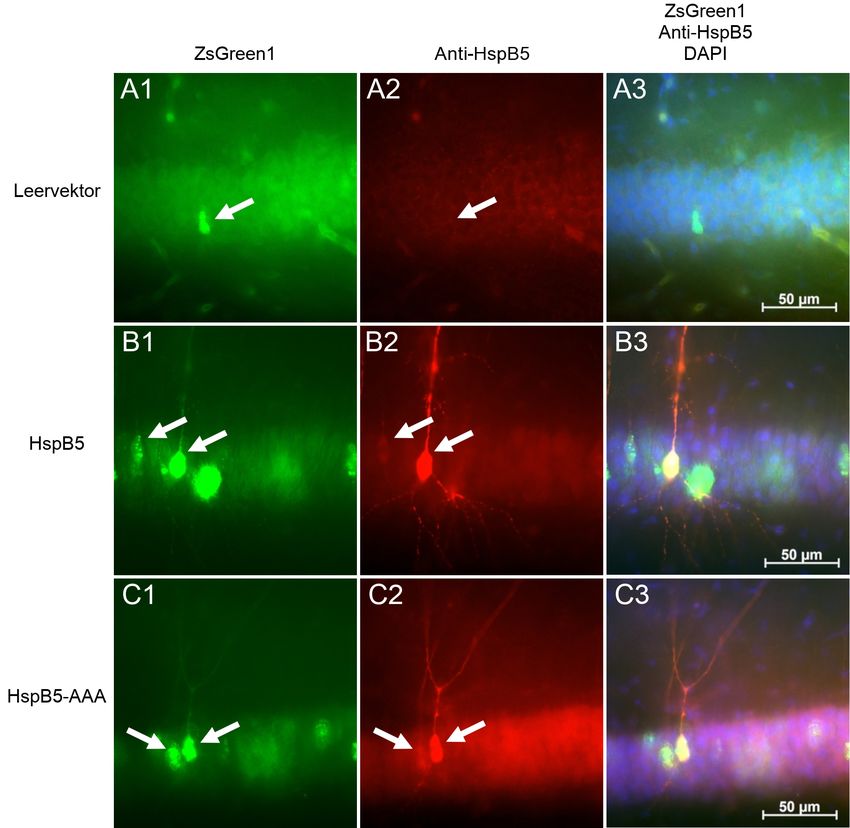
Abkürzungsverzeichnis
Hz Einheit: Hertz, Größe: Frequenz
IRES internal ribosomal entry site
l Einheit: Liter, Größe: Volumen
kg Einheit: Kilogramm, Größe: Masse
LB lysogeny broth
m Einheit: Meter, Größe: Länge
M Einheit: Molar, Größe: Stoffmengenkonzentration
MAP microtubule associated protein
MAPK mitogen-activated protein kinase
MgSO4 Magnesiumsulfat
mol Einheit: Mol, Größe: Stoffmenge
mTOR mammalian target of rapamycin
n Anzahl
NaHCO3 Natriumhydrogencarbonat
NaOH Natriumhydroxid
P Postnatal
p phospho
p-Wert Signifikanzniveau
PBS Phosphatgepufferte Salzlösung
PI3K phosphoinositide-3’ kinase
pLVX lentiviral expression vector
Ras G-Protein Ras
rpm rounds per minute
RT-PCR Real time - Polymerase chain reaction
S Serin
s Einheit: Sekunde, Größe: Zeit
sHsp small heat shock protein - kleines Hitzeschockprotein
Str. gr. Stratum granulosum
Str. mol. Stratum moleculare
viii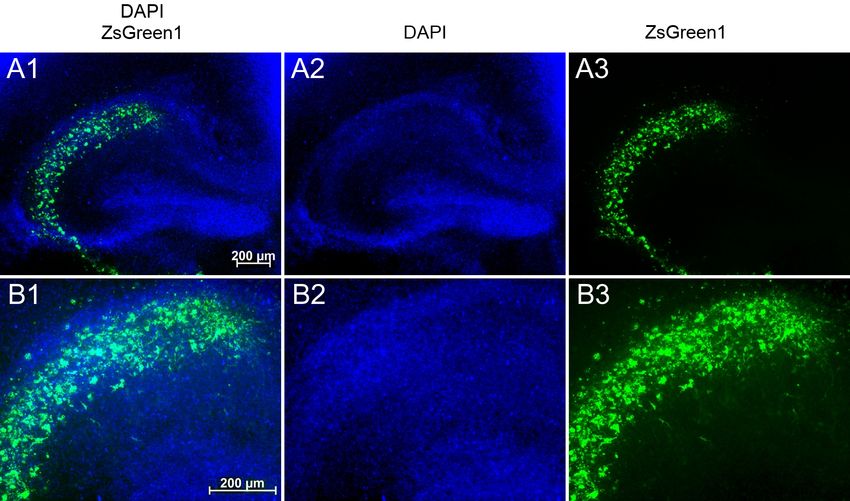
Abkürzungsverzeichnis
Str. or. Stratum oriens
Str. pyr. Stratum pyramidale
Str. rad. Stratum radiatum
TE Puffer TRIS EDTA Puffer
Ultra-LMP-Agarose Ultra-Low Melting Point-Agarose
V Einheit: Volt, Größe: elektrische Spannung
ZsGreen Zoanthus sp. green fluorescent protein
ix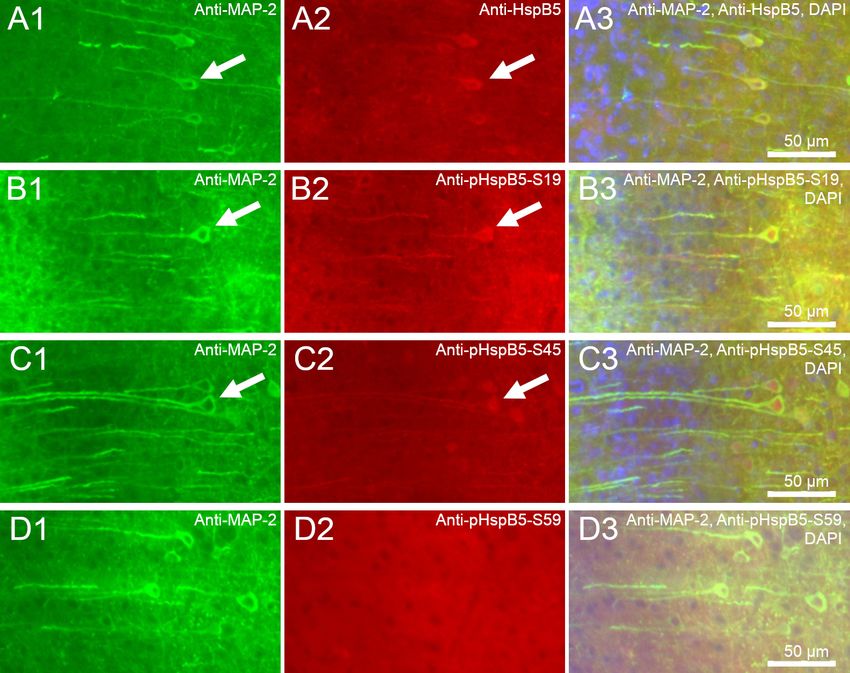
1 Einleitung
1.1 Aufbau des Nervengewebes und des
Hippocampus
1.1.1 Aufbau des Nervengewebes
Das Nervengewebe gliedert sich in Nervenzellen, auch Neurone genannt und Glia-
zellen. Die Neurone erfüllen wichtige Funktionen in der Verarbeitung und Weiter-
leitung von Information. Die Gliazellen sind eine Vielzahl verschiedener Zellen, die
wichtige Funktionen in der Aufrechterhaltung der Zellhomöostase in Nervengewe-
be erfüllen. So bilden sie beispielsweise die Myelinscheiden, die die schnelle Erre-
gungsleitung ermöglichen.
Durch die von Camillo Golgi 1883 entwickelte Silberfärbung von Neuronen konnten
Golgi [27, 22] und Cajal et al. [60] die Neurone, deren Dendritenbäume und ihre
Verzweigungen darstellen. Neurone enthalten ein Zellsoma mit Zellkern, Dendriten
und ein Axon. Die Signalaufnahme erfolgt an den Dendriten, die Signalweiterleitung
über das Axon. Die Dendriten bilden durch eine Vielzahl an Verzweigungen den
Dendritenbaum. Über Synapsen erfolgt die Signalaufnahme. Nach Signalverarbei-
tung erfolgt die Weiterleitung über das Axon, das an dessen Ende mit Synapsen
anderer Neurone verknüpft ist [36].
Das Grundgerüst der Dendriten bilden Mikrotubuli, die Wachstumsprozesse sowie
Stabilität bedingen [6]. Wichtige funktionelle Aspekte des Dendritenbaums sind die
dreidimensionale Ausbreitung durch Größe und Verzweigung und damit einherge-
hend die Anzahl der Synapsen über die Signale aufgenommen werden, sowie die
Anpassung des Dendritenbaums an sich ändernde Umgebungsbedingungen [36].
Wachstumsprozesse der Dendriten sind extrinsisch bspw. durch den Wachstums-
faktor Neurotrophin oder intrinsisch durch spezifische Transkriptionsfakoren, post-
translationale Degradation und weitere Signalwege reguliert [25].
11 Einleitung
1.1.2 Der Hippocampus
Abbildung 1: Schematische Darstellung des Hippocampus. GD: Gyrus denta-
tus, CA1-CA3: Cornu ammonis, Str. lac.-mol.: Stratum lacunosum-moleculare. In
blauen Punkten: Körnerzellen des Gyrus dentatus, in roten Dreiecken: Pyramiden-
zellen der CA4 / CA3-Region mit apikalem und basalem Dendritenbaum, in magenta
Dreiecken: Pyramidenzellen der CA2-Region, in grünen Dreiecken: Pyramidenzel-
len der CA1-Region mit apikalem und basalem Dendritenbaum, blau gepunktete
Linie: Moosfasern, rot gepunktete Linie: Schaffer-Kollateralen.
Der Hippocampus ist im zentralen Nervensystem paarig im Temporallappen gele-
gen, und hat als Teil des limbischen Systems wichtige Funktionen in Lernen und
Gedächtnis. Makroskopisch gliedert sich der Hippocampus in Gyrus dentatus, Cor-
nu ammonis (CA1 - CA4) sowie Subiculum, Präsubiculum, Parasubiculum und en-
torhinalen Kortex (Abbildung 1) [2]. Viele verschiedene Afferenzen sind bekannt.
Einen Hauptteil bilden die Projektionen des entorhinalen Kortex von Neuronen der
Schicht II, die als Tractus perforans in den Gyrus dentatus hineinziehen. Der Gyrus
dentatus bildet eine U-förmige Struktur und untergliedert sich in Stratum moleculare
(Synapsenschicht zwischen Afferenzen und Dendriten der Körnerzellen), Stratum
granulosum (Perikarya von Körnerzellen, Interneurone) und Hilus mit polymorpher
21 Einleitung
Zellschicht (verschiedene Zellen sowie Mooszellen enthaltend). Das Ammonshorn
(Cornu ammonis) gliedert sich in die Regionen CA1 - CA4. Im Stratum pyramida-
le sind die Somata der Pyramidenzellen der jeweiligen CA-Region lokalisiert. Der
basale Dendritenbaum erstreckt sich in das Stratum oriens und der apikale Den-
dritenbaum in das Stratum radiatum und lacunosum-moleculare. Axone der Pyra-
midenzellen der CA3-Region projizieren in die CA3- und CA2-Region sowie in die
CA1-Region. Letztere werden Schaffer-Kollateralen genannt und münden sowohl
im Stratum oriens an basale Dendritenbäume als auch im Stratum pyramidale an
apikale Dendritenbäume [2]. Den Hauptteil der Efferenzen bildet der Fornix über
den die Informationen zum Corpus mammillare weitergegeben werden [2]. In allen
Regionen und Schichten des Hippocampus sind auch eine Vielzahl verschiedener
Interneurone lokalisiert, die wichtige Funktionen in der Verschaltung und Verabei-
tung von Information erfüllen [2].
1.2 Hitzeschockproteine und Zellstress
Die Zelle als Grundbaustein erfüllt eine Vielzahl an Funktionen und ist ständigen
Umbauprozessen und äußeren Einflüssen unterworfen. Es sind eine Vielzahl an
Veränderungen der Umgebung bekannt, die je nach Intensität und Dauer, Zellen,
bspw. Neurone schädigen können. Unter Hitzestress versteht man die Erhöhung
der Temperatur in den für den Organismus unphysiologischen Bereich. Dies führt
zu einer sofortigen Induktion und Hochregulation der Hitzeschockproteine als Ant-
wort auf den Stressor [62, 75]. Neben Hitzestress sind viele andere Stressoren,
beispielsweise verschiedene Schwermetalle, Arsenit, Ethanol, und Hypoxie [65, 56]
bekannt, die ebenfalls eine Hochregulation der Hitzeschockproteine bewirken. Die
Hochregulation auf Proteinebene ist essentieller Bestandteil eines Schutzmechanis-
mus, die Zellfunktion zu erhalten und Apoptose zu verhindern. Die Klassifikation der
Hitzeschockproteine erfolgt anhand ihrer molekularen Größe in “HSPH (HSP110),
HSPC (HSP90), HSPA (HSP70), DNAJ (HSP40) und HSPB (kleine Hitzeschockpro-
teine)“ [38].
31 Einleitung
1.2.1 Die Gruppe der kleinen Hitzeschockproteine, HspBs
Die Gruppe der kleinen Hitzeschockproteine enthält zehn Mitglieder mit einem Mo-
lekulargewicht zwischen 12 bis 43 kDa. Das HUGO Gen Nomenklatur Kommitee
führte eine neue Nomenklatur ein und benannte sie HspB1 bis HspB10 [39, 44].
Ursprünglich wurden sie nach ihrem Molekulargewicht oder dem Gewebe ihrer Ent-
deckung bezeichnet, z.B. HspB1 als Hsp27 oder HspB5 als alpha-B-Crystallin. Al-
len kleinen Hitzeschockproteinen gemeinsam ist die konservierte alpha-Crystallin-
Domäne. C- und N-Terminus sind für jedes Hitzeschockprotein spezifisch und ent-
halten eine variable Anzahl an Aminosäuren. Die Aminosäuren variieren auch etwas
in der alpha-Crystallin-Domäne, bilden jedoch als wichtiges Merkmal beta-Faltblätter
in ihrer Sekundärstruktur aus [28, 43]. Am N-terminalen Ende führen posttransla-
tionale Modifikationen wie beispielsweise Phosphorylierungen zur Ausbildung der
spezifischen oligomeren Struktur der HspBs, die wiederum die Funktion und Loka-
lisation bestimmt [71, 28]. Des Weiteren ist die Ausbildung der Quartärstruktur ab-
hängig von Umgebungsbedingungen. Veränderungen oder auch Interaktionen mit
anderen Proteinen oder Substraten können durch Phosphorylierungen oder Oxidie-
rungen zur Strukturänderung oder auch Stabilisierung führen [15]. Die Ausbildung
der Quartärstruktur der HspBs ist als dynamischer Prozess auf sich ändernde phy-
siologische Bedingungen zu verstehen [66].
Die Gruppe der kleinen Hitzeschockproteine besitzt eine Vielzahl an verschiedenen
Funktionen. Hauptfunktion ist die bekannte Chaperon ähnliche Funktion. HspBs er-
kennen und binden an fehlgefaltete Proteine, verhindern deren Aggregation und De-
naturierung und ermöglichen mithilfe anderer Hsps die Rückfaltung [15]. Des Wei-
teren sind sie an einer Vielzahl zelleigener Prozesse wie dem Zellzyklus, Apoptose
und stressbedingten Anpassungsvorgängen beteiligt [15].
Die kleinen Hitzeschockproteine sind in verschiedenen Geweben des Körpers ex-
primiert. Im Muskelgewebe zeigen HspB1, HspB2, HspB3, HspB5, HspB6, HspB7
und HspB8 eine starke Expression und im Hodengewebe HspB9 und HspB10 [44].
HspB1, HspB5, HspB6 und HspB8 sind im Gehirn in verschiedenen Regionen ex-
41 Einleitung
primiert, allerdings in geringerer Menge verglichen mit anderen Geweben. HspB5
und HspB6 zeigen hier die höchste Expression [44].
1.3 HspB5 / alpha-B-Crystallin
1.3.1 Struktur von HspB5 / alpha-B-Crystallin
HspB5 wird nach seiner Entdeckung in der Augenlinse auch alpha-B-Crystallin ge-
nannt. Hier kommt es in hoher Konzentration vor und trägt zur lebenslangen Erhal-
tung der Transparenz der Augenlinse bei [17]. Später konnte HspB5 in verschiede-
nen anderen Geweben, z.B. in Skelettmuskulatur, Herzmuskulatur, glatten Muskel-
zellen und auch Gliazellen nachgewiesen werden [50] und wurde als Hitzeschock-
protein identifiziert.
Ein HspB5 Monomer enthält eine Sequenz von 175 Aminosäuren [18] und wiegt et-
wa 20 kDa [3]. Man unterscheidet die alpha-Crystallin-Domäne mit etwa 90 Amino-
säuren von jeweils angelagertem N- und C-terminalem Ende. Das C-terminale Ende
enthält ein I-X-I Motiv das die Ausbildung quartärer Strukturen ermöglicht [17, 24].
Am N-terminalen Ende wurden verschiedene Serin Reste identifiziert, die im Rah-
men posttranslationaler Modifikationen phosphoryliert werden können: S19, S45
und S59 [51]. Die Masse der HspB5 Oligomere ist sehr variabel, bei rekombinantem
humanen HspB5 liegt sie zwischen 530 - 684 kDa [3]. HspB5 liegt in dynamischen
Oligomeren vor, die durch den Austausch von Untereinheiten ihre Struktur ändern.
Beispielsweise führen Temperaturveränderungen zu Dissoziationen der Oligomere,
deren Masse in Experimenten zwischen 450 kDa und 600 kDa variiert [31]. Die dy-
namische Struktur ist als Zusammenspiel von Modifikationen in allen drei Domänen
zu verstehen und hat entscheidenden Einfluss auf die Funktionen [24].
1.3.2 Phosphorylierung von HspB5
Entscheidend für die Funktionen von HspB5 ist die Phosphorylierung an den Serin-
Resten S19, S45 und S59. Die Kinase, die S19 phosphoryliert ist noch unklar, S45
51 Einleitung
wird durch p44/42 MAP Kinase und S59 durch MAPKAP Kinase-2 phosphoryliert
[41, 7]. Die Phosphorylierung wird durch Zellstress induziert [32] und verändert
die oligomere Struktur, Größe, Stabilität und Lokalisation mit Auswirkung auf deren
Funktion [12, 7]. Verschiedene zytoprotektive Funktionen sind bekannt die durch
die Phosphorylierung reguliert werden [68, 7], allerdings ist es aktuell noch unklar,
welche Rolle die einzelnen Phosphorylierungsstellen dabei spielen.
1.4 HspB5 im Gehirn
HspB5 konnte in der Untersuchung von adulten Rattengehirnen mittels quantitati-
ver RT-PCR im gesamten Gehirn, Cortex, Cerebellum, Striatum und Hippocampus
nachgewiesen werden [44]. Jedoch ist die Expression im Gehirn deutlich niedriger
als in anderen Geweben, wie z.B. in der Herzmuskulatur [42].
HspB5 wurde zunächst in Gliazellen nachgewiesen [35, 59, 44]. Erst in der Unter-
suchung von Gehirnen mit neurodegenerativen Veränderungen, wurde eine erhöhte
Expression von HspB5 auch in Neuronen nachgewiesen neben der bereits bekann-
ten Lokalisation in Gliazellen [34, 35]. Beispielsweise ist HspB5 in sogenannten „bal-
looned neurons“ überexprimiert [14], dies sind pathologisch veränderte Neurone mit
aufgetriebenem Perikaryon. Diese Befunde deuten darauf hin, dass die Expression
von HspB5 in pathologischen Zuständen hochreguliert wird und auch in Neuronen
stattfindet.
1.4.1 HspB5 bei neurodegenerativen Erkrankungen
Neurodegenerative Erkrankungen bilden eine Gruppe von Erkrankungen, die mit
dem Verlust funktionsfähiger Neurone einhergehen. Dieser Verlust steht in engem
Zusammenhang mit der steigenden Lebenserwartung und damit Alterung in unserer
Gesellschaft. Des Weiteren spielen genetische Faktoren und Umwelteinflüsse eine
wichtige Rolle. Vielen Erkrankungen liegt neben einem Verlust an Hirnsubstanz eine
Proteinfehlfunktion, die Bildung von Proteinaggregaten und Einschlusskörperchen
zugrunde [77].
61 Einleitung
In der Untersuchung von Gehirnen an Morbus Alzheimer erkrankten Patienten konn-
te HspB5 in Astrozyten, Mikroglia und Oligodendrozyten nachgewiesen werden [26,
61]. Die Expression ist um etwa 20-30 % gesteigert und korreliert mit “Ablagerungen
von phosphorylierten Tau Proteinen und Neurofilamentknäueln“ [11].
Weiterhin wird HspB5 in einzelnen Neuronen verstärkt exprimiert, und zwar in so-
genannten „ballooned neurons“. Dabei wird HspB5 und HspB1 in den Somata und
Fortsätzen abgelagert [13, 14].
Die Rolle die HspB5 im Auftreten und Verlauf von neurodegenerativen Erkrankun-
gen spielt, ist aktuell noch nicht vollständig verstanden. Verschiedene zytoprotektive
Funktionen über seine aggregationshemmende [30], anti-inflammatorische und anti-
apoptotische [26] Wirkung sind denkbar. Die weitere Erforschung der Bedeutung
von HspB5 bei neurodegenerativen Erkrankungen nimmt einen wichtigen Stellen-
wert ein und könnte zu neuen therapeutischen Optionen führen.
1.4.2 HspB5 in Neuronen
Neben der Expression von HspB5 in einzelnen Neuronen bei neurodegenerativen
Erkrankungen, konnte HspB5 auch in dissoziierten hippocampalen Neuronenkul-
turen (Ratte) nachgewiesen werden [44]. Bei Hitzeschock oder anderen zellulären
Stressarten wird es hier, wie für ein Hsp typisch, hochreguliert. In diesen kultivierten
Neuronen war HspB5 überwiegend zytoplasmatisch lokalisiert. Interessanterweise
zeigte sich aber eine andere Lokalisation der phosphorylierten Form von HspB5,
und zwar in Dendriten, Axonen und Synapsen [64].
Weitere funktionelle Untersuchungen mittels Überexpressionsexperimenten zeig-
ten, dass HspB5 die Komplexität des Dendritenbaums beeinflusste, nicht aber Axon-
länge oder Synapsendichte [9]. Dieser Effekt auf den Dendritenbaum war phospho-
rylierungsabhängig, da eine nicht-phosphorylierbare Mutante von HspB5 diesen Ef-
fekt nicht zeigte.
Bei neurodegenerativen Erkrankungen kann oft eine Reduktion des Dendritenbaums
der Neurone beobachtet werden. Auch in kultivierten Neuronen lässt sich eine Re-
duktion der dendritischen Komplexität durch zellulären Stress, wie z.B. Hitzeschock
71 Einleitung
modellieren. Interessanterweise kann eine Überexpression von HspB5 solch einer
Hitzeschock-induzierten Rarefizierung der Neurone entgegen wirken. Auch dieser
Effekt zeigte sich abhängig von der Phosphorylierung von HspB5 [9].
Diese Befunde zeigen eine mögliche neue phosphorylierungsabhängige Schutz-
funktion von HspB5 auf den Dendritenbaum von Neuronen unter pathophysiologi-
schen Bedingungen.
1.5 Fragestellung
In dieser Arbeit sollte die Funktion von HspB5 in hippocampalen Neuronen in ihrem
organotypischen Umfeld weiter untersucht werden.
In einem ersten Projektteil wurde an Gehirnschnitten von Mäusen die Lokalisation
von HspB5 und seinen Phosphoformen (pHspB5-S19, pHspB5-S45, pHspB5-S59)
analysiert. Es sollte die Frage geklärt werden, in welchen Zelltypen HspB5 phos-
phoryliert und exprimiert wird und ob die Phosphorylierung seine subzelluläre Lo-
kalisation bestimmt, so wie es in dissoziierten hippocampalen Neuronen der Fall ist.
Dies wurde durch immunhistochemische Darstellung von HspB5 / phosphoHspB5
mit anschließender Fluoreszenzmikroskopie erreicht.
Im zweiten Projektteil sollte ein möglicher phosphorylierungsabhängiger Effekt von
HspB5 auf den Dendritenbaum hippocampaler Pyramidenzellen in Gehirnschnitten
untersucht werden. Dazu musste zunächst die Methode der Überexpression von
HspB5 in hippocampalen Pyramidenzellen der CA1-Region etabliert werden. Mäu-
seembryonen wurden dekapitiert, die entsprechenden Plasmide intraventrikulär inji-
ziert, hippocampale Neurone durch Elektroporation transfiziert und anschließend
organotypische Schnittkulturen angefertigt und über mehrere Tage kultiviert. Nach
Fixierung der Gehirnschnitte folgte die Immunmarkierung von ZsGreen1 zur Dar-
stellung der transfizierten Neurone.
Da diese Methode sich als nicht erfolgreich erwies, wurden die Plasmide HspB5,
die nicht phosphorylierbare Mutante HspB5-AAA und eine Kontrolle in vivo in em-
bryonalen Mäusen an Embryonaltag E15.5 mittels in utero Elektroporation in hippo-
campale Pyramidenzellen der CA1-Region transfiziert. Nach Expression bis Tag 15
81 Einleitung
postnatal erfolgte die Tötung der Embryonen, Entnahme und Fixierung des Gehirns,
Anfertigung von Gehirnschnitten und immunhistochemische Darstellung der Dendri-
tenbäume der transfizierten Neurone. Die Dendritenbäume der transfizierten Pyra-
midenzellen der CA1-Region wurden mit konfokaler Mikroskopie fotografiert und mit
der Imaris-Software analysiert. Es konnte in diesem Versuchssystem in vivo ähnlich
wie in den dissoziierten Neuronenkulturen eine Erhöhung der dendritischen Kom-
plexität durch HspB5, aber nicht durch die nicht phosphorylierbare HspB5-Mutante,
beobachtet werden.
92 Material und Methoden
2.1 Material
2.1.1 Allgemeine Reagenzien
Albumin Fraktion V, Carl Roth, Karlsruhe, Deutschland
Ampicillin Natriumsalz, Carl Roth, Karlsruhe, Deutschland
Basal Medium Eagle, Gibco Thermo Fisher Scientific, Waltham, USA
Calciumchlorid Dihydrat, Carl Roth, Karlsruhe, Deutschland
DAPI, Carl Roth, Karlsruhe, Deutschland
Endotoxin freier TE Puffer, Qiagen, Hilden, Deutschland
Fast Green FCF, Sigma-Aldrich, St. Louis, USA
D-(+)-Glucose, Sigma-Aldrich, Steinheim, Deutschland
L-Glutamin, Merck, Darmstadt, Deutschland
10x HBSS, Gibco Thermo Fisher Scientific, Waltham, USA
HEPES, Carl Roth, Karlsruhe, Deutschland
Horse Serum, Sigma-Aldrich, Steinheim, Deutschland
Kaliumchlorid, Merck, Darmstadt, Deutschland
Kaliumdihydrogenphosphat, Merck, Darmstadt, Deutschland
Kohlendioxid Flüssiggas, Linde, Dublin, Irland
Laminin 1 mg, Sigma-Aldrich, Steinheim, Deutschland
LB Medium (Luria/Miller), Carl Roth, Karlsruhe, Deutschland
Magnesiumsulfat Hydrat, Carl Roth, Karlsruhe, Deutschland
Mowiol 4-88, Merck, Darmstadt, Deutschland
Natriumchlorid, Sigma-Aldrich, Steinheim, Deutschland
Natriumhydrogencarbonat, Carl Roth, Karlsruhe, Deutschland
Natriumhydroxid, Merck, Darmstadt, Deutschland
di-Natriumhydrogenphosphat, Carl Roth, Karlsruhe, Deutschland
Paraformaldehyd, Merck, Darmstadt, Deutschland
102 Material und Methoden
Penicillin (10,000 Units/ml) / Streptomycin (10000 µg/ml), Gibco Thermo Fisher Scien-
tific, Waltham, USA
Poly-L Lysin, Sigma-Aldrich, Steinheim, Deutschland
Propan-2-ol, VWR Chemicals, Radnor, USA
D-(+)-Saccharose, Carl Roth, Karlsruhe, Deutschland
TopVision Low Melting Point Agarose, Thermo Fisher Scientific, Waltham, USA
Triton X-100, Merck, Darmstadt, Deutschland
UltraPure LMP Agarose, Invitrogen, Carlsbad, USA
2.1.2 Antikörper
Primärantikörper
Kaninchen anti-alphaB-Crystallin / HspB5 IgG (Anti-HspB5), monoklonal (Klon EPR2752),
GeneTex, Produktnummer: GTX62094
Kaninchen anti-phosphoSerin19-alphaB-Crystallin / HspB5 IgG (Anti-pHspB5-S19),
polyklonal, Enzo, Produktnummer: ADI-SPA-225
Kaninchen anti-phosphoSerin45-alphaB-Crystallin / HspB5 IgG (Anti-pHspB5-S45),
polyklonal, Enzo, Produktnummer: ADI-SPA-226
Kaninchen anti-phosphoSerin59-alphaB-Crystallin / HspB5 IgG (Anti-pHspB5-S59),
polyklonal, Enzo, Produktnummer: ADI-SPA-227
Kaninchen anti-ZsGreen1 IgG, polyklonal, Clontech, Produktnummer: 632474
Maus anti-MAP2 IgG1, monoklonal (Klon AP20), Millipore, Produktnummer: 2332553
Sekundärantikörper
Ziege anti-Maus IgG, Alexa Fluor 488, Thermo Fisher Scientific, Produktnummer:
A-11001
Ziege anti-Kaninchen IgG, Alexa Fluor 488, Thermo Fisher Scientific, Produktnum-
mer: A-11008
Ziege anti-Maus IgG, Alexa Fluor 568, Thermo Fisher Scientific, Produktnummer:
A-11031
112 Material und Methoden
Ziege anti-Kaninchen IgG, Alexa Fluor 568,Thermo Fisher Scientific, Produktnum-
mer: A-11036
2.1.3 Verbrauchsmaterial
Ampuwa Spüllösung 1000 ml Plastipur, Fresenius Kabi, Bad Homburg, Deutschland
Becherglas, Schott, Mainz, Deutschland
Borosilikat Glaskapillaren, World Precision Instruments, Sarasota, USA
Cellstar Petrischalen 6 cm, Greiner Bio-One, Kremsmünster, Österreich
Cellstar Petrischalen 12 cm, Greiner Bio-One, Kremsmünster, Österreich
Cellstar Serologische Pipette 25 ml, Greiner Bio-One, Kremsmünster, Österreich
Cellstar Serologische Pipette 10 ml, Greiner Bio-One, Kremsmünster, Österreich
Cellstar Serologische Pipette 5 ml, Greiner Bio-One, Kremsmünster, Österreich
Deckgläser Ø18 mm, Thermo Fisher Scientific, Waltham, USA
Einbettungs Form, Polysciences, Warrington, USA
Einkomponenten Sekundenkleber, Carl Roth, Karlsruhe, Deutschland
Einmalskalpell Cutifix Größe 11, B. Braun, Melsungen, Deutschland
Falcon 15 ml, 50 ml, Sarstedt, Nümbrecht, Deutschland
Feindosierungsspritze mit Luer-Ansatz 1 ml, B. Braun, Melsungen, Deutschland
Multiwell 6 Well, Corning Life Sciences, New York, USA
Multiwell 12 Well, Greiner Bio-One, Kremsmünster, Österreich
Nahtmaterial Perma-Hand Seide nicht resorbierbar 5-0, Ethicon, Somerville, USA
Objektträger, VWR, Radnor, USA
Parafilm M, Bemis, Neenah, USA
Pipettenspitzen (0,1 - 10 µl), Sarstedt, Nümbrecht, Deutschland
Pipettenspitzen (2 - 200 µl), Sarstedt, Nümbrecht, Deutschland
Pipettenspitzen (1000 µl), Sarstedt, Nümbrecht, Deutschland
Reaktionsgefäße 0,5 - 2,0 ml, Eppendorf, Hamburg, Deutschland
Rundfilter Ø90 mm, Schleicher & Schuell, Dassel, Deutschland
Saugrohr für Mikrokapillaren, Sigma-Aldrich, St. Louis, USA
Spritzenvorsatzfilter 0.20 µm, Sarstedt, Nümbrecht, Deutschland
122 Material und Methoden
Spritze 50 ml mit Luer-Lock Anschluss, Becton Dickinson, Drogheda, Irland
Standard Glas Kapillare, World Precision Instruments, Hertfordshire, England
Sterican Einmalkanüle 20 G x 1 “, B. Braun, Melsungen, Deutschland
TC-Schale 100 Standard, Sarstedt, Nümbrecht, Deutschland
Wilkinson Sword Classic Rasierklinge, Wilkinson Sword, High Wycombe, England
Zellkultureinsatz 0,4 µm 6-Wellformat, Corning Life Sciences, New York, USA
Zentrifugen Flasche Polycarbonat mit Deckel, Beckman Coulter, Brea, USA
2.1.4 Geräte
Abzug HLB 2448 GS, LaminAir Heraeus, Hanau, Deutschland
Axioskop 2 MOT Plus, Carl Zeiss, Oberkochen, Deutschland
Binokulares Mikroskop, Carl Zeiss, Oberkochen, Deutschland
Cytation 3, BioTek, Winooski, Vermont, USA
DNA Speed Vac Konzentrator DNA110, Thermo Fisher Scientific, Waltham, USA
Eismaschine, Ziegra Eismaschinen, Isenhagen, Deutschland
Elektroporator CUY21EDIT, Nepagene, Chiba, Japan
Fluovac Absorber, Harvard Apparatus, Holliston, USA
Galaxy 170S CO2 -Inkubator, Eppendorf, Hamburg, Deutschland
Gefrierschrank -20 ◦ C, Liebherr, Bulle, Schweiz
Gefrierschrank -80 ◦ C, Sanyo, Osaka, Japan
Gefrierschrank -80 ◦ C, Thermo Fisher Scientific, Waltham, USA
Heizplatte, Medax Nagel, Kiel, Deutschland
Isofluran Verdampfer, Harvard Apparatus, Holliston, USA
Kälteschrank, Nalge Nunc International, Rochester, New York, USA
Konfokales Mikroskop LSM 710 NLO mit Plan-Apochromat 20x/0,8 M27, Carl Zeiss,
Oberkochen, Deutschland
Kreisschüttler IKA KS260 basic, IKA-Werke, Staufen, Deutschland
Kühlschrank 4 ◦ C, Liebherr, Bulle, Schweiz
Magnetrührer mit Heizplatte MR 3001 K, Heidolph, Schwabach, Deutschland
Mikropipettenzieher P-97, Sutter Instrument, Novato, USA
132 Material und Methoden
Mikroschere, Fine Science Tools, Heidelberg, Deutschland
Mikrowelle, Bosch, Stuttgart, Deutschland
Mikrozentrifuge Biofuge pico Heraeus, Hanau, Deutschland
NanoDrop 2000 Spectrophotometer, Thermo Fisher Scientific, Waltham, USA
pH-Meter FiveEasy, Mettler Toledo, Columbus, USA
Picospritzer III, Parker Hannifin, Cleveland, USA
Pipetboy, Hirschmann, Eberstadt, Deutschland
Pipetten research plus P1-P1000, Eppendorf, Hamburg, Deutschland
Pinzette, Fine Science Tools, Heidelberg, Deutschland
Pinzette mit runden Elektroden Ø5 mm, Nepagene, Chiba, Japan
Präparierschere, Fine Science Tools, Heidelberg, Deutschland
Präparationstisch, Eigenbau der Universität Ulm, Ulm, Deutschland
Präzisionswaage CP4202S, Sartorius, Göttingen, Deutschland
Schleifgerät für Mikropipetten EG-44, Narishige, Tokio, Japan
Schüttler IKA-VIBRAX-VXR, IKA-Werke, Staufen, Deutschland
Spatel, Fine Science Tools, Heidelberg, Deutschland
Temperatur Kontroll Einheit HB 101/2, Panlab Harvard Apparatus, Holliston, USA
Thermo Heraeus BBD 6220 CO2 -Inkubator, Heraeus, Hanau, Deutschland
Thermomixer compact, Eppendorf, Hamburg, Deutschland
Vibratom Microm HM650 V, Thermo Fisher Scientific, Waltham, USA
Vortex-Genie 2, Scientific Industries, New York, USA
Wärmebad Microm SB 80, Walldorf, Deutschland
Wärmeschrank, Binder, Tuttlingen, Deutschland
Wasserbad, Grant Instruments, Shepreth, England
Wasserfilter Millipore Q-POD, Merck Millipore, Billerica, USA
Zentrifuge Avanti J-25, Breckman Coulter, Brea, USA
2.1.5 Kit
Endotoxin-freie Plasmid DNA Aufreinigung NucleoBond Xtra Midi EF, Macherey-
Nagel, Düren, Deutschland, Produktnummer: 740420.50
142 Material und Methoden
2.1.6 Plasmide
pLVX-IRES-ZsGreen1, Clontech Laboratories, Mountain View, USA, Produktnum-
mer: 632187
pLVX-IRES-ZsGreen1-HspB5, lag in der Arbeitsgruppe Golenhofen vor [9]
pLVX-IRES-ZsGreen1-HspB5-AAA, lag in der Arbeitsgruppe Golenhofen vor [9]
Das Plasmid pLVX-IRES-ZsGreen1 enthält ausschließlich ZsGreen1 und wird im
Folgenden zur Vereinfachung als “Leervektor“ bezeichnet. Die verwendeten Plas-
mide enthalten die HspB5 bzw. HspB5-AAA DNA, die in den Klonierungsabschnitt
eingebaut ist. HspB5-AAA ist die unphosphorylierbare Mutante, die an den Res-
ten S19, S45, S59 nicht phosphoryliert vorliegt. Weitere Sequenzen sind ein IRES
Abschnitt mit folgendem ZsGreen1 Abschnitt. Dies ermöglichte die unabhängige
Koexpression von HspB5, etc. und ZsGreen1, einem fluoreszierenden Protein der
Spezies Zoanthus sp. [52, 49].
2.1.7 Tiertötungen und Tierversuche
Alle Tiertötungen der Projekte “Lokalisation von HspB5 in Gehirnen der Maus“ und
“ex utero Elektroporation“ wurden in Übereinstimmung mit den Richtlinien der Tierschutz-
Versuchstierverordnung durch Dr. Britta Bartelt-Kirbach (Arbeitsgruppe Golenhofen,
Universität Ulm) an Mäusen der Linie C57BL6J von Janvier Labs, Le Genest-Saint-
Isle, Frankreich durchgeführt und durch das Regierungspräsidium Tübingen in dem
Hauptantrag Reg. O.103-8 genehmigt.
Alle Tierversuche des Projekts “in utero Elektroporation“ wurden nach den Richtlini-
en der Tierschutz-Versuchstierverordnung durchgeführt und durch das Regierungs-
präsidium Tübingen in dem Hauptantrag 1197 genehmigt. Die Durchführung der
Experimente erfolgte nach dem Ausbildungsantrag Reg. Nr. II. 193 / Arbeitsgruppe
Britsch durch Dr. Christoph Wiegreffe (Arbeitsgruppe Prof. Dr. Britsch, Universität
Ulm) und Dr. Britta Bartelt-Kirbach an Mäusen der Linie C57BL6J von Janvier Labs,
Le Genest-Saint-Isle, Frankreich.
152 Material und Methoden
2.1.8 Software
AutoQuant X3, Media Cybernetics, Cambridge, England
AxioVision 40, Carl Zeiss, Oberkochen, Deutschland
Imaris, Bitplane, Zürich, Schweiz
Zen 2010, Carl Zeiss, Oberkochen, Deutschland
2.2 Molekularbiologische Methoden
2.2.1 Plasmidpräparation
Zur Herstellung des Bakterienmediums wurden 16 g LB Medium in 800 ml Wasser
gelöst, autoklaviert und anschließend bei 4 ◦ C gekühlt gelagert. Vor Verwendung
des Bakterienmediums wurde Ampicillin (1 : 1000) hinzugefügt.
Die verwendeten bakteriellen Plasmide pLVX-IRES-ZsGreen1 Leervektor, pLVX-
IRES-ZsGreen1-HspB5 und pLVX-IRES-ZsGreen1-HspB5-AAA lagen in der Arbeits-
gruppe bereits vor. Mit einer sauberen Pipettenspitze wurde eine Probe des jeweili-
gen Plasmidstocks entnommen und in einem Erlenmeyerkolben in 100 ml Bakterien-
medium bei 37 ◦ C im Brutschrank unter ständigem Schütteln über Nacht inkubiert.
Am nächsten Morgen wurde die Bakterienlösung bei 15000 g, 4 ◦ C über 10 Minuten
zentrifugiert und das Pellet gesammelt. Das Pellet wurde mit dem Kit NucleoBond
Xtra Midi EF nach Protokoll des Herstellers endotoxinfrei aufgereinigt, um das Plas-
mid frei von anderen zellulären Bestandteilen zu erhalten. Als erster Schritt wurde
das Pellet in 8 ml Resuspensionspuffer, enthaltend RNase A, resuspendiert, bis
es vollständig gelöst war. Anschließend wurde 8 ml Lysepuffer hinzugegeben, fünf-
mal invertiert und nachfolgend die Lösung 5 Minuten bei Raumtemperatur inkubiert.
Währenddessen wurde der Rand der Säule mit 15 ml Äquilibrierpuffer befeuchtet.
Danach wurden 8 ml Neutralisationspuffer zur Lösung hinzugefügt und invertiert,
bis die nun blaue Lösung farblos wurde. Es folgten erneute 5 Minuten Inkubations-
zeit auf Eis. Durch dreimaliges Invertieren der Lösung erhielt man ein homogenes
Gemisch und befüllte damit die Säulen. Anschließend folgten drei Waschschritte.
162 Material und Methoden
Im ersten Schritt wurde die Säule mit 5 ml Filter-Waschpuffer gewaschen; nachdem
dieser Schritt beendet war, wurde der Säulenfilter entfernt. Im zweiten Waschschritt
wurde die Säule mit 35 ml Säulenwaschpuffer gewaschen. Als dritter und letzter
Waschschritt wurde die Säule mit 15 ml Waschpuffer gewaschen. Danach wurde
die Säule zur Elution mit 5 ml Elutionspuffer gewaschen, die Plasmidprobe wurde
in ein Zentrifugenröhrchen gewonnen. Zur Ausfällung wurden 3,5 ml Isopropanol
bei Raumtemperatur hinzugefügt; die Probe wurde geschüttelt und nach Hinzufü-
gen von 2 ml 70 % endotoxinfreiem Ethanol bei Raumtemperatur und 15000 g 5
Minuten zentrifugiert. Anschließend wurde durch vorsichtiges Auskippen das Etha-
nol aus dem Zentrifugenröhrchen entfernt. Die Probe wurde für ein paar Minuten bei
Raumtemperatur getrocknet, sodass das verbleibende Ethanol verdunsten konnte.
Als letzter Schritt wurde das verbleibende Pellet in etwa 50 bis 100 µl endotoxin-
freiem Wasser gelöst und die DNA Konzentration mit dem NanoDrop Photometer
bestimmt.
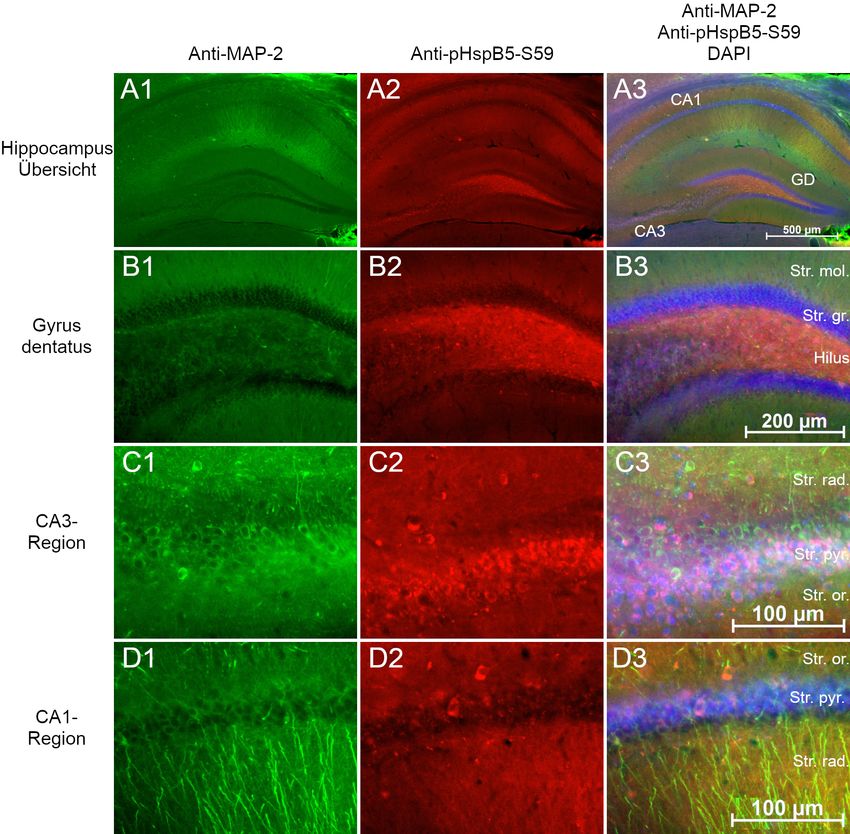
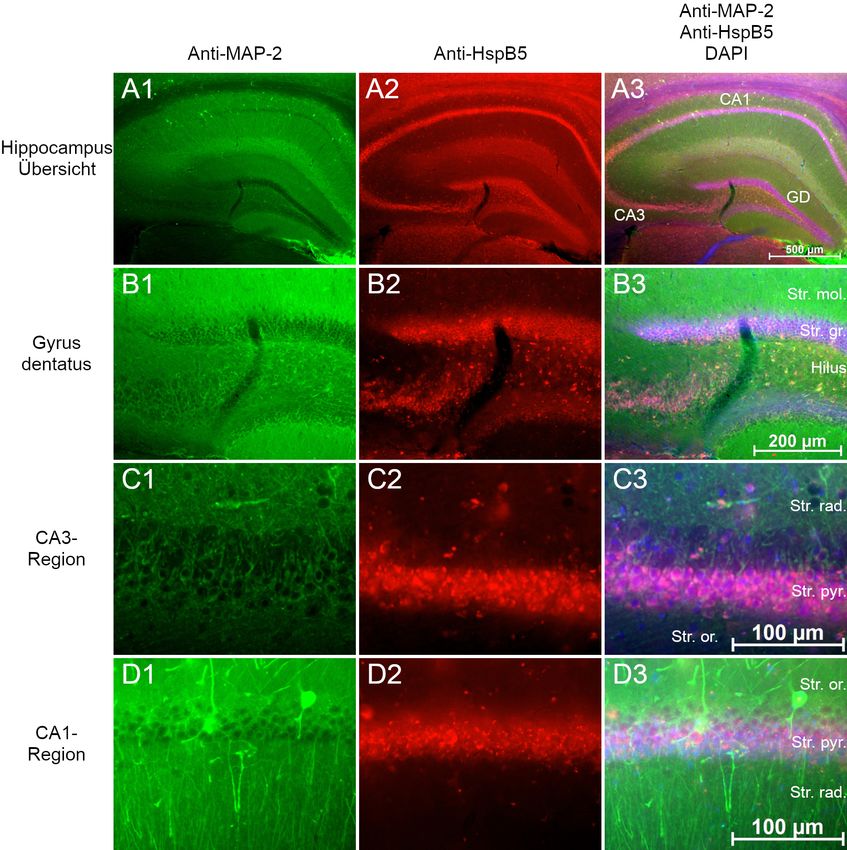
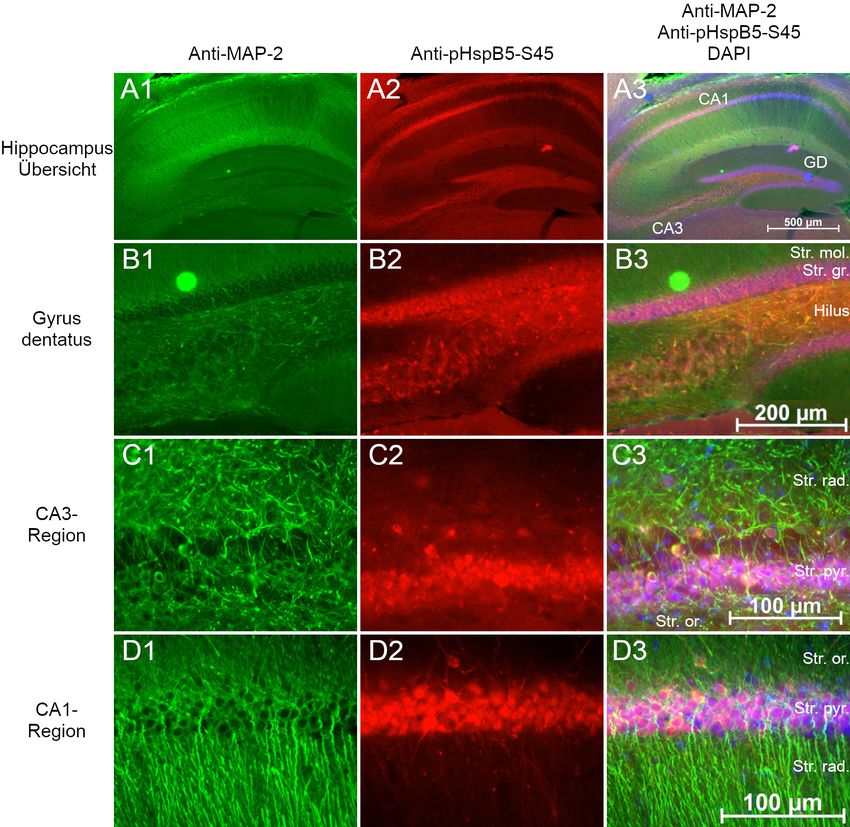
2.3 Lokalisation von HspB5 in Gehirnen der Maus
In diesem Projekt sollte die Lokalisation von HspB5 und seiner Phosphoformen
in Gehirnen der Maus mittels Immunmarkierung untersucht werden. Dazu wurden
adulte Mäuse der Linie C57BL6J mittels Kohlenstoffdioxidinhalation und folgender
Dekapitation getötet und die Gehirne aus der Schädelkalotte herauspräpariert. Die
Kalotte wurde mit einem medianen Schnitt eröffnet und anschließend das Gehirn
freipräpariert. Die entnommenen Gehirne wurden in PBS gewaschen und anschlie-
ßend in Fixierlösung (siehe Tabelle 6) über Nacht bei 4 ◦ C fixiert, am nächsten
Morgen mit PBS gewaschen und bei 4 ◦ C in PBS gelagert.
Die Anfertigung von frontalen und transversalen Gehirnschnitten erfolgte mit dem
Vibratom. Für die Herstellung der frontalen Gehirnschnitte wurde mit einer Rasier-
klinge in frontaler Schnittführung am rostralen Ende 3-4 mm Gehirnsubstanz ab-
geschnitten, sodass eine glatte Fläche entstand. Mit Sekundenkleber wurde das
Gehirn auf dem Schnittstempel so fixiert, dass nun kaudale Anteile nach oben zeig-
ten und rostrale Anteile nach unten. Das Schneiden erfolgte in gekühltem PBS. Für
172 Material und Methoden
die Herstellung der transversalen Schnitte wurden die Gehirne mit ihrer kaudalen
Fläche mit Sekundenkleber auf dem Schnittstempel fixiert. Die Gehirnschnitte, die
hippocampale Anteile enthielten, wurden mit folgenden Einstellungen hergestellt:
Schnittdicke: 100 µm; Frequenz: 60; Amplitude 0,7; Schnittgeschwindigkeit: 15-17.
Die Schnitte wurden in 6-Well-Platten mit PBS transferiert und dreimal in PBS unter
Schütteln bei 4 ◦ C gewaschen. Die Lagerung erfolgte bei 4 ◦ C unter Parafilmver-
schluss.
Die folgende Immundoppelmarkierung erfolgte wie in “Immunmarkierung von Ge-
hirnschnitten“ beschrieben. Immunmarkiert wurde HspB5, die Phosphoformen pHspB5-
S19, pHspB5-S45 und pHspB5-S59 in der Kombination mit MAP2. Die verwendeten
Antikörperkonzentrationen sind in Tabelle 9 aufgelistet. Zur Kontrolle wurde eine Im-
munmarkierung nur mit Sekundärantikörper durchgeführt.
Die Schnitte wurden mittels Fluoreszenzmikroskopie fotografiert und in AxioVision
dargestellt. Die Einstellungen Helligkeit und Kontrast wurden manuell optimiert.
2.4 Transfektion von hippocampalen Neuronen
mittels Elektroporation
Die Zielsetzung im Rahmen des Projekts bestand darin, hippocampale Neurone in
organotypischen Schnitten zu transfizieren und zu kultivieren. In Vorversuchen der
Arbeitsgruppe Golenhofen wurde versucht, nach Anfertigung von hippocampalen
Schnitten diese mittels eines viralen Transfektionssystems zu transduzieren. Aller-
dings war eine erfolgreiche Transduktion nicht möglich, da die Schnitte während der
Kultivierung sehr schnell eine Glianarbe bildeten.
Die Methode der ex utero Elektroporation bietet die Möglichkeit Zielregionen in Ge-
hirnen embryonaler Mäuse zu transfizieren und nachfolgend Gehirnschnitte anzu-
fertigen und zu kultivieren. Diese Methode wurde von Venkataramanappa et al. [72]
in der Arbeitsgruppe Prof. Dr. Britsch zur Untersuchung des Hippocampus etabliert.
In einem Kooperationsprojekt wurde die Methode erlernt und sollte auf die Untersu-
chung des Dendritenbaums hippocampaler Pyramidenzellen der CA1-Region über-
182 Material und Methoden
tragen werden. Dazu wurden trächtige Mäuse der Linie C57BL6J je nach Versuch
an den Tagen E13.5 - E18.5 oder deren Nachkommen postnatal an Tag P1 - P2
getötet, und die Embryonen bzw. die Nachkommen dekapitiert. Durch Injektion der
Plasmidlösung in den Seitenventrikel und anschließende Abgabe von Stromimpul-
sen mittels Elektroden wurde das Plasmid nach intrazellulär aufgenommen und die
Zielzellen transfiziert.
Die methodische Durchführung sowie Herstellung der Lösungen erfolgte adaptiert
nach Protokoll von F. Polleux et al. [58].
2.4.1 Beschichtung der Zellkultureinsätze
Lamininlösung
Lamininlösung wurde zu einer Konzentration von 1 mg/ml mit sterilem H2 O unter
sterilen Bedingungen angesetzt, zu jeweils 100 µl aliquotiert und bei -80 ◦ C gelagert.
Poly-L-Lysinlösung
Poly-L-Lysinlösung wurde zu einer Konzentration von 1 mg/ml mit sterilem Wasser
unter sterilen Bedingungen angesetzt, zu jeweils 1 ml aliquotiert und bei -20 ◦ C
gelagert.
Für die Beschichtung der Zellkultureinsätze wurden diese unter sterilen Bedingun-
gen in eine 6-Well-Platte positioniert. Jeweils 1 ml Poly-L-Lysin Lösung (1 mg/ml)
und 100 µl Laminin-Lösung (1 mg/ml) wurden in 12 ml sterilem Wasser gelöst. Je-
weils 2 ml steriles Wasser wurde in die 6-Well-Platte unter die Membraninserts pipet-
tiert und anschließend jeweils 1 ml der vorbereiteten Poly-L-Lysinlösung / Laminin-
Lösung auf jedes Membraninsert pipettiert. Die 6-Well-Platten wurden mit Parafilm
luftdicht verschlossen und anschließend über Nacht im Zellkulturinkubator inkubiert.
Am nächsten Morgen wurde unter sterilen Bedingungen die Poly-L-Lysinlösung /
Laminin-Lösung abpipettiert, die Membraninserts jeweils drei Mal mit sterilem Was-
ser gewaschen und anschließend getrocknet. Die getrockneten Membraninserts
konnten in 6-Well-Platten unter Parafilm Verschluss bei 4 ◦ C mehrere Wochen gela-
gert werden.
192 Material und Methoden
Vor Versuchsbeginn wurde die 6-Well-Platte, passend zu den Membraninserts vor-
bereitet. In jedes Well wurde 1,8 ml Kulturmedium pipettiert und anschließend eine
Membraninsert in die vorgesehene Verankerung eingepasst, sodass die Membran-
inserts von der Unterseite her mit Kulturmedium befeuchtet wurden. Während des
Versuchs erfolgte die Inkubation bei 37 ◦ C und 5 % Kohlenstoffdioxid / 95 % Luft.
2.4.2 Herstellen der Ultra LowMeltingPoint-Agarose
Die Ultra-LMP-Agarose wurde in einer Endkonzentration von 4 % in Complete HBSS
angesetzt. Anschließend wurde die Lösung in der Mikrowelle vorsichtig erhitzt. Durch
mehrmaliges Unterbrechen dieses Vorgangs und Umschwenken der Lösung konnte
der Siedepunkt sehr exakt erreicht werden und durch wiederholtes, kurzes Sieden
wurde die flüssige, gelartige Konsistenz erreicht. Durch geschlossenes Aufbewah-
ren bei 4 ◦ C konnte die Ultra-LMP-Agarose mehrmals wiederverwendet werden. Vor
Versuchsbeginn wurde der flüssige Zustand der Ultra-LMP-Agarose durch vorsichti-
ges Erhitzen in der Mikrowelle erreicht und durch Lagerung in einem Wärmeschrank
bei 42 ◦ C gewährleistet.
Tabelle 1: „HBSS complete“-Puffer (Complete Hank’s Balanced Salt Solution).
Zu der Lösung wurde destilliertes Wasser zu einem Gesamtvolumen von
500 ml hinzugefügt. Danach wurde die Lösung unter dem Abzug durch
einen 0.2 µm Filter steril filtriert. Die Aufbewahrung erfolgte bei 4 ◦ C.
Chemikalie Menge Endkonzentration
10 x HBSS 50 ml 1x
1 M HEPES (pH=7.4) 1.25 ml 2.5 mM
1 M D-Glukose 15 ml 30 mM
100 mM CaCl2 5 ml 1 mM
100 mM MgSO4 5 ml 1 mM
1 M NaHCO3 2 ml 4 mM
2.4.3 Ansetzen des Injektions-Plasmids
Fast Green wurde in endotoxinfreiem TE Puffer gelöst (0,1 %). Das aufgereinigte
endotoxinfreie Plasmid wurde mit endotoxinfreiem Wasser und Fast Green (End-
202 Material und Methoden
konzentration 0,01 %) zu der verwendeten Endkonzentration 500 ng/µl - 3000 ng/µl
verdünnt. Das Aufziehen in die Injektionspipetten erfolgte mittels eines Saugrohrs
für Mikrokapillaren.
2.4.4 Ausziehen und Schleifen der Injektionspipetten
Die Herstellung der Injektionspipetten erfolgte durch die Arbeitsgruppe Prof. Dr.
Britsch. Die verwendeten Kapillaren mit einem Durchmesser von 1,0 mm wurden
in einen Mikropipettenzieher eingespannt. Durch Erhitzen des Mittelpunkts der Ka-
pillare mit gleichzeitigem Zug an beiden Enden wurde jeweils eine spitze Injektions-
pipette mit einer Länge von 5,5 cm hergestellt. Folgende Einstellungen wurden ver-
wendet: Hitze: 540, Zug: 125, Geschwindigkeit: 20, Verzögerung 140. Anschließend
wurde die Spitze der Mikropipetten über eine Länge von 4 mm schräg angeschliffen,
sodass eine feine Injektionsspitze entstand. Anhand einer Vorlage wurden die Na-
deln in regelmäßigen Abständen markiert, sodass ein Abschnitt etwa 1 µl Volumen
entsprach. Die Lagerung erfolgte in einer Petrischale.
2.4.5 Transfektion und Elektroporation
Das trächtige Muttertier wurde je nach Experiment zwischen Embryonaltag E15.5
und Embryonaltag E18.5 mit Kohlenstoffdioxid narkotisiert und dekapitiert. Mit ei-
nem Longitudinalschnitt wurde die Bauchdecke eröffnet und der Uterus herausprä-
pariert. Der Uterus und folgend die Embryonen wurden in einer Petrischale mit
“HBSS complete“-Puffer auf Eis gekühlt gelagert. Die einzelnen Embryonen wur-
den herauspräpariert und mit einem zum Rumpf leicht schrägen Schnitt dekapitiert.
Die Tiere der Tage postnatal P0, P1 und P2 wurden ebenfalls dekapitiert und in
einer Petrischale mit “HBSS complete“-Puffer auf Eis gekühlt gelagert.
Es wurde jeweils ein Mauskopf auf eine Petrischale gelegt und ein leicht gebogener
Löffelspatel an die gegenüberliegende linke Hemisphäre als Gegengewicht ange-
legt und so fixiert. In die rechte Hemisphäre wurde die Injektionskapillare in einem
◦
45 Winkel von oben etwa 1 bis 1,5 mm tief durch die Kalotte in den rechten Sei-
212 Material und Methoden
Tabelle 2: Getestete Bedingungen zur Methodenoptimierung für die Transfek-
tion von pLVX-IRES-ZsGreen1 Leervektor mittels ex utero Elektro-
poration und folgender Überexpression in hippocampalen Pyrami-
denzellen
Zeitpunkte der Transfektion E13.5, E14.5, E15.5, E16.5, E18.5, P0, P1, P2
Plasmidkonzentrationen 500, 750, 1000, 1500, 3000
(ng/µl)
Schnittdicken (µm) 250, 300
Dauer der Kultivierung 2 Tage -17 Tage
tenventrikel eingeführt. Mithilfe des Fußtasters wurde der Picospritzer betätigt und
mit Pulsen von 20 ms Dauer 2 bis 3 µl Plasmid-Lösung injiziert.
Um das Gehirn zu elektroporieren wurde es mit wenigen Tropfen “HBSS complete“-
Puffer befeuchtet und die Elektroden folgendermaßen angelegt: die Minus-Elektrode
wurde Im Bereich des Kortex der injizierten rechten Seite angelegt und die Plus-
Elektrode unterhalb des Ohrs der gegenüberliegenden Seite. Durch leichten Druck
und “HBSS complete“-Puffer wurde die Leitfähigkeit gewährleistet. Mit Hilfe des
Fußtasters wurde der Elektroporator bedient und das Gehirn elektroporiert. Nach
Ende der Elektroporation wurde die vom Gerät angegebene gemessene Stromstär-
ke kontrolliert. Sie sollte 0,1 A > I > 0,05 A betragen.
Tabelle 3: Verwendete Einstellungen des Elektroporators zur ex utero Elektro-
poration
Parameter Einheit
Spannung 50 V
Anzahl Pulse 5
Zeit Puls “on“ 50 ms
Zeit Puls “off“ 950 ms
Nach Fertigstellung der Elektroporation wurden unter dem binokularen Mikroskop
die Gehirne aus dem Schädel herauspräpariert. Dazu wurde mit einer Präzisions-
schere die Kalotte von kaudal in einem mittigen Sagittalschnitt eröffnet und mithilfe
einer Präzisonspinzette weggebrochen. Anschließend wurde das Gehirn von rostral
222 Material und Methoden
Abbildung 2: Injektion und Elektroporation A Injektion von 2 bis 3 µl Plasmid-
Lösung in den rechten Seitenventrikel mit einer Injektionspipette. Anhand des blau-
en Farbstoffs in der Plasmid-Lösung konnte die korrekte intraventrikuläre Injektion
kontrolliert werden. B Positionierung der Elektroden. Die Minus-Elektrode wurde im
Bereich des Kortex der injizierten rechten Seite angelegt und die Plus-Elektrode
unterhalb des Ohrs der gegenüberliegenden Seite. Es folgte die Elektroporation.
nach kaudal mithilfe einer gebogenen Pinzette unterminiert und von der Schädel-
basis gelöst. Die freipräparierten Gehirne wurden mithilfe von Filterpapier an den
Rändern vorsichtig “getrocknet“ und anschließend in Einbettformen in flüssige 42
◦
C warme Ultra-LMP-Agarose eingelegt. Das Gehirn wurde mithilfe einer Kanüle
so positioniert, dass der rostrale Anteil mit Bulbus olfactorius nach oben zeigte und
kaudale Anteile mit Kleinhirn nach unten. Nach Erreichen der gewünschten Positi-
on wurde die Einbettform auf Eis gelegt, sodass die Ultra-LMP-Agarose anhärten
konnte. Dies dauerte etwa 5 Minuten.
2.4.6 Anfertigen von Schnittkulturen des Gehirns
10 % Formaldehyd (FA)-Lösung
10 g Paraformaldehyd wurden mit PBS in einem Gesamtvolumen von 100 ml gelöst
und unter Umrühren auf 60 ◦ C erhitzt. Nachfolgend wurde 1 M NaOH tropfenwei-
se hinzugegeben, bis die Flüssigkeit klar wurde. Der pH=7,4 wurde anschließend
kontrolliert und die Lösung abgekühlt. Die Lagerung erfolgte bei -20 ◦ C.
232 Material und Methoden
Tabelle 4: Kulturmedium. Die Lösung wurde unter dem Abzug durch einen 0.2 µm
Filter steril filtriert. Danach wurde hitzeinaktiviertes Pferdeserum zu einer
Gesamtkonzentration von 5 % hinzugefügt. Die Lagerung erfolgte bei 4
◦
C.
Chemikalie Menge für 50 ml Endkonzentration
Basal Medium Eagle 35 ml
Complete HBSS 12.9 ml
1 M D-Glukose 1.35 ml 20 mM
200 mM L-Glutamin 0.25 ml 1 mM
Penicillin / Streptomycin 0.5 ml Penicillin: 100 Einhei-
ten/ml;
Streptomycin: 0.1 mg/ml
Tabelle 5: 10x Phosphatgepufferte Salzlösung (10x PBS). Die Chemikalien wur-
den in aufgereinigtem Wasser in einem Gesamtvolumen von 2500 ml
gelöst. Die Lagerung erfolgte bei Raumtemperatur und die Verdünnung
zu PBS erfolgte ebenfalls mit aufgereinigtem Wasser.
Chemikalie Menge für 2500 ml Endkonzentration
Natriumchlorid 200,15 g 1,37 M
Kaliumchlorid 5,0 g 26,8 mM
di-Natriumhydrogenphosphat 28,7 g 95,8 mM
Kaliumdihydrogenphosphat 5,0 g 14,7 mM
Tabelle 6: Fixierlösung: Formaldehyd 4 %. Die Fixierlösung wurde bei 4 ◦ C auf-
bewahrt.
Chemikalie Menge
10 % FA-Lösung 20 ml
PBS 20 ml
Saccharose 50 % 10 ml
Im nächsten Schritt wurden mit dem Vibratom Frontalschnitte der Gehirne ange-
fertigt. Dazu wurde die Einbettform mit einer Rasierklinge aufgeschnitten und der
Agaroseblock herausgelöst. Anschließend wurde überschüssige Agarose mit einer
Rasierklinge abgeschnitten, sodass ein rechteckiger Block mit etwa 2-4 mm Ab-
stand zwischen Gehirn und Schnittrand entstand. Dieser Block wurde auf die Ober-
fläche des Schnittstempels aufgeklebt, sodass die kaudalen Anteile des Gehirns
242 Material und Methoden
nach oben und die rostralen Anteile nach unten zeigten. Nach etwa 1 Minute Klebe-
zeit wurde der Schnittstempel in dem Schneidebecken, das mit gekühltem “HBSS
complete“-Puffer gefüllt war, in der Magnetvorrichtung positioniert. Das gesamte Be-
cken wurde justiert um möglichst exakt positionierte Frontalschnitte zu erhalten.
Folgende Einstellungen des Vibratoms wurden verwendet: die Schnittdicke wurde
zwischen 250 µm - 300 µm variiert, Frequenz 60 Hz und Amplitude 0,7. Bis zum Er-
reichen erster hippocampaler Anteile von kaudal betrug die Schnittgeschwindigkeit
20-25, danach wurde sie auf 15-17 verlangsamt. Nach Erreichen des Hippocampus
wurden die folgenden Gehirnschnitte mithilfe eines Pinsels vorsichtig in eine auf Eis
liegende 6-Well-Platte gefüllt, die pro Well 3 ml “HBSS complete“-Puffer enthielt. Für
die Kultivierung wurden unter dem binokularen Mikroskop geeignete Gehirnschnitte
ausgewählt, die alle Hippocampusareale mit guter Schnittqualität enthielten.
Die anschließende Positionierung auf den Zellkultureinsätzen erfolgte unter sterilen
Bedingungen unter dem Abzug. Dazu wurden die vorbereiteten 6-Well-Platten aus
dem Inkubator entnommen und in Kreuzform wurden aus einem Gesamtvolumen
von 100 µl Kulturmedium (37 ◦ C), fünf Punkte auf den Zellkultureinsatz pipettiert. Mit
einem gebogenen Spatel und einer Präzisionspinzette wurde jeder einzelne Gehirn-
schnitt auf einen vorbereiteten Punkt positioniert. Insgesamt lagen so maximal fünf
Gehirnschnitte auf einem Zellkultureinsatz. Nach Fertigstellung eines Zellkulturein-
satzes wurde vorsichtig die überbleibende Flüssigkeit auf der Membran abpipettiert,
sodass die Membran und die Gehirnschnitte trocken lagen. Ein Versuchsexperiment
umfasste einen Zeitrahmen von zwei bis zweieinhalb Stunden vom Töten des em-
bryonalen beziehungsweise postnatalen Maustiers bis zum Beginn der Kultivierung
der Gehirnschnitte.
Die nachfolgende Kultivierung erfolgte bei 37 ◦ C und 5 % Kohlenstoffdioxid / 95
% Luft. Alle 2 Tage wurden 0,9 ml der insgesamt 1,8 ml Kulturmedium unter der
Membran abpipettiert und durch frisches, vorher auf 37 ◦ C erwärmtes Kulturmedium
ersetzt. Die Dauer der Kultivierung variierte in den Experimenten zwischen zwei
Tagen und 17 Tagen.
252 Material und Methoden
Zur Fixierung der Gehirnschnitte wurde das Kulturmedium unterhalb des Zellkultur-
einsatzes vollständig abpipettiert und auf die Membranoberfläche 2 ml Fixierlösung
pipettiert. Die Gehirnschnitte wurden über Nacht bei 4 ◦ C gelagert. Am nächsten
Morgen wurde die Fixierlösung abpipettiert und die Membraninserts dreimal jeweils
15 Minuten vorsichtig in 1x PBS gewaschen. Die Lagerung bis zur Immunmarkie-
rung erfolgte in 2 ml PBS je Membraninsert bei 4◦ C.
2.5 Überexpression von HspB5 mittels in utero
Elektroporation
In dem dritten Projektteil wurden hippocampale Neurone in vivo mittels in utero Elek-
troporation transfiziert und verschiedene Proteine überexprimiert. Die Durchführung
der in utero Elektroporation erfolgte durch Dr. rer. nat. Christoph Wiegreffe aus der
Arbeitsgruppe Prof. Dr. Britsch. Die darauffolgende Entnahme der Gehirne, Anfer-
tigung und Analyse der Gehirnschnitte wurde selbst durchgeführt. Zur Transfekti-
on wurden die Plasmide pLVX-IRES-ZsGreen1-HspB5 sowie pLVX-IRES-ZsGreen1
Leervektor und pLVX-IRES-ZsGreen1-HspB5-AAA verwendet. In Experiment 1 und
Experiment 3 wurden Konzentrationen von 1 µg/µl verwendet; in Experiment 2 0,5
µg/µl. Es wurden an drei Versuchstagen jeweils drei trächtige Muttertiere der Linie
C57BL6J operiert; die Anzahl und das Überleben der elektroporierten Tiere sind in
Kapitel 3 Tabelle 10 beschrieben. Die Experimente wurden an Embryonaltag E15.5
durchgeführt und an Tag postnatal 15 durch Töten der Tiere beendet. Der operative
Eingriff an einem Muttertier dauerte zwischen 20 - 30 Minuten.
Zu Beginn wurde das Muttertier mit Isofluran narkotisiert und in Rückenlage fixiert
(Abbildung 3 A). Nach Desinfektion mit 70 % Ethanol wurde die Bauchdecke in ei-
nem medianen Längsschnitt eröffnet, und der Uterus freigelegt. Mittels Picospritzer
wurde mit einer Injektionspipette und Pulsen von 20 ms Dauer 1-2 µl der vorbe-
reiteten Plasmid-Lösung in die Seitenventrikel der Gehirne der Mäuseembryonen
injiziert (Abbildung 3 B). Anschließend folgte die Elektroporation mit den in Tabelle
7 beschriebenen Einstellungen. Die Positionierung der Elektroden erfolgte wie be-
26Sie können auch lesen

















































