Falk Gastro-Kolleg Leber und Gallenwege - Dr. Falk Pharma GmbH
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Falk
Gastro-Kolleg
Leber und
Gallenwege
Primär biliäre Zirrhose (PBC) und Prof. Dr. U. Leuschner,
Interdisziplinäres
Facharztzentrum
Autoimmuncholangitis (AIC) Frankfurt/Main
Zusammenfassung
Bei der primär biliären Zirrhose handelt es sich um eine autoimmune, cholestatische
und chronisch verlaufende Krankheit der Leber. Sie befällt zu 90 % Frauen und endet
unbehandelt in der kompletten Zirrhose. Die Cholestaseparameter steigen signifikant
an, die Transaminasen weniger stark. Typisch sind eine Vermehrung der IgM-Globuline
und das Auftreten von antimitochondrialen Antikörpern (AMA) gegen die E2-Unterein-
heit des Pyruvatdehydrogenasekomplexes (PDC). Histologisch wird die Krankheit in 4
Stadien, vom entzündlich-progressiven bis zum zirrhotischen Stadium eingeteilt. Die
wichtigsten Symptome und Befunde sind Müdigkeit, Pruritus und autoimmune Begleit-
symptome. Die Therapie besteht in der Verabreichung von Ursodeoxycholsäure, die
Behandlung wird so früh wie möglich und so lange wie möglich durchgeführt. Spricht
die Behandlung auf Ursodeoxycholsäure nur unvollständig an, kann eine Kombination
mit den Immunsuppressiva Prednison, Azathioprin und Budesonid versucht werden. Die
letzte Therapieoption stellt die Lebertransplantation dar. Die Behandlung der Symptome
und Befunde, wie Pruritus, Müdigkeit und Osteoporose gestaltet sich schwierig.
Bei der Autoimmuncholangitis handelt es sich histologisch um das Bild der primär
biliären Zirrhose, antimitochondriale Antikörper lassen sich bei diesem Krankheitsbild
aber nicht nachweisen. Die Autoimmuncholangitis wird daher auch als antimitochondri-
ale Antikörper-negative PBC bezeichnet. Wie bei der PBC besteht auch bei der AIC die
Behandlung in der Verabreichung von Ursodeoxycholsäure, in manchen Fällen kann eine
Fragebeantwortung unter
Kombination mit einem Immunsuppressivum weiterhelfen.
www.falkfoundation.de
Schlüsselwörter
Falk Gastro-Kolleg
Primär biliäre Zirrhose | Autoimmunkrankheiten der Leber | Therapie der PBC |
Autoimmuncholangitis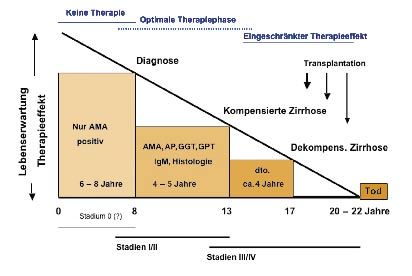
A. Primär biliäre Zirrhose (PBC)
1. Definition
Die primär biliäre Zirrhose ist eine chronische, progrediente Leberkrankheit. Sie be- Die primär biliäre Zirrhose (PBC) ist
ginnt an den interlobulären und proximal-septalen Gallengängen. Die Entzündung eine chronische, cholestatische
dehnt sich über das ganze Portalfeld aus und kann auf das Läppchenparenchym über- Autoimmunkrankheit der Leber.
greifen. Die Krankheit führt zur Verödung der Gallengänge und endet in der komplet-
ten Leberzirrhose. Die PBC gehört somit zu den „Vanishing bile duct syndromes“. Die
primär biliäre Zirrhose ist eine klassische Autoimmunkrankheit in der Leber.
2. Epidemiologie
Die primär biliäre Zirrhose befällt zu 90 % Frauen im Alter von 25 bis 50 Jahren. In Die Prävalenz der PBC
der letzten Zeit wurden aber auch einige Kinder mit PBC beobachtet. Die primär beträgt bei Europäern
biliäre Zirrhose galt vor 50 Jahren als eine seltene Krankheit. Ihre Inzidenz wurde mit wahrscheinlich 25-40
2/100.000, ihre Prävalenz mit 2 − 4/100.000 Einwohner angegeben. Auf Grund von pro 100.000 Einwohner.
Untersuchungen aus Großbritannien und den Vereinigten Staaten rechnet man
heute mit einer Inzidenz von 3,2/100.000 Einwohner und einer Prävalenz von
25/100.000 Einwohner. Berücksichtigt man, dass die PBC zu 90 % Frauen befällt,
dann liegt die Prävalenz in England bei 94/100.000 Frauen im Alter über 40 Jahre
(Tab. 1). Für Bewohner der nördlichen Halbkugel kann man wohl mit einer durch-
schnittlichen Prävalenz von 25 – 40 pro 100.000 Einwohner und 70 − 90 pro 100.000
Frauen rechnen.
Tab. 1
Mittlere jährliche Inzidenz und Prävalenz für vermutete
und gesicherte Fälle mit PBC in Nordengland
Gesamtpopulation Erwachsene ≥ 20 Jahre Frauen ≥ 40 Jahre
Inzidenz
1987 2,3 3,1 9,1
1994 3,2 4,3 10,0
Prävalenz
1987 14,9 20,1 54,1
1994 25,0 33,5 94,0
O.F.W. James, Hepatology 1999; 30: 390-394
Die Zunahme von Inzidenz und Prävalenz der PBC ist wahrscheinlich nur auf die
einfachere Diagnostik zurückzuführen. Es ist unbekannt, ob die Krankheit an sich
zunimmt.
Die PBC findet sich weltweit und bei allen Völkern, allerdings mit unterschiedlicher
Inzidenz und Prävalenz. Wie alle anderen Autoimmunkrankheiten tritt sie aber am
häufigsten auf der nördlichen Erdhalbkugel auf.
3. Ätiologie
Wie von allen anderen Autoimmunkrankheiten ist die Ätiologie der PBC unbekannt. Wie bei allen Autoimmunkrank-
Für die PBC besteht offenbar nur eine schwache genetische Suszeptibilität, eine ge- heiten ist die Ätiologie unbekannt.
netische Assoziation besteht mit dem Chromosom 6p 21.3. Immungenetische Un- Als Trigger werden Bakterien, Viren
tersuchungen haben gezeigt, dass offenbar eine besondere, wenn auch nur schwa- und Toxine diskutiert.
che Assoziation mit HLA-DR8 vorkommt. Eine Erbkrankheit ist die primär biliäre
Zirrhose aber nicht.
Besteht eine genetische Suszeptibilität dann können exogene oder endogene Fakto-
ren die Krankheit auslösen. Derartige Faktoren sind bei der PBC nicht bekannt, disku-
tiert werden Bakterien, Viren, toxische Gallensäuren und Umwelteinflüsse (Tab. 2). So
hat sich z. B. gezeigt, dass Frauen mit PBC häufiger Harnwegsinfekte entwickeln als
Lebergesunde. Lebergesunde Frauen mit rezidivierenden Harnwegsinfekten hatten
leicht erhöhte AMA-Konzentrationen im Serum. Mit Hilfe der PCR ließen sich Propi-Tab. 2
Ätiologie der primär biliären Zirrhose (PBC)
Hypothetische Faktoren
Bakterielle Ätiologie Virale Ätiologie Andere Ursachen
• Mycobakterien • Enteroviren • Pilzinfektionen
• Escherichia coli • Epstein-Barr-Viren • Toxische Gallensäuren
• Salmonella Typhimurium • Cytomegalie-Viren • Umweltfaktoren
• Hepatitisviren
onibakterien in der Nähe von epitheloidzelligen Granulomen und in der Nachbar-
schaft destruierter Gallengänge nachweisen. Eine pathogenetische Bedeutung
könnten sie vielleicht für die Entwicklung von Granulomen haben, für die Ätiologie
der PBC spielen sie keine Rolle.
Auch für eine Virusätiologie finden sich keine eindeutigen Hinweise, doch können
Viren als Auslöser der PBC nicht ausgeschlossen werden. Neuere Untersuchungen
halten endogene Retroviren, die man früher mit der Ätiologie der PBC in Verbin-
dung gebracht hatte, heute eher wieder für unwahrscheinlich. Auch Mikrochimäris-
mus kommt offenbar als Ursache der PBC nicht infrage. Unter Mikrochimärismus
versteht man den Übertritt fötaler Zellen des Cytotrophoblasten während der
Schwangerschaft in das mütterliche Blut, die Haut, die Lymphknoten, Milz und Le-
ber. Man nahm an, dass die kindliche DNA einen Autoimmunprozess initiieren kann.
Diese Hypothese wird heute nicht mehr vertreten.
4. Klinik
a) Symptome und Befunde
Die PBC verläuft über Jahre asymptomatisch. 60 % der Patienten sind bei Diagnose- Charakteristische Symptome sind
stellung asymptomatisch, 10 Jahre später nur noch 15 % und nach 15 − 20 Jahren Müdigkeit (Lethargie) und Pruritus.
nur noch 5 %. Der Krankheitsverlauf ist recht gut charakterisiert. Sowohl für asymp-
tomatische als auch symptomatische Patienten wurde ein durchschnittlicher Krank- An Befunden finden sich Kratzspuren,
heitsverlauf von 10 – 20 Jahren ermittelt. Xanthelasmen, Osteoporose,
Sicca-Syndrom.
Häufige Symptome sind Müdigkeit, Lethargie und Leistungsknick. 5 Jahre nach Dia-
gnosestellung geben 60 − 70 % der Patienten Müdigkeit an. Diese besser mit dem
Terminus Lethargie bezeichnete Müdigkeit beeinflusst bei mehr als der Hälfte der
Patienten das Familien- und Berufsleben. Das Symptom korreliert weder mit den
Laborwerten noch mit dem histologischen Befund. Häufig ist die Lethargie mit de-
pressiven Verstimmungen assoziiert.
Ein weiteres richtungsweisendes Symptom stellt der Pruritus dar, der als das Bedürfnis
des Patienten sich zu kratzen definiert ist. Der Pruritus tritt hauptsächlich nachts auf,
unstillbarer Pruritus kann eine Indikation zur Lebertransplantation sein (Tab. 3). Ist der
Pruritus stark, dann findet man Kratzmale auf dem Rücken und an den Armen. Wichtig
ist die Beobachtung, dass Pruritus zum ersten Mal bei schwangeren Frauen während
einer Schwangerschaftscholestase auftritt, post partum verschwindet, und nach eini-
ger Zeit wieder als Symptom einer wahrscheinlich neu entstandenen PBC rezidiviert.
Pruritus bei primär biliärer Zirrhose (PBC) Tab. 3
• Beginnt schleichend: 20 – 70 % der Patienten entwickeln Pruritus
• Kann zu jedem Zeitpunkt der Krankheit auftreten
• Kann spontan wieder verschwinden
• Häufig perianal und perigenital
• Häufig an Handflächen und Fußsohlen
• Bei Wärme verstärkt
• Nimmt im Winter zu (assoziiert mit trockener Haut)
• Verstärkt in Schwangerschaft und während HormontherapieXanthome an den inneren Augenlidern werden als Xanthelasmen bezeichnet. Xan-
thome findet man auch an den Fersen, am Gesäß und auf den Handflächen. Sie
werden bei 10 − 20 % der PBC-Patienten beschrieben. Sie können spontan wieder
verschwinden. Xanthome korrelieren nicht mit den Cholesterinwerten oder den Tri-
glyzeridkonzentrationen im Serum.
Die PBC-bezogene post menopausal-bereinigte Prävalenz der Osteoporose beträgt
25 − 35 %. Begünstigt wird die Entwicklung der Osteoporose durch die Immobilität
des Leberpatienten, verringerte Muskelmasse, durch falsche Ernährung und Störun-
gen des Hormonstoffwechsels. Frakturen finden sich hauptsächlich an der Wirbel-
säule und den Rippen, während die Röhrenknochen und das Becken seltener be-
troffen sind. Eine Abnahme der Körpergröße um mindestens 4 cm nach dem 25.
Lebensjahr ist ein Hinweis auf eine Wirbelkörperfraktur.
Die Diagnose der Osteoporose wird häufig vor der Diagnose der PBC gestellt und
führt den Patienten daher nicht selten zum Orthopäden. Patienten im Spätstadium
der PBC haben eine fortgeschrittenere Osteoporose als solche im Frühstadium, al-
lerdings sind sie zu dieser Zeit auch älter. Neueren Studien zufolge ist nicht ganz
klar, ob es sich bei der Osteoporose tatsächlich um eine PBC-assoziierte Skelettver-
änderung handelt oder ob hier nicht geschlechts- und altersspezifische Faktoren
die entscheidende Rolle spielen.
Bei dem Sicca-Syndrom kommt es zum Versiegen der Drüsensekrete mit Trocken-
heits- und Reibegefühl an den Augen (Xerophthalmie), mit Schluckstörungen durch
einen trockenen Ösophagus, und wegen verringerter Speichelsekretion zu Karies.
Die Verringerung des Pankreassekretes kann im Extremfall zu einer exokrinen Pan-
kreasinsuffizienz führen. Hierdurch und durch die PBC-bedingte Cholestase kommt
es zur Malabsorption von Fetten und fettlöslichen Vitaminen (A, D, E, K), was Stea-
torrhoe, Nachtblindheit, neurologische und Gerinnungsstörungen zur Folge haben
kann. Störungen der Fettresorption und der Vitaminmangel können auch auf eine
die PBC gelegentlich begleitende Sprue zurückgeführt werden.
Das hepatozelluläre Karzinom (HCC) findet sich wie bei den meisten anderen chro-
nischen Leberkrankheiten erst im Zirrhosestadium. Es ist nicht gesichert, ob es bei
der PBC häufiger vorkommt. Risikofaktoren sind höheres Alter und männliches Ge-
schlecht. Das kumulative Risiko des hepatozellulären Karzinoms beeinflusst die Le-
benserwartung der PBC-Patienten nicht, da es sich spät entwickelt und nur wenig
progredient ist.
Tab. 4
PBC-assoziierte Krankheiten
• Arthropathie • Keratoconjunctivitis sicca
• Arthritis • Hautveränderungen
• Sicca-Syndrom - Lichen planus
• Sjögren-Syndrom - Pemphigoid
• Raynaud-Syndrom - Dermatomyositis
• Sklerodermie • Zöliakie
• CREST-Syndrom • Colitis ulcerosa
(oder einzelne Komponenten*) • Lungenfibrose
• System. Lupus erythemat. • Cholelithiasis
• Glomerulonephritis • Leberzellkarzinom
• Hashimoto-Thyreoiditis
• Osteoporose
*Calcinosis cutis, Raynaud-Syndrom, Dysmotilität
des Ösophagus, Sklerodaktylie, Teleangiektasien
Wie alle Autoimmunkrankheiten ist auch die PBC von Autoimmunsyndromen begleitetet (Tab.
4). Etwa 30 % aller PBC-Patienten weisen eine oder mehrere dieser Begleitkrankheiten auf. Am
häufigsten ist die Hashimoto-Thyreoiditis, häufig findet man ein solitäres Raynaud-Syndrom,
seltener dagegen rheumatische Arthritiden. Im Gegensatz zur primär sklerosierenden Cholan-
gitis (PSC) ist die PBC wesentlich seltener mit chronisch entzündlichen Darmkrankheiten asso-
ziiert. Wenn dies der Fall ist, dann hauptsächlich mit der Colitis ulcerosa, seltener mit dem Mor-
bus Crohn. Bei etwa 15 – 20 % der Patienten kann sich auch eine Zöliakie entwickeln.b) Labordiagnostik
Da es sich bei der PBC um eine cholestatische Leberkrankheit handelt, kann die al- Charakteristische Laborbefunde:
kalische Phosphatase (AP) 10 − 15fach, die Gammaglutamyltranspeptidase (GGT) 20 Erhöhung der Cholestaseparameter
− 25fach erhöht sein, während die Transaminasen GOT und GPT geringer ansteigen. (AP, GGT), Nachweis von AMA-M2,
Typisch für die PBC sind erhöhte Konzentrationen des Immunglobulins IgM sowie ANA-MND, erhöhte IgM-Konzentration
der Nachweis von antimitochondrialen Antikörpern mit einer Titerhöhe von min- im Serum.
destens 1: 80.
Die antimitochondrialen Antikörper (AMA) sind typisch für die PBC und finden sich
bei 90% der Patienten. Sie sind nicht spezies- oder organspezifisch. AMA stellen ei-
nen sehr frühen Marker der PBC dar, der gelegentlich schon Jahre vor der ersten
Diagnose beobachtet wird. Typisch für die PBC sind AMA-M2, während die Subty-
pen M1, 3, 5 oder 6 zum Beispiel auch bei der Syphilis, dem Pseudolupus, bei Kolla-
genosen oder medikamentösen Leberschäden auftreten können. Das Zielantigen
für AMA-M2 bei der PBC liegt auf der inneren Mitochondrienmembran und ist die
E2-Untereinheit des Pyruvatdehydrogenasekomplexes (PDC).
Ob AMA bei der Pathogenese der PBC eine Rolle spielen, ist ungeklärt. So fand man
nämlich die E2-Untereinheit des PDC in hoher Konzentration auf Gallengangsepithel-
zellen von PBC-Patienten exprimiert und als Ausdruck einer Immunreaktion T-Zell-
reiche Entzündungsinfiltrate in der Umgebung. Natürlich kann es sich dabei auch
um ein Sekundärphänomen handeln.
PBC-typisch sind auch spezielle antinukleäre Antikörper (ANA). Hier kann man so
genannte Multiple nuclear dot-Antikörper (MND) von Ring-staining-ANA unter-
scheiden. Beide Subtypen sind beweisend für eine PBC, besonders wenn AMA feh-
len. MND-ANA sollen besonders bei Patienten mit Xerophthalmie vorkommen. An-
dere Autoantikörper, wie zum Beispiel p-ANCA, Schilddrüsenantikörper, Antikörper
gegen Thrombozyten, Histon- und Ribonukleoprotein-Autoantikörper spielen eine
untergeordnete Rolle. Sie sind Ausdruck einer unspezifischen Reaktion des Immun-
systems.
c) Histologie
Histologisch wird die PBC in die Stadien I − IV unterteilt. Stadium I ist das portale I: Portale Entzündung mit
Stadium, bei dem sich lockere lympho-plasmazelluläre Infiltrate in den Portalfeldern Gallengangsdestruktion.
finden. CD4-Lymphozyten dominieren CD8-Zellen im Verhältnis 4:1.
Tab. 5
Histologie und Staging der PBC und der AIC
Stadium I: Portales Stadium
Portale Hepatitis, floride Gallengangsdestruktion,
nur geringe entzündliche Veränderungen im Leberläppchen
Stadium II: Periportales Stadium
Periportale Hepatitis, floride Gangsdestruktion, Gallengangsproliferation,
Piecemealnekrosen, Granulome, geringe Veränderungen im Läppchen
Stadium III: Septales Stadium
Brückenfibrose, Veränderungen wie in Stadium II, Ductopenie, Cholestase,
Galletröpfchen in den Hepatozyten
Stadium IV: Zirrhotisches Stadium
Biliäre Zirrhose, Bild wie im Stadium III, noduläre Regeneration,
Umbau der Gefäßarchitektur, Gallengänge fehlen, Cholestase
Das Stadium II wird als periportales Stadium bezeichnet. Das Gallengangsepithel, als II: Periportale Entzündung plus
eigentliche Zielstruktur, wird von Lymphozyten segmental, später zirkulär zerstört. Proliferation der Gallengänge,
Es können sich histiozytäre und ephiteloidzellige Granulome entwickeln. Als Kom- Entzündung greift auf Leber-
pensation für die destruierten Gallengänge kommt es zur Gallengangsproliferation. gewebe über.
Entzündungszellen greifen auf das periportale Läppchenparenchym über, und es
entwickeln sich Piecemealnekrosen.Das Stadium III bezeichnet man als septales Stadium. Hier sind zahlreiche Gallen- III: Septales Stadium
gänge eines Portalfeldes zerstört, neue proliferiert. Die entzündlichen Infiltrate grei- mit zunehmender Fibrose.
fen weiter auf das Leberparenchym über, und im Portalfeld nimmt der Bindege-
websgehalt allmählich zu. Bindegewebsstränge nehmen Kontakt zu benachbarten
Portalfeldern auf, das heißt, es entwickelt sich eine progressive Fibrose.
Im Stadium IV, das als Zirrhosestadium bezeichnet wird, handelt es sich um die kom- IV: Zirrhosestadium mit totalem
plette Leberzirrhose mit fast vollständiger Rarefizierung kleinerer Gallengänge Umbau des Lebergewebes.
(Tab. 5).
Die beschriebenen morphologischen Befunde sind für die PBC nicht pathognomo-
nisch, sondern können auch bei anderen Autoimmunkrankheiten der Leber auftre-
ten. Die Gallengangsveränderungen der PBC sind damit nur als typisch, aber nicht
als spezifisch zu bezeichnen. Da sich bei einem einzigen Patienten mehrere histolo-
gische Stadien in einer Leber gleichzeitig nachweisen lassen, man spricht bei der
PBC von einer fokalen Krankheit, kann die histologische Diagnose, besonders aber
das Grading und Staging schwer sein. In den meisten Fällen genügt es, wenn der
Pathologe bei charakteristischen Laborbefunden den mikroskopischen Befund als
mit einer PBC vereinbar beschreibt. Eine Zuordnung von Symptomen und Labor-
werten zu den histologischen Stadien findet sich Tabelle 6 (Tab. 6).
Tab. 6
Symptome, Labor und Histologie bei PBC
Symptome Labor Histologie
Lethargie, Pruritus AP, GGT, (IgM), AMA Stadium I:
(GOT, GPT) Port. Entzündung
Lethargie, Pruritus AP, GGT, IgM, AMA Stadium II:
(GOT, GPT) dto.
Lethargie, Pruritus, AP, GGT, IgM, AMA, Stadium III:
abdom. Beschwerden (GOT, GPT, Bilirubin) Fibrose
Lethargie, Pruritus, AP, GGT, IgM, AMA Stadium IV:
Komplikationen der GOT, GPT, Bilirubin Zirrhose
Zirrhose Abfall der Synthese-
parameter
d) Bildgebende Verfahren
Bildgebende Verfahren spielen in der Diagnostik der PBC eine untergeordnete Rolle. Sonogra-
phisch kann das Echomuster der Leber verstärkt sein, die Gallengänge sind sonographisch und
bei der ERC unauffällig. Erst im Spätstadium der Krankheit erscheinen sie rarefiziert, das Bild
gleicht dem eines entlaubten Baumes. Sonographie, ERC, CT und MNR dienen lediglich dem
Ausschluss anderer, hauptsächlich tumoröser Lebererkrankungen, nicht aber der Diagnose-
stellung bei PBC.
e) Differenzialdiagnose
Sind die Cholestaseparameter erhöht, lassen sich AMAs und eine erhöhte IgM-Konzentration Differenzialdiagnostisch kommen
nachweisen, kann die Diagnose als gesichert angesehen werden. Für die Erstdiagnose ist die die Autoimmunhepatitis, die
Leberbiopsie zur Sicherung erforderlich, histologische Verlaufskontrollen sind nur bei Verdacht primär sklerosierende Cholangitis
auf ein Malignom sinnvoll, oder wenn der Verdacht auf ein Overlapsyndrom oder eine Autoim- und die Autoimmuncholangitis
muncholangitis (AIC, s. unten) besteht. Dann wird die Diagnose nämlich schwieriger. in Frage.
Differenzialdiagnostisch müssen hauptsächlich eine Autoimmunhepatitis und eine primär sklero-
sierende Cholangitis (PSC) abgegrenzt werden, aber auch die meisten anderen chronischen Le-
berkrankheiten müssen bei weniger PBC-typischen Befunden ausgeschlossen werden (Tab. 6).
f ) Natürlicher Verlauf
Aufgrund epidemiologischer Untersuchungen gilt heute, dass vom ersten Auftreten
antimitochondrialer Antikörper bis zur endgültigen Diagnose 6 − 8 Jahre vergehen.Bis zum 13. Jahr, also 5 − 7 Jahre lang, folgt das entzündlich-progressive Stadium.
Dieses Stadium wird auch als reversibles Stadium bezeichnet und stellt die Phase
der besten Therapiechancen dar. Vom 13. − 17. Jahr nach dem ersten Auftreten an-
timitochondrialer Antikörper folgen das Stadium der kompensierten Zirrhose und
das 4-jährige Stadium der dekompensierten Zirrhose, das schließlich nach etwa 22
Jahren zum Tode führt (Abb. 1).
Abb. 1
5. Therapie der primär biliären Zirrhose und extrahepatischer Manifestationen
a) Therapie der PBC
Da die Ätiologie der PBC unbekannt ist, gibt es keine kausale Therapie. Auch die Die Standardtherapie erfolgt
Lebertransplantation ist nicht in der Lage, die Krankheit in jedem Fall zu heilen, da mit Ursodeoxycholsäure (UDC),
es bei etwa 15% der Transplantierten zu Rezidiven im Transplantat kommt. 12 − 16 mg/kg KG/Tag oder in Kombi-
nation mit niedrigen Dosen Prednison.
Die Monotherapie mit D-Penicillamin, Glukokortikoiden, Colchizin und anderen Me-
dikamenten hat sich nicht bewährt. 1985 haben wir Ursodeoxycholsäure (UDC) in
die Behandlung der PBC eingeführt, UDC ist heute das einzige Medikament, für das
eindeutig positive Effekte nachgewiesen werden konnten. UDC ist eine physiolo-
gische, beim Menschen vorkommende Gallensäure. Als dihydroxylierte Gallensäure
ist sie gut wasserlöslich und so gut wie untoxisch. Nur bei etwa 2% der Patienten
kommt es zur Diarrhoe.
Die Indikation zur UDC-Therapie ist vom Tag der Diagnosestellung an gegeben. Bei 30 % kommt es zur
Frühstadien sprechen besser auf die Behandlung an als Spätstadien. Unter der Dosis Normalisierung der Laborwerte,
von 12 − 16 mg pro kg KG pro Tag ist UDC in der Lage, die Progression der Krankheit bei 70 % zur signifikanten Verbesserung.
in das nächst höhere Stadium zu verzögern oder sogar zu verhindern. Bei 30 % der
Patienten im Stadium I und Stadium II kommt es nach 3 − 5 Jahren zur völligen Nor- Die Therapie verlängert
malisierung der Laborwerte, die Histologie verbessert sich signifikant, der Pruritus das transplantationsfreie Intervall
nimmt ab oder verschwindet. Die antimitochondrialen Antikörper bleiben aber wei- und führt offenbar
terhin nachweisbar. zur Lebensverlängerung.
Bei 70 % der Patienten kommt es zu einer signifikanten Verbesserung der Laborwer-
te, aber nicht zur Normalisierung. 16 randomisierten kontrollierten Studien mit ins-
gesamt 1422 Patienten sind folgende Ergebnisse zu entnehmen: Gegenüber Plaze-
bo führte UDC zu einer signifikanten Verringerung der cholestaseanzeigenden
Laborparameter, von Bilirubin und der Inzidenz der Lebertransplantation (p < 0,04).
Aszites und Ösophagusvarizen entwickeln sich seltener, das transplantationsfreie
Intervall wird verlängert. Der positive Einfluss von UDC auf die Leberhistologie kann
heute für die Frühstadien I und II der Krankheit als gesichert gelten. Wird die UDC-
Therapie frühzeitig begonnen, kommt es zur signifikanten (p < 0,05) Lebensverlän-
gerung. Seit Einführung der UDC in die Therapie der PBC hat die Mortalität bei PBC-
Patienten unter 65 Jahren abgenommen, desgleichen die Zeit der HospitalisierungTab. 7
Therapieeffekte mit Ursodeoxycholsäure (UDC)
bei primär biliärer Zirrhose im Vergleich zu Plazebo
Daten bis April 2002
• Randomisierte kontrollierte Studien: 16
• Nur Studien mit adäquater Randomisierung eingeschlossen
• UDC mit allen Dosierungen versus Plazebo
• Patientenzahl: 1422
• Ergebnisse: UDC verbesserte signifikant (p < 0,05)
- Aszites
- Ikterus
- Bilirubin
- Leberenzyme
- Inzidenz der Transplantation (< 0,04)
- Hatte keine Nebenwirkungen
- Fraglicher Einfluss auf die Mortalität ± Transplantation
und die Behandlungskosten (Tab. 7). Die PBC-bedingte Indikation zur Lebertrans-
plantation ging um 30 % zurück. Die Behandlung mit UDC ist nebenwirkungsfrei.
Bei Therapieunterbrechung oder völligem Absetzen des Medikamentes kommt es
sofort zum Rezidiv. Die Behandlung muss daher kontinuierlich, lebenslang oder bis
zur Lebertransplantation durchgeführt werden.
Bei 70 % der Patienten führt die Therapie zwar zur Verbesserung, aber nicht zur Nor-
malisierung der Laborparameter, das heißt, die Krankheit wird nicht geheilt. Da es
sich bei der PBC um eine Autoimmunkrankheit und um eine cholestatische Leber-
krankheit handelt, wurden Kombinationstherapien eingesetzt: UDC und Prednison
(z. B. 10 – 15 mg/Tag), UDC plus Prednison plus Azathioprin (z. B. 100 mg/Tag) und
UDC plus Budesonid (3 x 3 mg/Tag).
In allen Studien war die Kombinationstherapie der Monotherapie mit UDC überle-
gen. Wegen der noch nicht gesicherten Nebenwirkungen ist die Kombinationsthe-
rapie mit Budesonid derzeit noch nicht generell zu empfehlen. Behandlungsversu-
che bei Patienten, die nicht oder nicht ausreichend auf die Monotherapie mit UDC
angesprochen haben, wurden auch mit Methotrexat, Colchizin, Silymarin, Bezafibrat
und Sulindac, einem nichtsteroidalen Antirheumatikum, durchgeführt. Auch hierbei
wird über Einzelerfolge berichtet.
b) Therapie der extrahepatischen Manifestationen
Gegen die Müdigkeit oder Lethargie gibt es keine spezifische Therapie. Ruhepausen Die Therapie extrahepatischer
im Wechsel mit dosierter körperlicher Aktivität können hilfreich sein. Vor jedem Be- Manifestationen kann
handlungsversuch müssen eine Anämie, eine Schilddrüsenunterfunktion, eine Ne- Probleme bereiten.
benniereninsuffizienz und eine endogene Depression ausgeschlossen, gegebenen-
falls einer spezifischen Therapie zugeführt werden. Schwer ist besonders
die Behandlung des Pruritus,
Zur Behandlung des Pruritus werden Medikamente eingesetzt, die in den Gallen- für die chronische Müdigkeit (Lethargie)
säurenstoffwechsel eingreifen, Steroide, Antihistaminika und Opioid-Rezeptoranta- gibt es keine Behandlung.
gonisten (Tab. 8). An erster Stelle der Therapie stehen Cholestyramin bzw. Colesti-
pol, die bei bis zu 90 % der Patienten mit leichtem oder mittelschwerem Pruritus
erfolgreich sind. Die beiden ersten Dosen sollen vor und nach dem Frühstück verab-
reicht werden. Colestipol wirkt weniger obstipierend als Cholestyramin. Die Opioid-
Rezeptorantagonisten Naloxon und Naltrexon sind bei 50 − 60 % der Patienten er-
folgreich. Nachteile sind ihr hoher Preis, ihre Lebertoxizität und gelegentlich
Symptome des Opiatentzuges. Weiterhin wurden Anorexie, Nausea, Koliken und
Bluthochdruck beschrieben. Auch der Enzyminduktor Rifampicin wurde erfolgreich
eingesetzt, gelegentlich wirkt es hepatotoxisch und führt zu hämolytischer Anämie.
Antihistaminika machen nur müde, sind aber unwirksam.Tab. 8
Therapiemöglichkeiten bei Pruritus
• Ursodeoxycholsäure: 10 – 20 mg/kg KG/Tag.
• Cholestyramin: 4 – 24 g tgl., oder Colestipol: 5 – 30 g/Tag
• Naloxon: 0,2 μg/kg/min i.v. oder 2 – 3 x 0,4 mg/Tag i.v.
• Naltrexon: 50 mg/Tag oral
• Nalmefen: 10 mg, steigern bis 120 mg/Tag
• Ondansetron: 3 x 4 bis 3 x 8 mg/Tag
• Rifampicin: 300 – 500 mg/Tag, bis 10 mg/kg/Tag
• Phenobarbital: 3 – 5 mg/kg/Tag
• Antihistaminika: keine Wirkung, machen nur müde
• Plasmapherese: in Erprobung
• Manchmal hilft nur eine Kombination verschiedener Methoden
• MARS-Therapie (Albumin-Dialyse)
In der Frühphase der Osteoporose genügen eine regelmäßige, gemischte Kost und
körperliche Aktivität im Freien. Aktiviertes Vitamin D und Kalzium sind nur als Kom-
binationstherapie hilfreich. Auch die Behandlung mit Bisphosphonaten wie Alen-
dronat und Etidronat sind hilfreich. Wichtig ist, dass die anti-osteoporotische Thera-
pie prophylaktisch eingesetzt wird, eine vorhandene Osteoporose ist schwerlich zu
beseitigen.
Die Therapie der seltenen Steatorrhoe besteht hauptsächlich in der Reduktion des
Nahrungsfettes und bei deutlicher Symptomatik in der Gabe von mittelkettigen Tri-
glyceriden (Ceres-Margarine). Bei gleichzeitiger Therapie eines Pruritus mit Chole-
styramin muss die Cholestyramin-Dosis reduziert oder aber auf ein anderes Präparat
übergegangen werden, da Cholestyramin Gallensäuren bindet, und Gallensäuren
für eine suffiziente Fettresorption verantwortlich sind. Ist die Pankreasinsuffizienz
ausgeprägt, so gibt man Pankreasenzyme in ausreichend hoher Dosierung.
Vitaminmangelzustände sind selten und bedürfen meist keiner Therapie. Das Sicca-
Syndrom kann mit falschen Tränen und Vaginalgelen behandelt werden, reguläre
Zahn- und Mundhygiene sowie die Einnahme von Medikamenten in aufrechter Po-
sition mit reichlich Flüssigkeit sind angezeigt. Hilfreich kann bei schwerem Sicca-
Syndrom auch die Gabe von Salagen sein (Pilocarpin-Präparat), das aber zu starkem
Schwitzen führen kann und noch zahlreiche andere Nebenwirkungen hat.
c) Lebertransplantation
Eine Indikation zur Lebertransplantation ist dann gegeben, wenn das Bilirubin über Die Indikation ist bei
6 mg (102 μmol/l) aber noch unter 9 mg % (153 μmol/l) liegt. Auch der Abfall des nachlassender Leberfunktion gegeben.
Quick-Wertes unter 60 % der Norm und die nachlassende Syntheseleistung für Al- Die Ergebnisse sind sehr gut.
bumin stellen gute Indikatoren für die Notwendigkeit einer Transplantation dar.
Die 1-Jahresüberlebensrate
Die Lebertransplantation ist die letzte therapeutische Option bei kompletter Zirrho- beträgt 93 %,
se. Mit 16 % liegt die Häufigkeit der Lebertransplantation bei PBC mit deutlichem die 8-Jahresüberlebensrate
Abstand hinter der Transplantationsrate der Virushepatitis C und der Alkoholzirrho- beträgt 71 %.
se. Die Transplantationsergebnisse sind sehr gut. Die Überlebensrate beträgt nach
1 Jahr 81 %, nach 5 Jahren 76 % und nach 8 Jahren 71 %. Neuere Zahlen geben für
die PBC sogar eine 1-Jahresüberlebensrate von 93 % an.
Bei etwa 10 − 15 % der transplantierten Patienten rezidiviert die PBC im Transplantat.
Laborwerte sind dabei nur ein schwacher Hinweis auf ein Rezidiv. Eine erhöhte AP fin-
det sich z. B. nur bei 46 % der Patienten, geringe Transaminasen- und Bilirubinerhöhun-
gen bei jeweils 20 %. Die antimitochondrialen Antikörper können persistieren, oder sie
werden wenige Wochen nach der Transplantation wieder nachweisbar. Histologisch ist
das PBC-Rezidiv nur schwer von einer Abstoßungsreaktion, von postoperativen biliären
Komplikationen und von einer chronischen Hepatitis-C-Infektion abzugrenzen.B. Autoimmuncholangitis (AIC) 1. Definition und Epidemiologie der AIC Bei der Autoimmuncholangitis handelt es sich um eine Krankheit, die klinisch und Bei der Autoimmuncholangitis (AIC) histologisch einer PBC entspricht, bei der sich serologisch aber keine antimitochon- handelt es sich um drialen Antikörper (AMA) nachweisen lassen. Nicht selten finden sich bei diesen Pa- eine AMA-negative PBC tienten aber antinukleäre Antikörper (ANA) oder Antikörper gegen glatte Muskelfa- oder ein Überlappungssyndrom. sern (SMA), wie sie für die Autoimmunhepatitis typisch sind. Es handelt sich hier also um eine Krankheit mit einem gemischten hepatitischen und cholestatischen Bild. Synonyme für die AIC sind Immuncholangitis, Autoim- muncholangitis, AMA-negative PBC und auch Overlapsyndrom zwischen PBC und Autoimmunhepatitis. Die Definition der AIC ist also nicht eindeutig, da unklar ist, ob es sich hier um ein Überlappungssyndrom, eine selbständige Krankheit, eine Variante der Autoimmun- hepatitis oder um eine Sonderform der PBC handelt. Die AIC findet sich bei 5 − 16 % aller Patienten mit PBC, bei denen die AMA-Testung mit molekular definierten Testsystemen (ELISA, Westernblot) durchgeführt wurde. Obwohl sich bei der AIC wie bei der PBC die PDC-E2 im apikalen Bereich des Gallen- gangsepithels nachweisen lässt, fehlen antimitochondriale Antikörper, die gegen diesen Enzymkomplex gerichtet sind. Die bei der AIC beobachteten ANA unter- scheiden sich von den ANAs der PBC häufig dadurch, dass sie bei Spezialfärbung keine Multiple nuclear dots und kein Ring staining aufweisen. 2. Diagnostik und Differenzialdiagnosen Die Autoimmuncholangitis wird hauptsächlich durch die Leberhistologie diagnosti- Die Histologie entspricht der der PBC, ziert. Histologisch entspricht die AIC vollständig einer PBC. Auch die Symptome der AMA fehlen. Dafür lassen sich häufig AIC sind mit denen der PBC identisch, weswegen sich heute für die AIC immer mehr ANA und SMA nachweisen. der Begriff AMA-negative PBC durchsetzt. Differenzialdiagnostisch muss die AIC von der PBC, der PSC und der Typ-1-Autoim- munhepatitis abgegrenzt werden. Von der PBC unterscheidet sie sich durch das Feh- len der antimitochondrialen Antikörper bei häufigerem Vorkommen von ANA und SMA, von der PSC durch den cholangiographischen und histologischen Befund, und von der Autoimmunhepatitis durch die für die PBC typischen Gallengangsdestruktio- nen und die Gallengangsproliferation bei fehlender Hypergammaglobulinämie. 3. Therapie der AIC Die Therapie besteht wie bei der PBC in der Verabreichung von Ursodeoxycholsäure Wie bei der klassischen PBC wird (UDC) in einer Dosis von 12 − 16 mg/kg Körpergewicht täglich. Sollte die Behand- UDC in der Dosis 12 − 16 mg/kg KG/Tag lung nach 6 − 8 Monaten nicht zu einem Abfall der Laborparameter um 70 − 80 % verabreicht, bei unvollständigem geführt haben, kann UDC mit Glukokortikoiden und/oder Azathioprin in der übli- Therapieergebnis Kombination mit chen Dosierung kombiniert werden. Über die Prognose der Autoimmuncholangitis einem Immunsuppressivum möglich. mit und ohne Therapie ist nichts bekannt.
Zu empfehlende Literatur Literatur 1. Leuschner U. Autoimmunkrankheiten der Leber und Overlapsyndrome. 2., vollständig aktualisierte Auflage. UNI-MED Verlag AG, Bremen, ISBN 3–89599–883-4, 172 Seiten , 2005. 2. Hübscher SG. Histological classification of autoimmune cholestatic liver diseases. In: Manns MP, Paumgartner G, Leuschner U (Hrsg.), Immunology and Liver. Kluwer Academic Publishers, Dordrecht, Boston, London, 2000: 223-243. 3. Strassburg CP, Manns MP. Primär biliäre Zirrhose und primär sklerosierende Cholangitis. Internist. Prax., 1996; 36: 57-74. 4. Parés A, Caballeria L, Rodés J et al. Long-term effects of ursodeoxycholic acid in primary biliary cirrhosis: Results of a double-blind controlled multicentric trial. J Hepatol 2000; 32: 561-566.
Fragen zur primär biliären Zirrhose (PBC) Falk
und Autoimmuncholangitis (AIC) Gastro-Kolleg
Frage 1:
Ein Patient klagt über eine seit 2 − 3 Jahren ständig zunehmende Leber und
Müdigkeit, die alkalische Phosphatase (AP) sei bei seinem Hausarzt
einige Male erhöht beobachtet worden. Koliken seien nicht auf- Gallenwege
getreten. Woran denken Sie?
Wahrscheinlich psychische Belastung
Verdacht auf cholestatische Lebererkrankung
Wahrscheinlich liegt ein Skelettschaden vor
AP-Erhöhung weist auf Cholelithiasis hin
Bedeutungslos. Kontrolle ist nicht erforderlich Bitte beachten Sie:
Bei der Beantwortung der Fragen
Frage 2: ist immer nur 1 Antwort möglich.
Ein Patient legt Ihnen folgende Laborkonstellation vor: AP erhöht, Die Beantwortung der Fragen und
GGT erhöht, Gamma-Globuline und IgG normal, SMA negativ, AMA Erlangung des Fortbildungszertifikats
positiv, LKM negativ, p-ANCA positiv. Woran denken Sie? ist nur online möglich.
Autoimmunhepatitis
Primär sklerosierende Cholangitis
Primär biliäre Zirrhose
Sarkoidose der Leber
Frühform eines M. Wilson
Frage 3:
Was ist richtig?
Die PBC ist sehr selten, ihre Prävalenz beträgt 5 pro 100.000 Einwohner
Die PBC hat in den letzten Jahren absolut zugenommen
Die PBC ist eine typische Erkrankung des Mittelmeerraumes
Die PBC ist eine typische Männerkrankheit
Bei Kindern und Jugendlichen wurde die PBC noch nicht beobachtet
Frage 4:
Welche Laborkonstellation ist charakteristisch für eine
primär biliäre Zirrhose?
AMA-M4, AP erhöht, Gamma-Globuline grenzwertig erhöht
ANA-MND (multiple nuclear dots) erhöht, AP, GGT erhöht, IgM leicht erhöht
Bilirubin erhöht, p-ANCA nachweisbar, LKM-Antikörper positiv
AP und GLDH stark erhöht, IgA deutlich erhöht, Leukozytose
Coeruloplasmin erniedrigt, ANA und LKM
Frage 5:
Welche von den genannten Befunden sind für die PBC
am wenigsten typisch?
Ulnarabduktion der Finger IV und V, Striae abdominales
Xanthelasmen, Xanthome
Osteopenie, Osteoporose
Xerophthalmie, Schluckbeschwerden
Totenfinger, Sicca-SyndromFrage 6:
Typisch für die PBC ist folgender histologischer Befund?
Falk
Rosettenformation der Hepatozyten
Gastro-Kolleg
Mallory bodies, Schaumzellen
Gallengangsproliferation, portale Rundzelleninfiltrate
Lobuläre entzündliche Infiltrate, Portalfelder unauffällig
Fibrose in Zone 3 des Leberläppchens
Frage 7:
Bei pathologischen Werten von GPT, AP, GGT, ANA und einem AMA-
Titer von 1:320 klagt ein Patient über ständig rezidivierende wei- Stadium I
che Stühle. Woran denken Sie? Stadium II
PBC mit Zöliakie (Sprue)
Primär sklerosierende Cholangitis (PSC) mit CED
Primär biliäre Zirrhose (PBC) mit Kurzdarmsyndrom
Choledocholithiasis mit Gallensäurenverlustsyndrom
Schistosomiasis
Frage 8:
Sie therapieren einen Patienten mit PBC mit 12 mg UDC pro kg KG
täglich, aber die sehr hohen Laborwerte bessern sich nach 6 − 8
Monaten nicht weiter. Was machen Sie?
Stadium III
Diagnose überprüfen
UDC-Dosis auf 20 − 25 mg/kg täglich erhöhen Ectet, sit vero ero dolorer sis nulluptatem
iriustrud tat lore dolore tem in elis ad erosto
UDC mit einem niedrigdosierten Immunsuppressivum,
dunt praese feuguercipis
z. B. Prednison kombinieren
UDC-Dosis verringern, da UDC hier möglicherweise toxisch wirkt
Therapieversuch mit Methotrexat einleiten
➤ Ut alit, commodi psuscipit wisis
Frage 9:
Wie ist die Prognose der unbehandelten primär biliären Zirrhose
(PBC)?
Sehr gut, Todesfälle nicht bekannt
Geht in 10 − 20 Jahren in eine komplette Zirrhose über
Entwickelt in 15 % ein hepatozelluläres Karzinom (HCC) ➤ Alis adignim quissequat
Die Lebertransplantation heilt die PBC vollständig aus
➤ Ut alit, commodi psuscipit wisis
Bei fulminantem Verlauf Letalität 60 %
Frage 10:
In welchem Krankheitsstadium ist die PBC konservativ am besten
zu therapieren ? ➤ Ut alit, commodi
Wenn außer AMA alle anderen Laborparameter noch normal sind
Im Stadium III, um den Übergang in die Zirrhose zu verhindern
In den entzündlich-progressiven Stadien I und II
Im Stadium IV durch Lebertransplantation
In allen Stadien gleich gute Erfolgsaussichten Abb. 1
Lit lor irit autatum in henit iurer ilit lam do-
lut lore faccum amcommy nos euis nullaor
suscip et, sumsan hent nit veliquatio eugia-
met laorpero do diam delit wisl eniam euisi
bla conse ting eugait vel irit, qui et velis alisi
etuero et, vel dolobore velit ip et nonsenitSie können auch lesen